
Inflammatory Cells in Atherosclerosis


Abstract
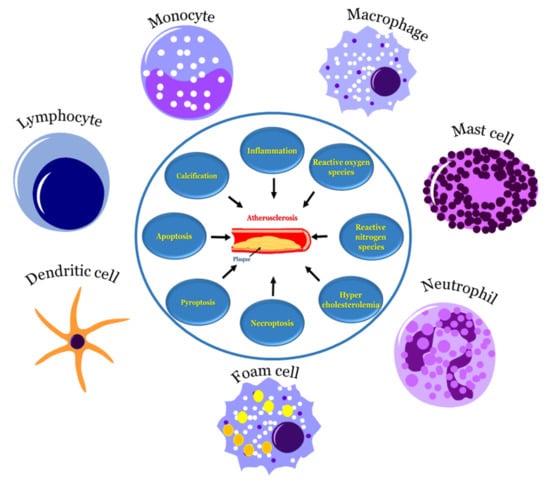
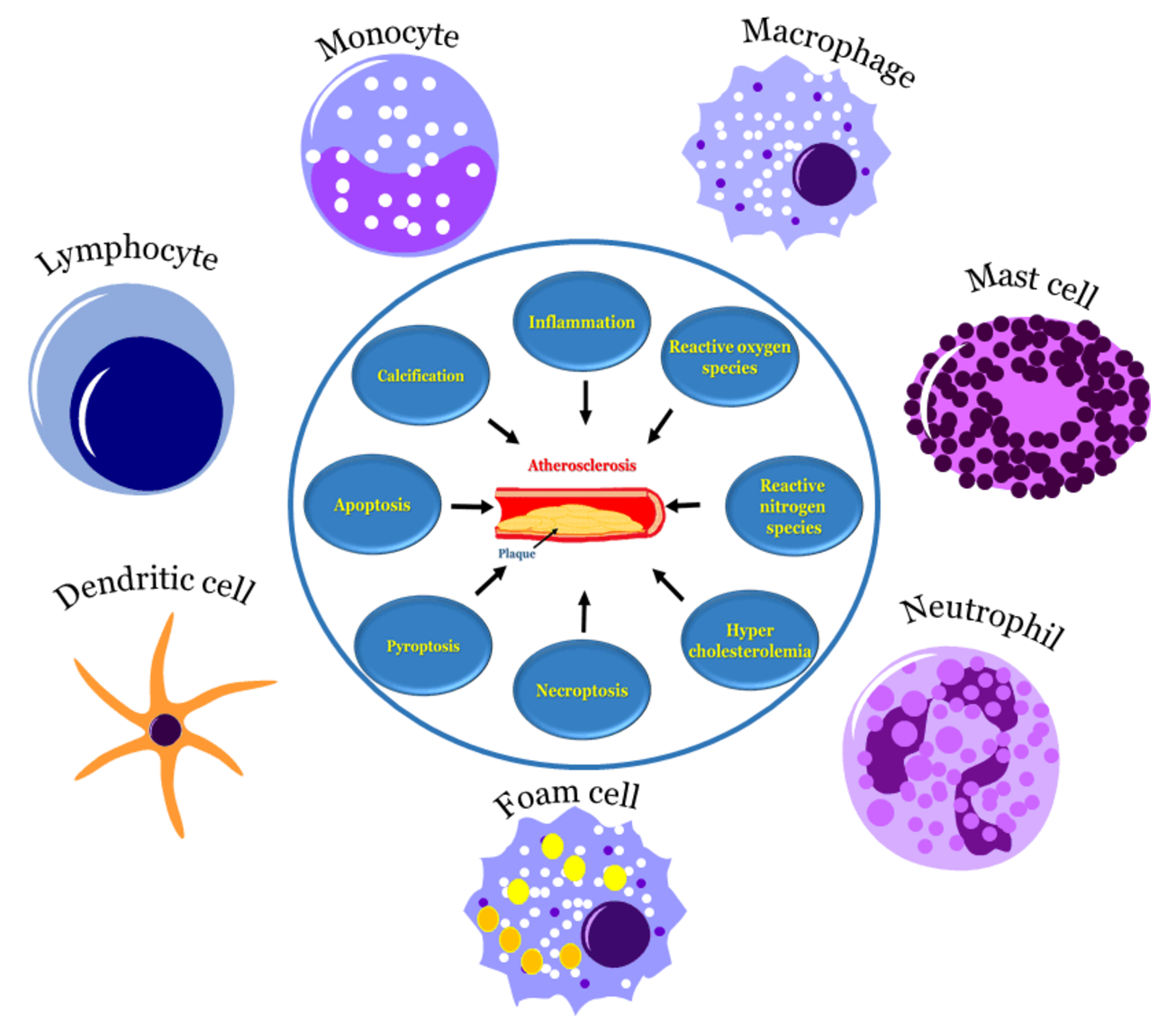
:
1. Introduction
2. Monocytes
3. Macrophages
4. Foam Cells
5. Lymphocytes
6. Neutrophils
7. Dendritic Cells
8. Mast Cells
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Glass, C.K.; Witztum, J.L. Atherosclerosis. The road ahead. Cell 2001, 104, 503–516. [Google Scholar] [CrossRef] [Green Version]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef]
- Hansson, G.K.; Robertson, A.K.; Söderberg-Nauclér, C. Inflammation and atherosclerosis. Annu. Rev. Pathol. 2006, 1, 297–329. [Google Scholar] [CrossRef] [PubMed]
- Rafieian-Kopaei, M.; Setorki, M.; Doudi, M.; Baradaran, A.; Nasri, H. Atherosclerosis: Process, indicators, risk factors and new hopes. Int. J. Prev. Med. 2014, 5, 927–946. [Google Scholar] [PubMed]
- Libby, P. The changing landscape of atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef]
- Libby, P. Inflammation during the life cycle of the atherosclerotic plaque. Cardiovasc. Res. 2021, 117, 2525–2536. [Google Scholar] [CrossRef] [PubMed]
- Saba, L.; Brinjikji, W.; Spence, J.D.; Wintermark, M.; Castillo, M.; Borst, G.J.D.; Yang, Q.; Yuan, C.; Buckler, A.; Edjlali, M.; et al. Roadmap Consensus on Carotid Artery Plaque Imaging and Impact on Therapy Strategies and Guidelines: An International, Multispecialty, Expert Review and Position Statement. Am. J. Neuroradiol. 2021, 42, 1566–1575. [Google Scholar] [CrossRef]
- Soehnlein, O.; Libby, P. Targeting inflammation in atherosclerosis—From experimental insights to the clinic. Nat. Rev. Drug Discov. 2021, 20, 589–610. [Google Scholar] [CrossRef]
- Ilhan, F.; Kalkanli, S.T. Atherosclerosis and the role of immune cells. World J. Clin. Cases 2015, 3, 345–352. [Google Scholar] [CrossRef]
- Hansson, G.K.; Hermansson, A. The immune system in atherosclerosis. Nat. Immunol. 2011, 12, 204–212. [Google Scholar] [CrossRef]
- Gerrity, R.G. The role of the monocyte in atherogenesis: I. Transition of blood-borne monocytes into foam cells in fatty lesions. Am. J. Pathol. 1981, 103, 181–190. [Google Scholar] [PubMed]
- Schaffner, T.; Taylor, K.; Bartucci, E.J.; Fischer-Dzoga, K.; Beeson, J.H.; Glagov, S.; Wissler, R.W. Arterial foam cells with distinctive immunomorphologic and histochemical features of macrophages. Am. J. Pathol. 1980, 100, 57–80. [Google Scholar] [PubMed]
- Steinberg, D.; Witztum, J.L. Lipoproteins and atherogenesis. Current concepts. Jama 1990, 264, 3047–3052. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. The pathogenesis of atherosclerosis: A perspective for the 1990s. Nature 1993, 362, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Current concepts of the pathogenesis of the acute coronary syndromes. Circulation 2001, 104, 365–372. [Google Scholar] [CrossRef] [Green Version]
- Virmani, R.; Ladich, E.R.; Burke, A.P.; Kolodgie, F.D. Histopathology of carotid atherosclerotic disease. Neurosurgery 2006, 59, S219–S227. [Google Scholar] [CrossRef]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef]
- Steinberg, D.; Lewis, A. Conner Memorial Lecture. Oxidative modification of LDL and atherogenesis. Circulation 1997, 95, 1062–1071. [Google Scholar] [CrossRef]
- Itabe, H. Oxidative modification of LDL: Its pathological role in atherosclerosis. Clin. Rev. Allergy Immunol. 2009, 37, 4–11. [Google Scholar] [CrossRef]
- Chisolm, G.M.; Steinberg, D. The oxidative modification hypothesis of atherogenesis: An overview. Free Radic. Biol. Med. 2000, 28, 1815–1826. [Google Scholar] [CrossRef]
- Matsuura, E.; Kobayashi, K.; Tabuchi, M.; Lopez, L.R. Oxidative modification of low-density lipoprotein and immune regulation of atherosclerosis. Prog. Lipid Res. 2006, 45, 466–486. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, S.; Raghavamenon, A.; Garelnabi, M.O.; Santanam, N. Oxidized low-density lipoprotein. Methods Mol. Biol. 2010, 610, 403–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riad, A.; Narasimhulu, C.A.; Deme, P.; Parthasarathy, S. A Novel Mechanism for Atherosclerotic Calcification: Potential Resolution of the Oxidation Paradox. Antioxid. Redox Signal. 2018, 29, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Porter, N.A. New insights regarding the autoxidation of polyunsaturated fatty acids. Antioxid. Redox Signal. 2005, 7, 170–184. [Google Scholar] [CrossRef]
- Bergt, C.; Pennathur, S.; Fu, X.; Byun, J.; O’Brien, K.; McDonald, T.O.; Singh, P.; Anantharamaiah, G.M.; Chait, A.; Brunzell, J.; et al. The myeloperoxidase product hypochlorous acid oxidizes HDL in the human artery wall and impairs ABCA1-dependent cholesterol transport. Proc. Natl. Acad. Sci. USA 2004, 101, 13032–13037. [Google Scholar] [CrossRef] [Green Version]
- Bergt, C.; Oram, J.F.; Heinecke, J.W. Oxidized HDL. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1488–1490. [Google Scholar] [CrossRef] [Green Version]
- Tabas, I.; Bornfeldt, K.E. Macrophage Phenotype and Function in Different Stages of Atherosclerosis. Circ. Res. 2016, 118, 653–667. [Google Scholar] [CrossRef] [Green Version]
- Poznyak, A.V.; Nikiforov, N.G.; Markin, A.M.; Kashirskikh, D.A.; Myasoedova, V.A.; Gerasimova, E.V.; Orekhov, A.N. Overview of OxLDL and Its Impact on Cardiovascular Health: Focus on Atherosclerosis. Front. Pharmacol. 2021, 11, 1–11. [Google Scholar] [CrossRef]
- Khatana, C.; Saini, N.K.; Chakrabarti, S.; Saini, V.; Sharma, A.; Saini, R.V.; Saini, A.K. Mechanistic Insights into the Oxidized Low-Density Lipoprotein-Induced Atherosclerosis. Oxid. Med. Cell. Longev. 2020, 2020, 5245308. [Google Scholar] [CrossRef]
- Borén, J.; Chapman, M.J.; Krauss, R.M.; Packard, C.J.; Bentzon, J.F.; Binder, C.J.; Daemen, M.J.; Demer, L.L.; Hegele, R.A.; Nicholls, S.J.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: Pathophysiological, genetic, and therapeutic insights: A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2020, 41, 2313–2330. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Goncalves, I.; Tengryd, C.; Nitulescu, M.; Persson, A.F.; To, F.; Bengtsson, E.; Volkov, P.; Orho-Melander, M.; Nilsson, J.; et al. Reduced oxidized LDL in T2D plaques is associated with a greater statin usage but not with future cardiovascular events. Cardiovasc. Diabetol. 2020, 19, 214. [Google Scholar] [CrossRef] [PubMed]
- Bays, H.E.; Taub, P.R.; Epstein, E.; Michos, E.D.; Ferraro, R.A.; Bailey, A.L.; Kelli, H.M.; Ferdinand, K.C.; Echols, M.R.; Weintraub, H.; et al. Ten things to know about ten cardiovascular disease risk factors. Am. J. Prev. Cardiol. 2021, 5, 100149. [Google Scholar] [CrossRef] [PubMed]
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update From the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef]
- Narasimhulu, C.A.; Litvinov, D.; Jiang, X.; Yang, Z.; Parthasarathy, S. Atherosclerosis: Oxidation Hypothesis. In Molecular Basis of Oxidative Stress; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013. [Google Scholar] [CrossRef]
- Skerfving, S.; Bergdahl, I.A. Chapter 43—Lead. In Handbook on the Toxicology of Metals, 4th ed.; Nordberg, G.F., Fowler, B.A., Nordberg, M., Eds.; Academic Press: San Diego, SD, USA, 2015; pp. 911–967. [Google Scholar] [CrossRef]
- Harari, F.; Barregard, L.; Östling, G.; Sallsten, G.; Hedblad, B.; Forsgard, N.; Borné, Y.; Fagerberg, B.; Engström, G. Blood Lead Levels and Risk of Atherosclerosis in the Carotid Artery: Results from a Swedish Cohort. Environ. Health Perspect. 2019, 127, 127002. [Google Scholar] [CrossRef]
- Lanphear, B.P.; Rauch, S.; Auinger, P.; Allen, R.W.; Hornung, R.W. Low-level lead exposure and mortality in US adults: A population-based cohort study. Lancet Public Health 2018, 3, e177–e184. [Google Scholar] [CrossRef]
- Obeng-Gyasi, E.; Armijos, R.X.; Weigel, M.M.; Filippelli, G.M.; Sayegh, M.A. Cardiovascular-Related Outcomes in U.S. Adults Exposed to Lead. Int. J. Environ. Res. Public Health 2018, 15, 759. [Google Scholar] [CrossRef] [Green Version]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef]
- Hansson, G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef] [Green Version]
- Bobryshev, Y.V. Transdifferentiation of smooth muscle cells into chondrocytes in atherosclerotic arteries in situ: Implications for diffuse intimal calcification. J. Pathol. 2005, 205, 641–650. [Google Scholar] [CrossRef]
- Oh, E.S.; Na, M.; Rogers, C.J. The Association Between Monocyte Subsets and Cardiometabolic Disorders/Cardiovascular Disease: A Systematic Review and Meta-Analysis. Front. Cardiovasc. Med. 2021, 8, 640124. [Google Scholar] [CrossRef] [PubMed]
- Tahir, S.; Steffens, S. Nonclassical monocytes in cardiovascular physiology and disease. Am. J. Physiol.-Cell Physiol. 2021, 320, C761–C770. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.; Mack, C.D.; Li, S.C.H.; Fletcher, J.P.; Medbury, H.J. Nature versus Number: Monocytes in Cardiovascular Disease. Int. J. Mol. Sci. 2021, 22, 9119. [Google Scholar] [CrossRef] [PubMed]
- Mehta, N.N.; Reilly, M.P. Monocyte Mayhem. Circ. Cardiovasc. Genet. 2012, 5, 7–9. [Google Scholar] [CrossRef]
- Ghattas, A.; Griffiths, H.R.; Devitt, A.; Lip, G.Y.; Shantsila, E. Monocytes in coronary artery disease and atherosclerosis: Where are we now? J. Am. Coll. Cardiol. 2013, 62, 1541–1551. [Google Scholar] [CrossRef] [Green Version]
- Ley, K.; Miller, Y.I.; Hedrick, C.C. Monocyte and macrophage dynamics during atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1506–1516. [Google Scholar] [CrossRef] [Green Version]
- Badimon, L.; Padró, T.; Vilahur, G. Atherosclerosis, platelets and thrombosis in acute ischaemic heart disease. Eur. Heart J. Acute Cardiovasc. Care 2012, 1, 60–74. [Google Scholar] [CrossRef]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Inflammation in atherosclerosis: From pathophysiology to practice. J. Am. Coll. Cardiol. 2009, 54, 2129–2138. [Google Scholar] [CrossRef] [Green Version]
- Libby, P.; Lichtman, A.H.; Hansson, G.K. Immune effector mechanisms implicated in atherosclerosis: From mice to humans. Immunity 2013, 38, 1092–1104. [Google Scholar] [CrossRef] [Green Version]
- Sampath, P.; Moideen, K.; Ranganathan, U.D.; Bethunaickan, R. Monocyte Subsets: Phenotypes and Function in Tuberculosis Infection. Front. Immunol. 2018, 9, 1726. [Google Scholar] [CrossRef] [Green Version]
- Tacke, F.; Alvarez, D.; Kaplan, T.J.; Jakubzick, C.; Spanbroek, R.; Llodra, J.; Garin, A.; Liu, J.; Mack, M.; van Rooijen, N.; et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J. Clin. Investig. 2007, 117, 185–194. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.W.; Ivanov, S.; Williams, J.W. Monocyte Recruitment, Specification, and Function in Atherosclerosis. Cells 2020, 10, 15. [Google Scholar] [CrossRef] [PubMed]
- Auffray, C.; Fogg, D.; Garfa, M.; Elain, G.; Join-Lambert, O.; Kayal, S.; Sarnacki, S.; Cumano, A.; Lauvau, G.; Geissmann, F. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science 2007, 317, 666–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintar, A.; McArdle, S.; Wolf, D.; Marki, A.; Ehinger, E.; Vassallo, M.; Miller, J.; Mikulski, Z.; Ley, K.; Buscher, K. Endothelial Protective Monocyte Patrolling in Large Arteries Intensified by Western Diet and Atherosclerosis. Circ. Res. 2017, 120, 1789–1799. [Google Scholar] [CrossRef]
- Williams, J.W.; Randolph, G.J.; Zinselmeyer, B.H. A Polecat’s View of Patrolling Monocytes. Circ. Res. 2017, 120, 1699–1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, R.N.; Carlin, L.M.; Hubbeling, H.G.; Nackiewicz, D.; Green, A.M.; Punt, J.A.; Geissmann, F.; Hedrick, C.C. The transcription factor NR4A1 (Nur77) controls bone marrow differentiation and the survival of Ly6C-monocytes. Nat. Immunol. 2011, 12, 778–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Bai, Y.; Zhao, X.; Tian, T.; Tang, L.; Ru, J.; An, Y.; Wang, J. NR4A1 contributes to high-fat associated endothelial dysfunction by promoting CaMKII-Parkin-mitophagy pathways. Cell Stress Chaperones 2018, 23, 749–761. [Google Scholar] [CrossRef]
- Marcovecchio, P.M.; Zhu, Y.P.; Hanna, R.N.; Dinh, H.Q.; Tacke, R.; Wu, R.; McArdle, S.; Reynolds, S.; Araujo, D.J.; Ley, K.; et al. Frontline Science: Kindlin-3 is essential for patrolling and phagocytosis functions of nonclassical monocytes during metastatic cancer surveillance. J. Leukoc. Biol. 2020, 107, 883–892. [Google Scholar] [CrossRef]
- Liu, Y.; Reynolds, L.M.; Ding, J.; Hou, L.; Lohman, K.; Young, T.; Cui, W.; Huang, Z.; Grenier, C.; Wan, M.; et al. Blood monocyte transcriptome and epigenome analyses reveal loci associated with human atherosclerosis. Nat. Commun. 2017, 8, 393. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Qian, N.; Xu, J.; Wang, Y. The Roles of Macrophages in Heart Regeneration and Repair After Injury. Front. Cardiovasc. Med. 2021, 8, 1–10. [Google Scholar] [CrossRef]
- Nasser, M.I.; Zhu, S.; Huang, H.; Zhao, M.; Wang, B.; Ping, H.; Geng, Q.; Zhu, P. Macrophages: First guards in the prevention of cardiovascular diseases. Life Sci. 2020, 250, 117559. [Google Scholar] [CrossRef] [PubMed]
- Khoury, M.K.; Yang, H.; Liu, B. Macrophage Biology in Cardiovascular Diseases. Arterioscler. Thromb. Vasc. Biol. 2021, 41, e77–e81. [Google Scholar] [CrossRef] [PubMed]
- Chinetti-Gbaguidi, G.; Colin, S.; Staels, B. Macrophage subsets in atherosclerosis. Nat. Rev. Cardiol. 2015, 12, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef] [Green Version]
- Winkels, H.; Ehinger, E.; Vassallo, M.; Buscher, K.; Dinh, H.Q.; Kobiyama, K.; Hamers, A.A.J.; Cochain, C.; Vafadarnejad, E.; Saliba, A.-E.; et al. Atlas of the Immune Cell Repertoire in Mouse Atherosclerosis Defined by Single-Cell RNA-Sequencing and Mass Cytometry. Circ. Res. 2018, 122, 1675–1688. [Google Scholar] [CrossRef]
- Kim, K.; Shim, D.; Lee, J.S.; Zaitsev, K.; Williams, J.W.; Kim, K.-W.; Jang, M.-Y.; Jang, H.S.; Yun, T.J.; Lee, S.H.; et al. Transcriptome Analysis Reveals Nonfoamy Rather Than Foamy Plaque Macrophages Are Proinflammatory in Atherosclerotic Murine Models. Circ. Res. 2018, 123, 1127–1142. [Google Scholar] [CrossRef]
- Cochain, C.; Vafadarnejad, E.; Arampatzi, P.; Pelisek, J.; Winkels, H.; Ley, K.; Wolf, D.; Saliba, A.E.; Zernecke, A. Single-Cell RNA-Seq Reveals the Transcriptional Landscape and Heterogeneity of Aortic Macrophages in Murine Atherosclerosis. Circ. Res. 2018, 122, 1661–1674. [Google Scholar] [CrossRef]
- Cole, J.E.; Park, I.; Ahern, D.J.; Kassiteridi, C.; Danso Abeam, D.; Goddard, M.E.; Green, P.; Maffia, P.; Monaco, C. Immune cell census in murine atherosclerosis: Cytometry by time of flight illuminates vascular myeloid cell diversity. Cardiovasc. Res. 2018, 114, 1360–1371. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.D.; Nishi, H.; Poles, J.; Niu, X.; McCauley, C.; Rahman, K.; Brown, E.J.; Yeung, S.T.; Vozhilla, N.; Weinstock, A.; et al. Single-cell analysis of fate-mapped macrophages reveals heterogeneity, including stem-like properties, during atherosclerosis progression and regression. JCI Insight 2019, 4, e124574. [Google Scholar] [CrossRef] [Green Version]
- Willemsen, L.; de Winther, M.P. Macrophage subsets in atherosclerosis as defined by single-cell technologies. J. Pathol. 2020, 250, 705–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, M.; Zhuo, X.; Ma, A. STAT6 Upregulation Promotes M2 Macrophage Polarization to Suppress Atherosclerosis. Med. Sci. Monit. Basic Res. 2017, 23, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Xia, X.D.; Gu, H.M.; Zhang, D.W. Proprotein Convertase Subtilisin/Kexin-Type 9 and Lipid Metabolism. Adv. Exp. Med. Biol. 2020, 1276, 137–156. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Melnichenko, A.A.; Myasoedova, V.A.; Grechko, A.V.; Orekhov, A.N. Mechanisms of foam cell formation in atherosclerosis. J. Mol. Med. 2017, 95, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Javadifar, A.; Rastgoo, S.; Banach, M.; Jamialahmadi, T.; Johnston, T.P.; Sahebkar, A. Foam Cells as Therapeutic Targets in Atherosclerosis with a Focus on the Regulatory Roles of Non-Coding RNAs. Int. J. Mol. Sci. 2021, 22, 2529. [Google Scholar] [CrossRef]
- Allahverdian, S.; Pannu, P.S.; Francis, G.A. Contribution of monocyte-derived macrophages and smooth muscle cells to arterial foam cell formation. Cardiovasc. Res. 2012, 95, 165–172. [Google Scholar] [CrossRef] [Green Version]
- Aluganti Narasimhulu, C.; Fernandez-Ruiz, I.; Selvarajan, K.; Jiang, X.; Sengupta, B.; Riad, A.; Parthasarathy, S. Atherosclerosis—Do we know enough already to prevent it? Curr. Opin. Pharmacol. 2016, 27, 92–102. [Google Scholar] [CrossRef]
- Wang, Y.; Dubland, J.A.; Allahverdian, S.; Asonye, E.; Sahin, B.; Jaw, J.E.; Sin, D.D.; Seidman, M.A.; Leeper, N.J.; Francis, G.A. Smooth Muscle Cells Contribute the Majority of Foam Cells in ApoE (Apolipoprotein E)-Deficient Mouse Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 876–887. [Google Scholar] [CrossRef]
- Wang, D.; Yang, Y.; Lei, Y.; Tzvetkov, N.T.; Liu, X.; Yeung, A.W.K.; Xu, S.; Atanasov, A.G. Targeting Foam Cell Formation in Atherosclerosis: Therapeutic Potential of Natural Products. Pharmacol. Rev. 2019, 71, 596–670. [Google Scholar] [CrossRef]
- Nus, M.; Tsiantoulas, D.; Mallat, Z. Plan B (-cell) in atherosclerosis. Eur. J. Pharmacol. 2017, 816, 76–81. [Google Scholar] [CrossRef]
- Ammirati, E.; Moroni, F.; Magnoni, M.; Camici, P.G. The role of T and B cells in human atherosclerosis and atherothrombosis. Clin. Exp. Immunol. 2015, 179, 173–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pattarabanjird, T.; Li, C.; McNamara, C. B Cells in Atherosclerosis. JACC Basic Transl. Sci. 2021, 6, 546–563. [Google Scholar] [CrossRef] [PubMed]
- Poznyak, A.V.; Bezsonov, E.E.; Popkova, T.V.; Starodubova, A.V.; Orekhov, A.N. Immunity in Atherosclerosis: Focusing on T and B Cells. Int. J. Mol. Sci. 2021, 22, 8379. [Google Scholar] [CrossRef] [PubMed]
- Adamo, L.; Rocha-Resende, C.; Mann, D.L. The Emerging Role of B Lymphocytes in Cardiovascular Disease. Annu. Rev. Immunol. 2020, 38, 99–121. [Google Scholar] [CrossRef]
- Engelbertsen, D.; Lichtman, A.H. Innate lymphoid cells in atherosclerosis. Eur. J. Pharmacol. 2017, 816, 32–36. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Santos-Zas, I.; Lemarié, J.; Zlatanova, I.; Cachanado, M.; Seghezzi, J.-C.; Benamer, H.; Goube, P.; Vandestienne, M.; Cohen, R.; Ezzo, M.; et al. Cytotoxic CD8+ T cells promote granzyme B-dependent adverse post-ischemic cardiac remodeling. Nat. Commun. 2021, 12, 1483. [Google Scholar] [CrossRef]
- Wigren, M.; Nilsson, J.; Kolbus, D. Lymphocytes in atherosclerosis. Clin. Chim. Acta 2012, 413, 1562–1568. [Google Scholar] [CrossRef] [Green Version]
- Available online: www.rndsystems.com/resources/cell-markers/immune- (accessed on 9 February 2019).
- Butcher, M.J.; Gjurich, B.N.; Phillips, T.; Galkina, E.V. The IL-17A/IL-17RA axis plays a proatherogenic role via the regulation of aortic myeloid cell recruitment. Circ. Res. 2012, 110, 675–687. [Google Scholar] [CrossRef] [Green Version]
- Taleb, S.; Romain, M.; Ramkhelawon, B.; Uyttenhove, C.; Pasterkamp, G.; Herbin, O.; Esposito, B.; Perez, N.; Yasukawa, H.; Van Snick, J.; et al. Loss of SOCS3 expression in T cells reveals a regulatory role for interleukin-17 in atherosclerosis. J. Exp. Med. 2009, 206, 2067–2077. [Google Scholar] [CrossRef] [Green Version]
- Saigusa, R.; Winkels, H.; Ley, K. T cell subsets and functions in atherosclerosis. Nat. Rev. Cardiol. 2020, 17, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Döring, Y.; Drechsler, M.; Soehnlein, O.; Weber, C. Neutrophils in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 288–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trtica Majnarić, L.; Guljaš, S.; Bosnić, Z.; Šerić, V.; Wittlinger, T. Neutrophil-to-Lymphocyte Ratio as a Cardiovascular Risk Marker May Be Less Efficient in Women Than in Men. Biomolecules 2021, 11, 528. [Google Scholar] [CrossRef] [PubMed]
- Lacy, P. Mechanisms of degranulation in neutrophils. Allergy Asthma Clin. Immunol. 2006, 2, 98–108. [Google Scholar] [CrossRef] [Green Version]
- Soehnlein, O. Multiple roles for neutrophils in atherosclerosis. Circ. Res. 2012, 110, 875–888. [Google Scholar] [CrossRef] [Green Version]
- Meng, L.B.; Yu, Z.M.; Guo, P.; Wang, Q.Q.; Qi, R.M.; Shan, M.J.; Lv, J.; Gong, T. Neutrophils and neutrophil-lymphocyte ratio: Inflammatory markers associated with intimal-media thickness of atherosclerosis. Thromb. Res. 2018, 170, 45–52. [Google Scholar] [CrossRef] [Green Version]
- Sanda, G.E.; Belur, A.D.; Teague, H.L.; Mehta, N.N. Emerging Associations Between Neutrophils, Atherosclerosis, and Psoriasis. Curr. Atheroscler. Rep. 2017, 19, 53. [Google Scholar] [CrossRef]
- Silvestre-Roig, C.; Braster, Q.; Wichapong, K.; Lee, E.Y.; Teulon, J.M.; Berrebeh, N.; Winter, J.; Adrover, J.M.; Santos, G.S.; Froese, A.; et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature 2019, 569, 236–240. [Google Scholar] [CrossRef]
- Gomez, I.; Ward, B.; Souilhol, C.; Recarti, C.; Ariaans, M.; Johnston, J.; Burnett, A.; Mahmoud, M.; Luong, L.A.; West, L.; et al. Neutrophil microvesicles drive atherosclerosis by delivering miR-155 to atheroprone endothelium. Nat. Commun. 2020, 11, 214. [Google Scholar] [CrossRef] [Green Version]
- Geng, S.; Zhang, Y.; Lee, C.; Li, L. Novel reprogramming of neutrophils modulates inflammation resolution during atherosclerosis. Sci. Adv. 2019, 5, eaav2309. [Google Scholar] [CrossRef] [Green Version]
- Zernecke, A. Dendritic Cells in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 763–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, C.; Zernecke, A.; Libby, P. The multifaceted contributions of leukocyte subsets to atherosclerosis: Lessons from mouse models. Nat. Rev. Immunol. 2008, 8, 802–815. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, M.; Tabas, I. Dendritic cells in atherosclerosis. Semin. Immunopathol. 2014, 36, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Christ, A.; Temmerman, L.; Legein, B.; Daemen, M.J.A.P.; Biessen, E.A.L. Dendritic Cells in Cardiovascular Diseases. Circulation 2013, 128, 2603–2613. [Google Scholar] [CrossRef] [Green Version]
- Van Vré, E.A.; Van Brussel, I.; Bosmans, J.M.; Vrints, C.J.; Bult, H. Dendritic cells in human atherosclerosis: From circulation to atherosclerotic plaques. Mediat. Inflamm. 2011, 2011, 941396. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Zhang, J.; Zhang, W.; Xu, Y. A myriad of roles of dendritic cells in atherosclerosis. Clin. Exp. Immunol. 2021, 206, 12–27. [Google Scholar] [CrossRef]
- Kovanen, P.T.; Bot, I. Mast cells in atherosclerotic cardiovascular disease—Activators and actions. Eur. J. Pharmacol. 2017, 816, 37–46. [Google Scholar] [CrossRef]
- Conti, P.; Shaik-Dasthagirisaeb, Y. Atherosclerosis: A chronic inflammatory disease mediated by mast cells. Cent. Eur. J. Immunol. 2015, 40, 380–386. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, J.B.; Harlan, C.W.; Harlan, G.C.; Virmani, R. The association of mast cells and atherosclerosis: A morphologic study of early atherosclerotic lesions in young people. Hum. Pathol. 1994, 25, 154–159. [Google Scholar] [CrossRef]
- Kaartinen, M.; Penttilä, A.; Kovanen, P.T. Mast cells of two types differing in neutral protease composition in the human aortic intima. Demonstration of tryptase- and tryptase/chymase-containing mast cells in normal intimas, fatty streaks, and the shoulder region of atheromas. Arterioscler. Thromb. 1994, 14, 966–972. [Google Scholar] [CrossRef] [Green Version]
- Kovanen, P.T. Mast Cells as Potential Accelerators of Human Atherosclerosis-From Early to Late Lesions. Int. J. Mol. Sci. 2019, 20, 4479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patella, V.; Marinò, I.; Lampärter, B.; Arbustini, E.; Adt, M.; Marone, G. Human heart mast cells. Isolation, purification, ultrastructure, and immunologic characterization. J. Immunol. 1995, 154, 2855–2865. [Google Scholar] [PubMed]
- Kritikou, E.; Depuydt, M.A.C.; de Vries, M.R.; Mulder, K.E.; Govaert, A.M.; Smit, M.D.; van Duijn, J.; Foks, A.C.; Wezel, A.; Smeets, H.J.; et al. Flow Cytometry-Based Characterization of Mast Cells in Human Atherosclerosis. Cells 2019, 8, 834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pussinen, P.J.; Paju, S.; Koponen, J.; Viikari, J.S.A.; Taittonen, L.; Laitinen, T.; Burgner, D.P.; Kähönen, M.; Hutri-Kähönen, N.; Raitakari, O.T.; et al. Association of Childhood Oral Infections With Cardiovascular Risk Factors and Subclinical Atherosclerosis in Adulthood. JAMA Netw. Open 2019, 2, e192523. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Hazen, S.L. The contributory role of gut microbiota in cardiovascular disease. J. Clin. Investig. 2014, 124, 4204–4211. [Google Scholar] [CrossRef]
- Gupta, K.; Idahosa, C.; Roy, S.; Lee, D.; Subramanian, H.; Dhingra, A.; Boesze-Battaglia, K.; Korostoff, J.; Ali, H. Differential Regulation of Mas-Related G Protein-Coupled Receptor X2-Mediated Mast Cell Degranulation by Antimicrobial Host Defense Peptides and Porphyromonas gingivalis Lipopolysaccharide. Infect. Immun. 2017, 85, e00246-17. [Google Scholar] [CrossRef] [Green Version]
- Lassila, R.; Lindstedt, K.; Kovanen, P.T. Native macromolecular heparin proteoglycans exocytosed from stimulated rat serosal mast cells strongly inhibit platelet-collagen interactions. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 3578–3587. [Google Scholar] [CrossRef]
- Lassila, R.; Jouppila, A. Mast cell-derived heparin proteoglycans as a model for a local antithrombotic. Semin. Thromb. Hemost. 2014, 40, 837–844. [Google Scholar] [CrossRef] [Green Version]

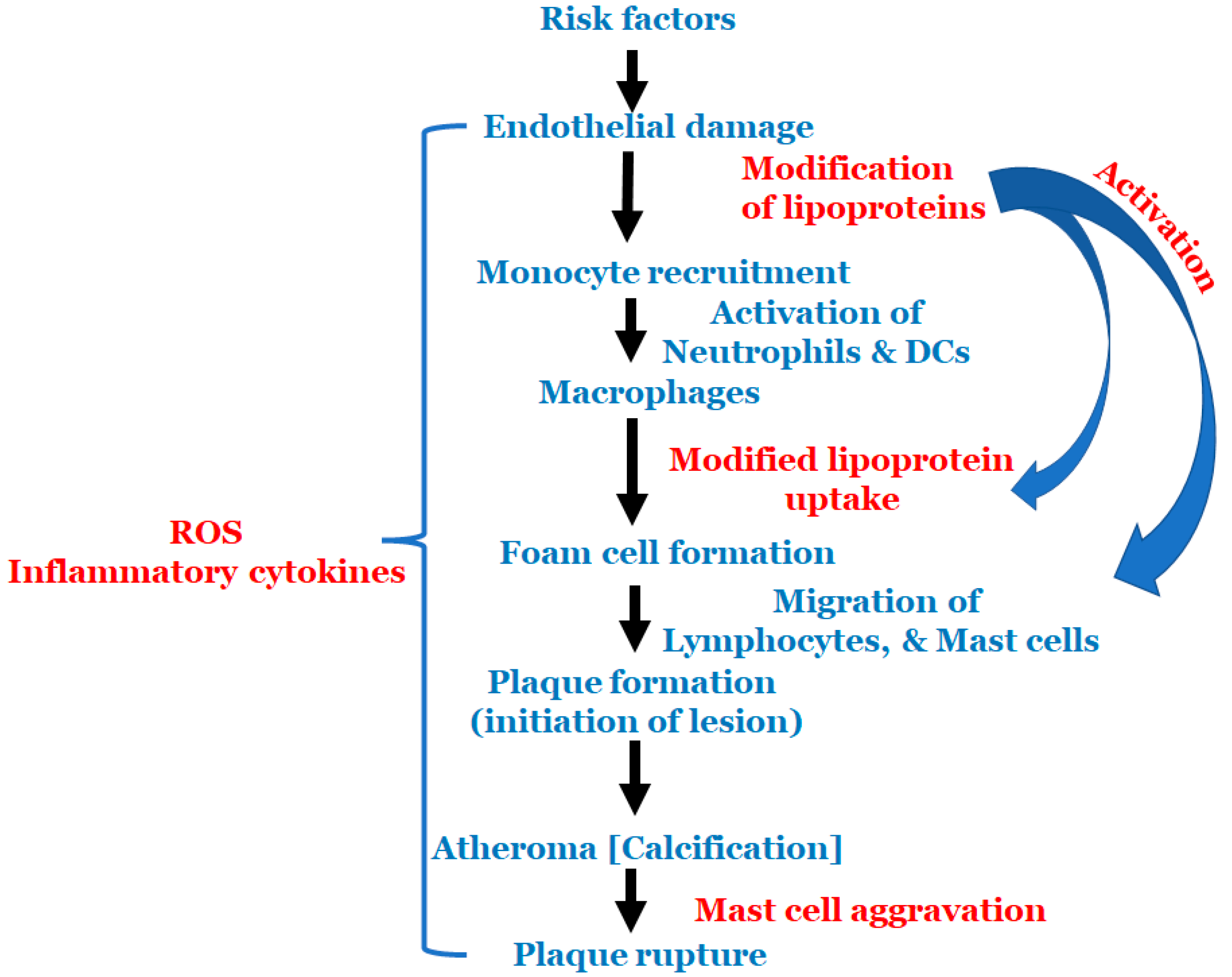

{kind=link}
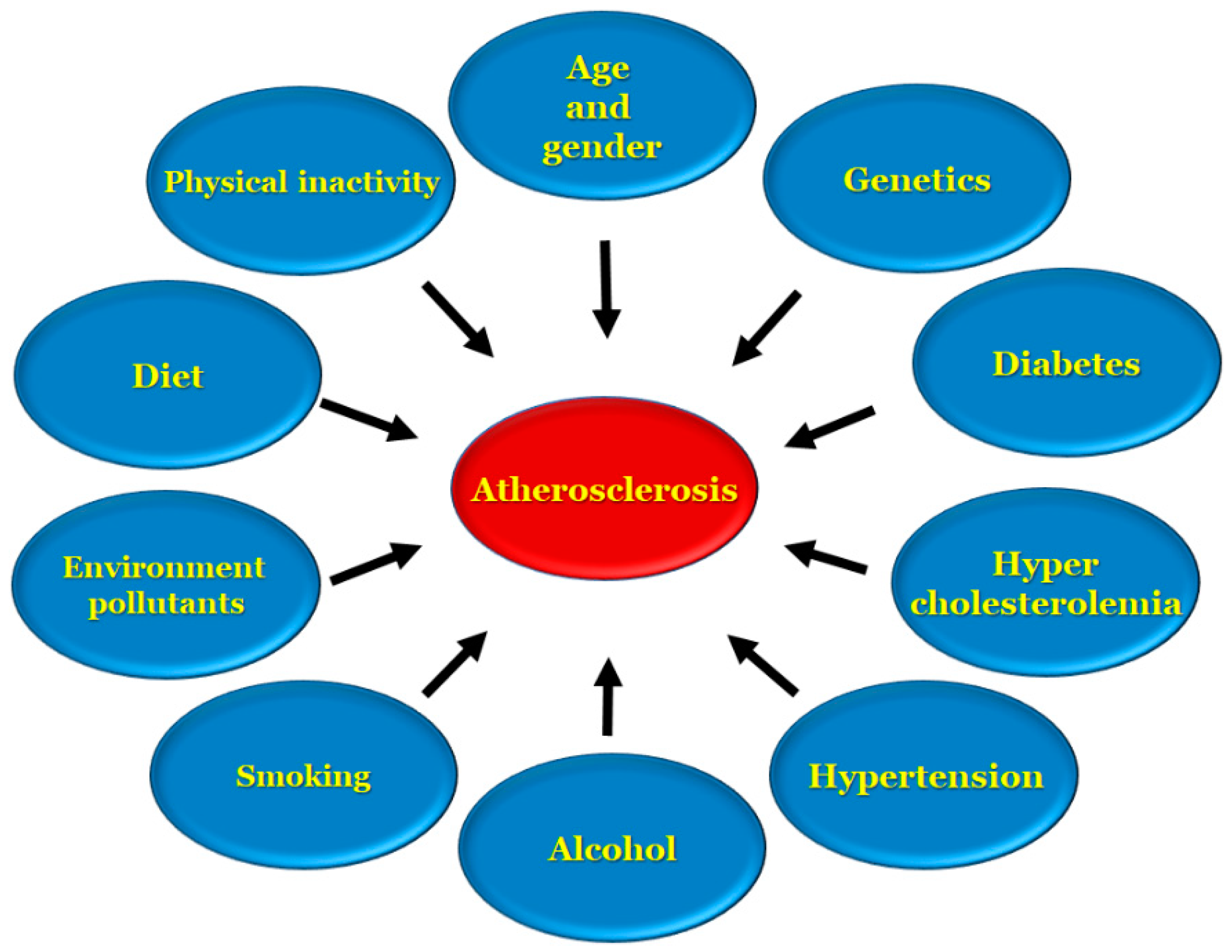{kind=link}
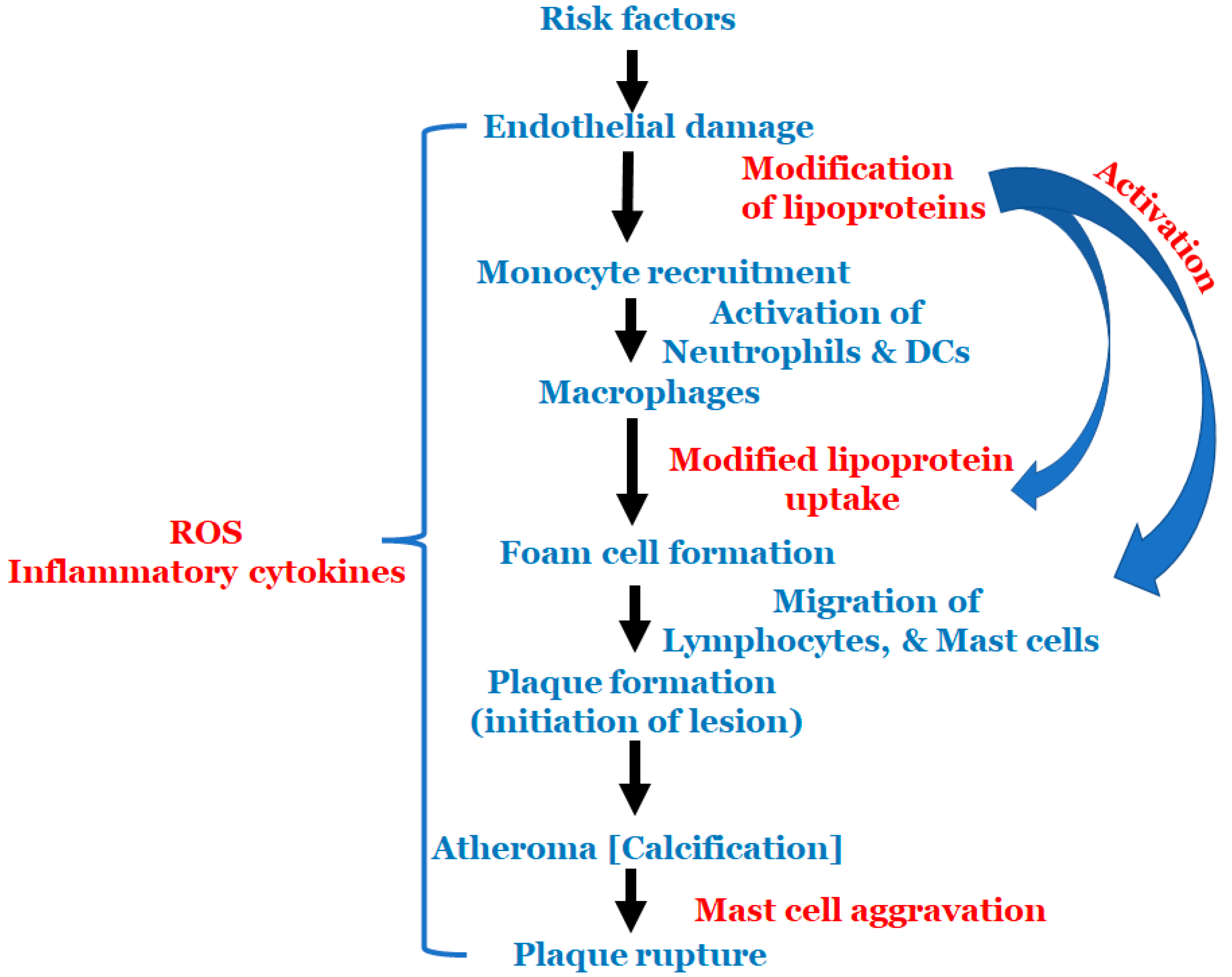{kind=link}
| Monocyte Type | Human Markers | Mice Markers | Properties and Function | References |
|---|---|---|---|---|
| Classical (Mon 1) | CD14++ and CD16− | Ly6C++ and CD43+ | Highly phagocytic and vital scavenger cells | [46,47] |
| Intermediate (Mon 2) | CD14++ and CD16+ | Ly6Cint and CD43+ | Endothelial adherence Antigen presentation Major role in proliferation and stimulation of T cells, inflammatory responses and angiogenesis Pro-inflammatory in nature and secretes inflammatory cytokines | |
| Non-classical (Mon 3) | CD14+ and CD16++ | Ly6C+ and CD43++ | Very mobile Monitors endothelial injury Anti-inflammatory in nature |
| Macrophage Type | Stimulus for Differentiation | Role in Inflammation | Mice Markers | Human Markers | References |
|---|---|---|---|---|---|
| M1 | Ox-LDL, proinflammatory cytokines, lipopolysaccharides | Proinflammatory | IL-1β, TNF, IL-6, IL-12, IL-23, CXCL9, CXCL10, CXCL11, arginase II | IL-1β, TNF, IL-6, IL-12, IL-23, CXCL9, CXCL10, CXCL11, arginase II | [47,62,64,65] |
| M2 (a,b,c) | IL-4, IL-10, IL-1β | Anti-inflammatory, resistant to lipid accumulation | Arginase I, resistin-like α, Ym1, Ym2, MMGL, stabilin-1, CD163, IL-10high, IL-12low | MMR, IL-1RA, factor XIIIa, CD200R, CCL18, stabilin-1, CD163, IL-10high, IL-12low | |
| Mox | Oxidized phospholipids | Antioxidant | HO-1 (heme oxygenase sulfiredoxin-1), TR, NFE2L2 (nuclear factor (erythroid-derived 2)-like 2) | HO-1, sulfiredoxin-1, TR, NFE2L2 | |
| M4 | CXCL4 (C-X-C motif chemokine 4) | Proinflammatory, reduces phagocytosis | MMP-7, S100-A8, MMR (macrophage mannose receptor) | MMP-7, S100-A8, MMR |
| Lymphocyte Type | Human Markers | Mice Markers | Characteristics | References |
|---|---|---|---|---|
| B1 cells | High levels of IgM | High levels of IgM | Active in innate immunity housed in the peritoneal and pleural cavities | |
| low levels of IgD, detectable levels of CD5 | Low levels of IgD, detectable levels of CD5 | [88,89,90,91] | ||
| B2, follicular B cells (FOB) | CD10, CD19+ CD20, CD21mid, CD22, CD23, CD24low | CD1dlow, CD19mid, CD21mid, CD22, CD23, CD24low, CD38+ | Housed in the spleen and lymph nodes Participate in T-cell-dependent immune responses | |
| B2, marginal zone B cells (MZB) | CD1c, CD19+, CD20, CD21high, CD27+, IgM+ | CD1dhigh, CD19mid, CD21high, CD22, CD23, CD35+, CD43− | Housed in the spleen and lymph nodes Active in early immune response and can uptake ox-LDL | |
| T cells,Th1 | CCR1+, CCR5+, CD3+, CD4+, CD8−, CD14−, CD19−, CXCR3+ | CCR1+, CCR5+, CD3+, CD4+, CD8−, CD14−, CD19−, CXCR3+ | Promote lesion destabilization Alter endothelial function Most abundant type at lesion sites | |
| T cells,Th2 | CCR3+, CCR4+, CCR8+, CD3+ CD4+, CD8−, CD14+, CD19+ | CCR3+, CCR4+, CCR8+, CD3+, CD4+, CD8−, CD14+, CD19+ | Inhibit TH1 differentiation and promote the survival and proliferation of mast cells |
| Mice Markers | Human Makers | Function | References | |
|---|---|---|---|---|
| Neutrophils | Lin-, CD11b, CD45+, Ly-6C | CD11b, CD14low/int, CD15+, CD16, CD32 | Central role in innate immunity by destroying foreign particles Ratio of neutrophils to lymphocytes acts as an atherosclerosis progression tool | [91,95,97,98,99] |
| Types of Dendritic Cells | Mice Markers | Human Marker | Characteristics | References |
|---|---|---|---|---|
| Classical, cDC1 | CD8+CD103+, CD11c, CD45, CD40, CD83, CX3CR1 | CD141/BDCA-3+, CD11c, CD14, CCR7 | Activate CD8+ T cells | [90,91,104] |
| Classical, cDC2 | CD11b+, CD11c, CD45, CD40, CD83, CX3CR1 | CD1c/BDCA-1+, CD11c, CD14, CCR7 | Promote Th2/Th17-mediated immune responses | |
| Plasmacytoid | CD1a-, CD11clow, Lin-, IL-3 R alpha, CD123+ | Lin-, CD11c+, Ly-6C+ | Regulate MHCI/MHCII to activate naïve CD4+ T cells |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehu, M.; Narasimhulu, C.A.; Singla, D.K. Inflammatory Cells in Atherosclerosis. Antioxidants 2022, 11, 233. https://doi.org/10.3390/antiox11020233
Mehu M, Narasimhulu CA, Singla DK. Inflammatory Cells in Atherosclerosis. Antioxidants. 2022; 11(2):233. https://doi.org/10.3390/antiox11020233
Chicago/Turabian StyleMehu, Marcelle, Chandrakala Aluganti Narasimhulu, and Dinender K. Singla. 2022. "Inflammatory Cells in Atherosclerosis" Antioxidants 11, no. 2: 233. https://doi.org/10.3390/antiox11020233
APA StyleMehu, M., Narasimhulu, C. A., & Singla, D. K. (2022). Inflammatory Cells in Atherosclerosis. Antioxidants, 11(2), 233. https://doi.org/10.3390/antiox11020233