
The Full-Length Transcriptome Sequencing and Identification of Na+/H+ Antiporter Genes in Halophyte Nitraria tangutorum Bobrov

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. RNA Extraction, Library Construction and SMRT Sequencing
2.3. SSR Analysis
2.4. CDS, TF, and lncRNA Analysis
2.5. Gene Functional Annotation
2.6. Identification and Multi-Segment Alignments of Na+/H+ Antiporter Genes
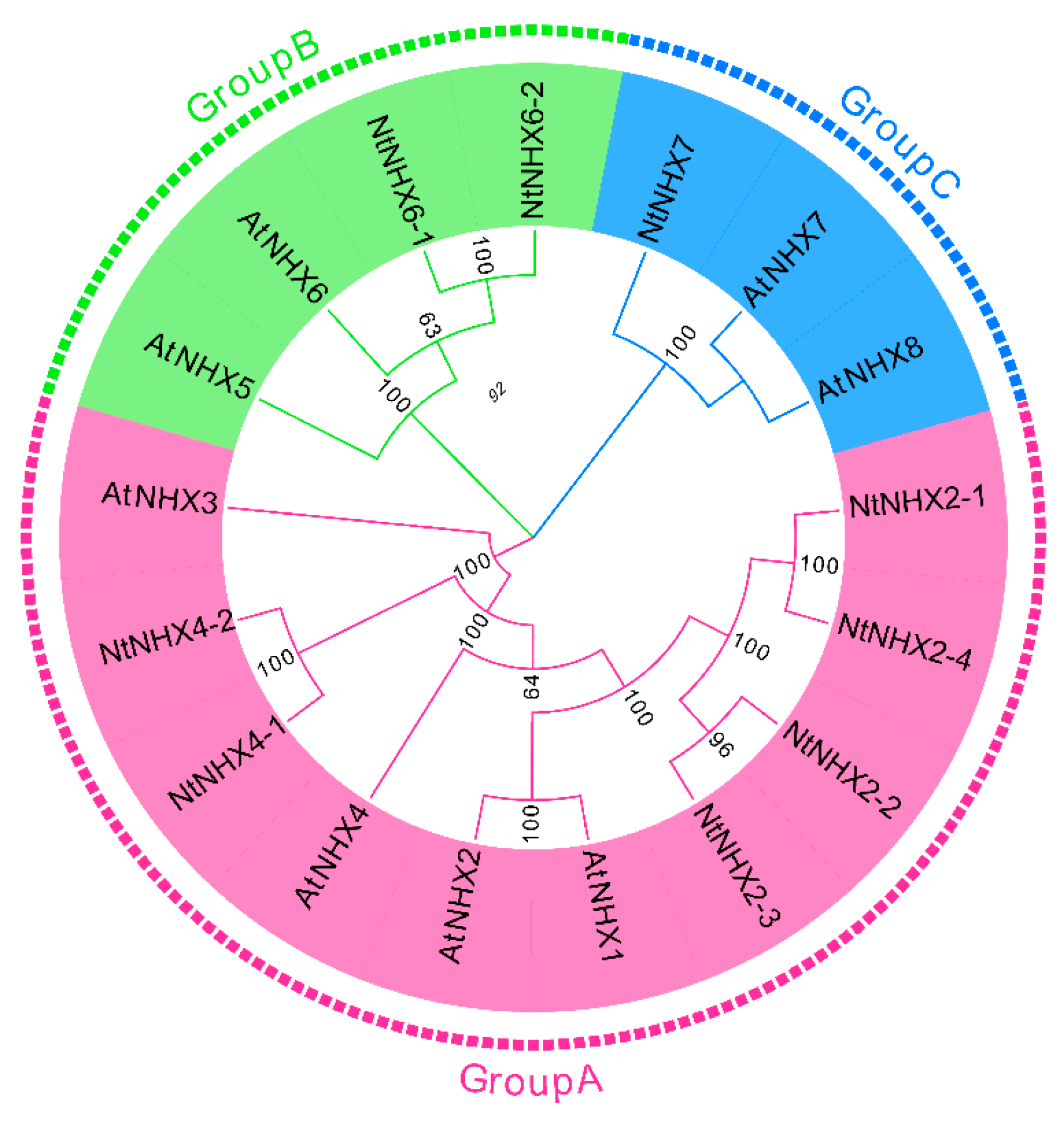
2.7. Phylogenetic and Motif Analyses
2.8. Gene Cloning
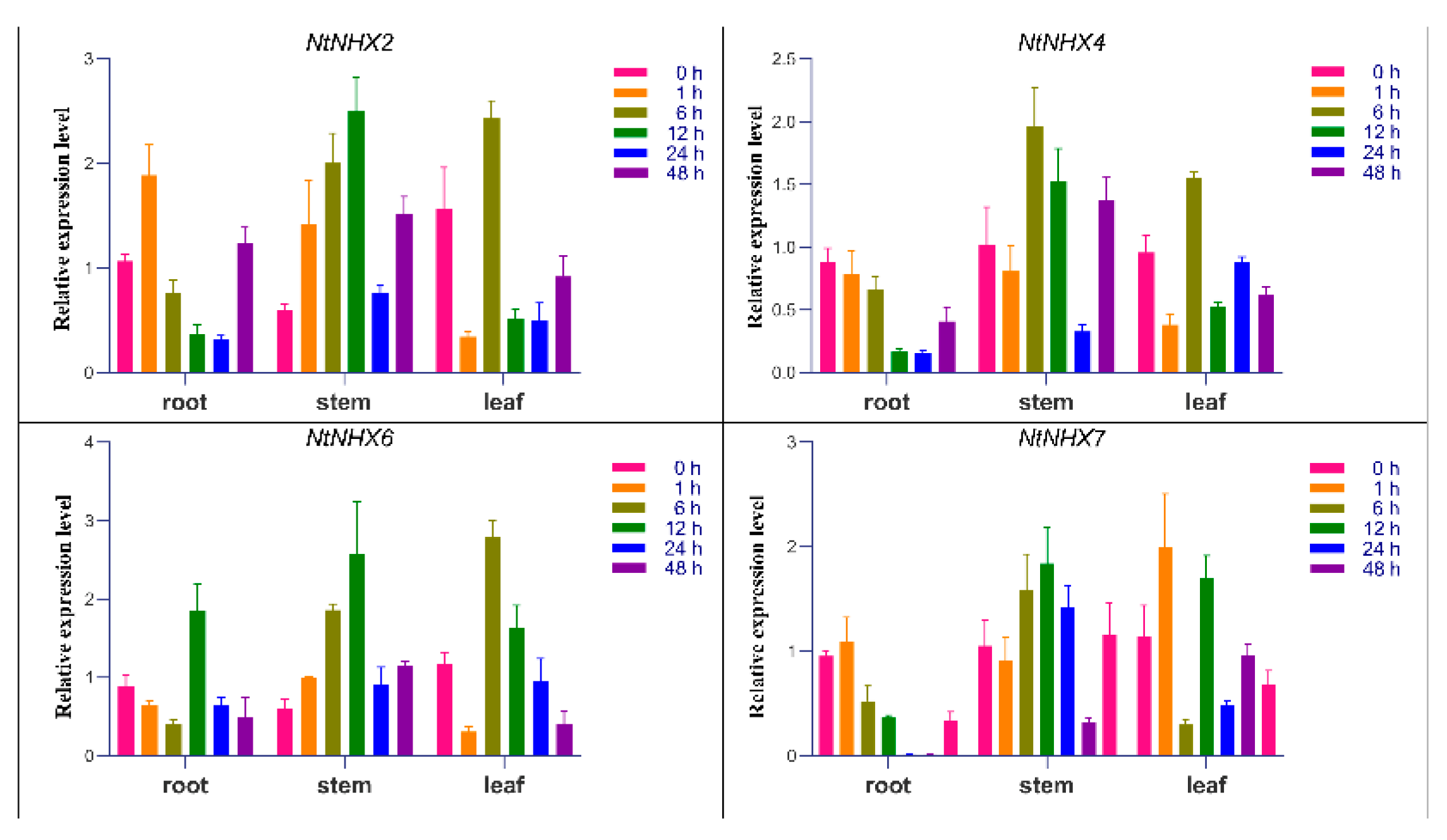
2.9. QRT-PCR Analysis
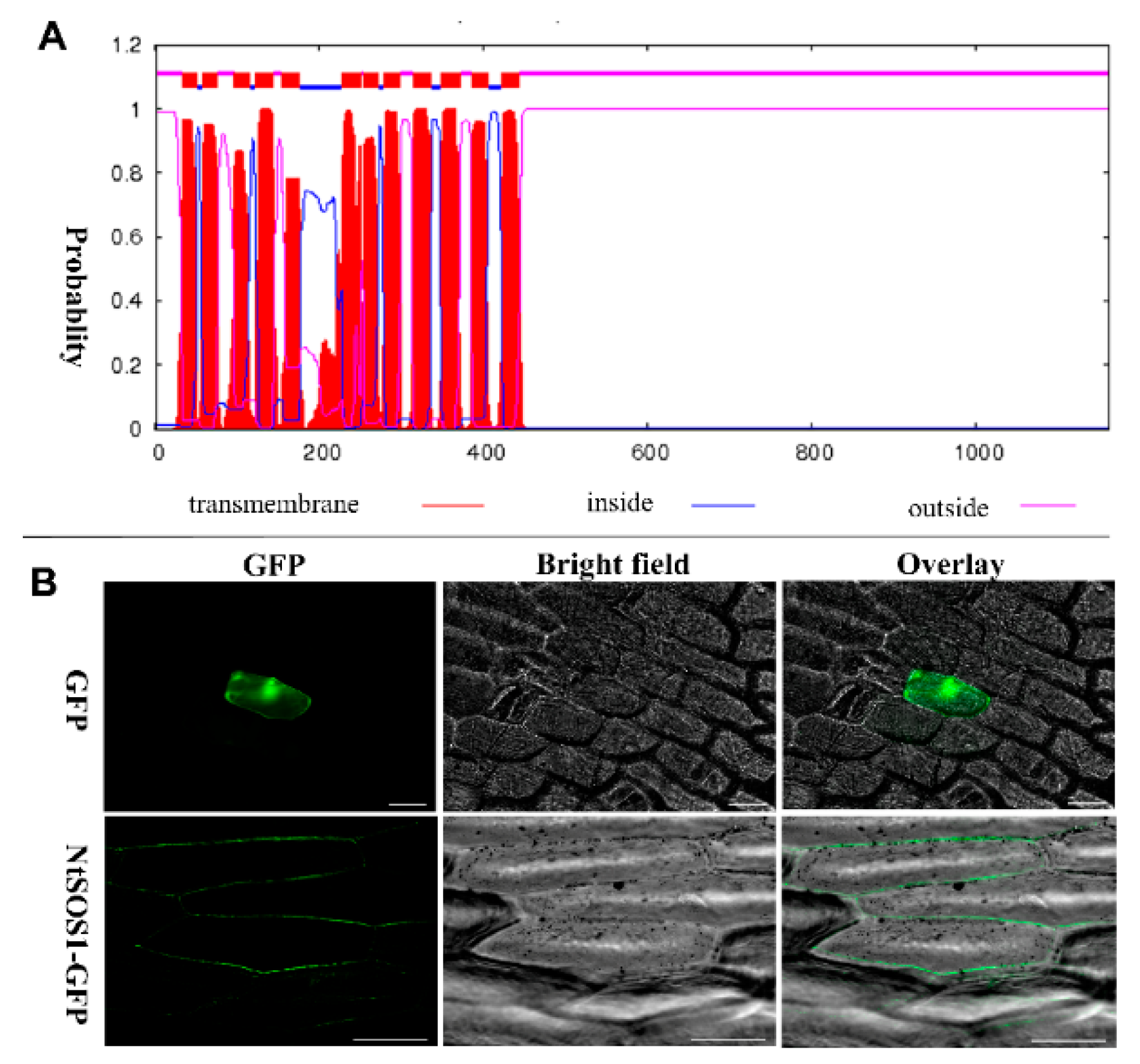
2.10. Subcellular Localization
3. Results
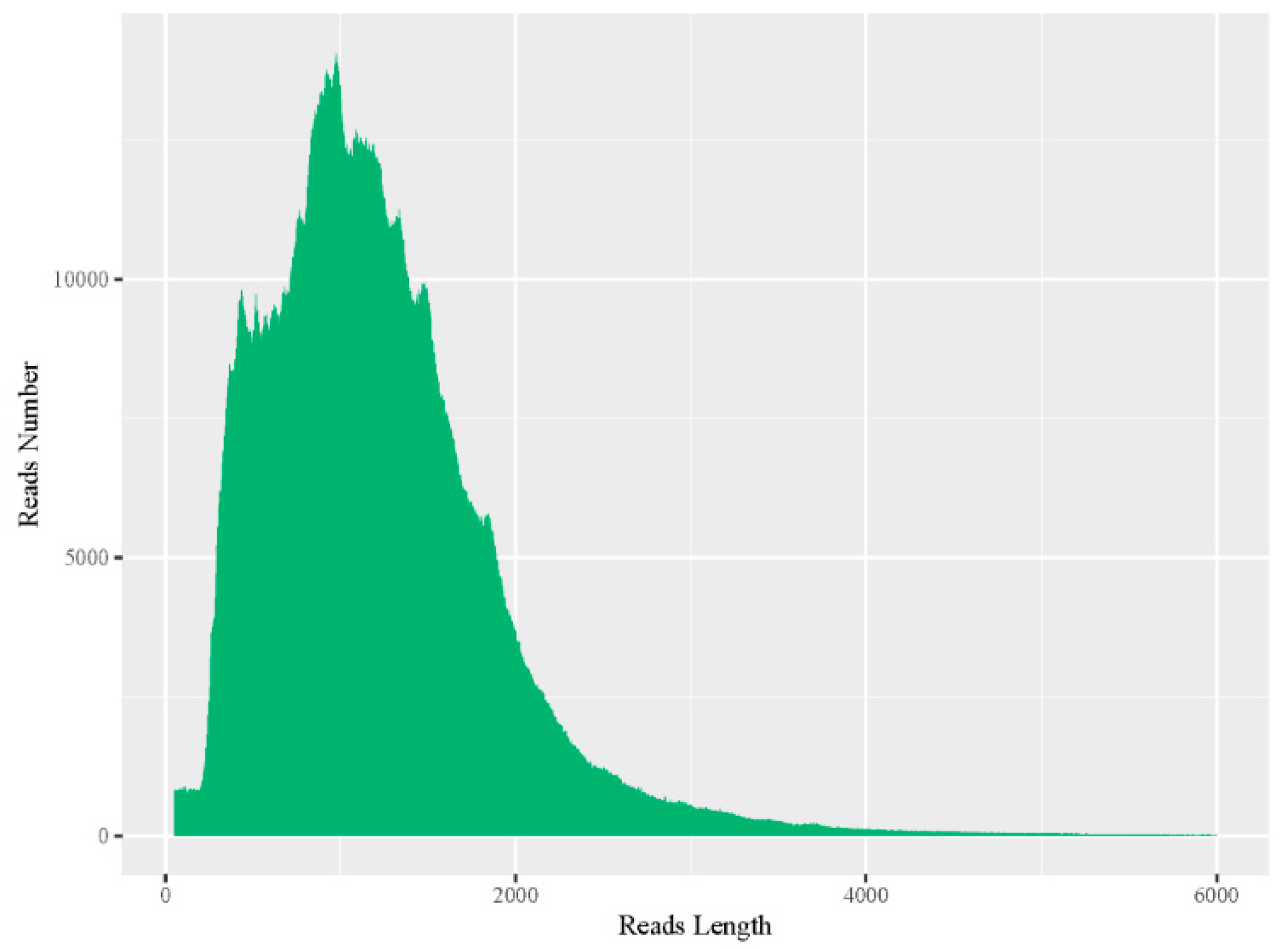
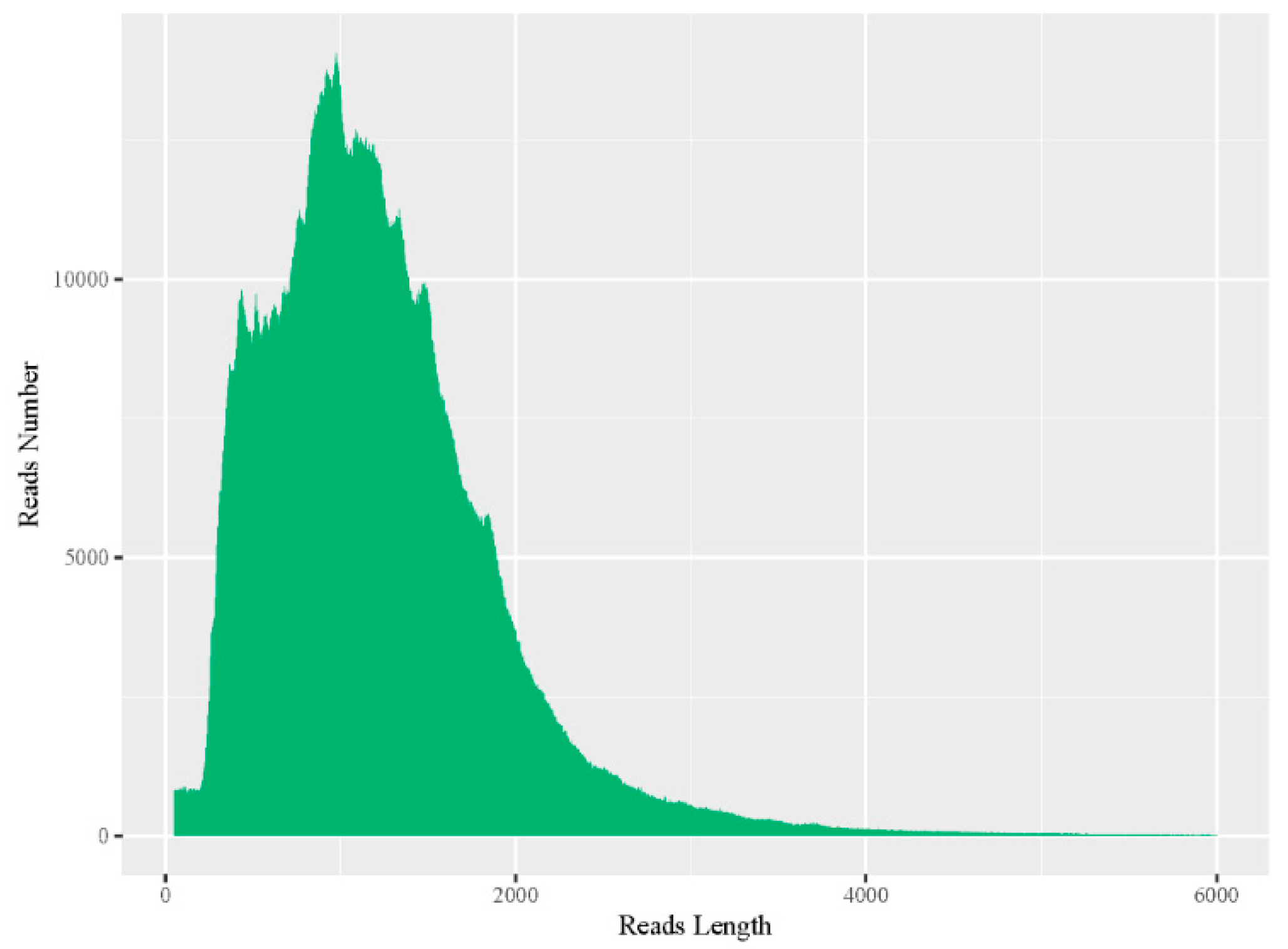
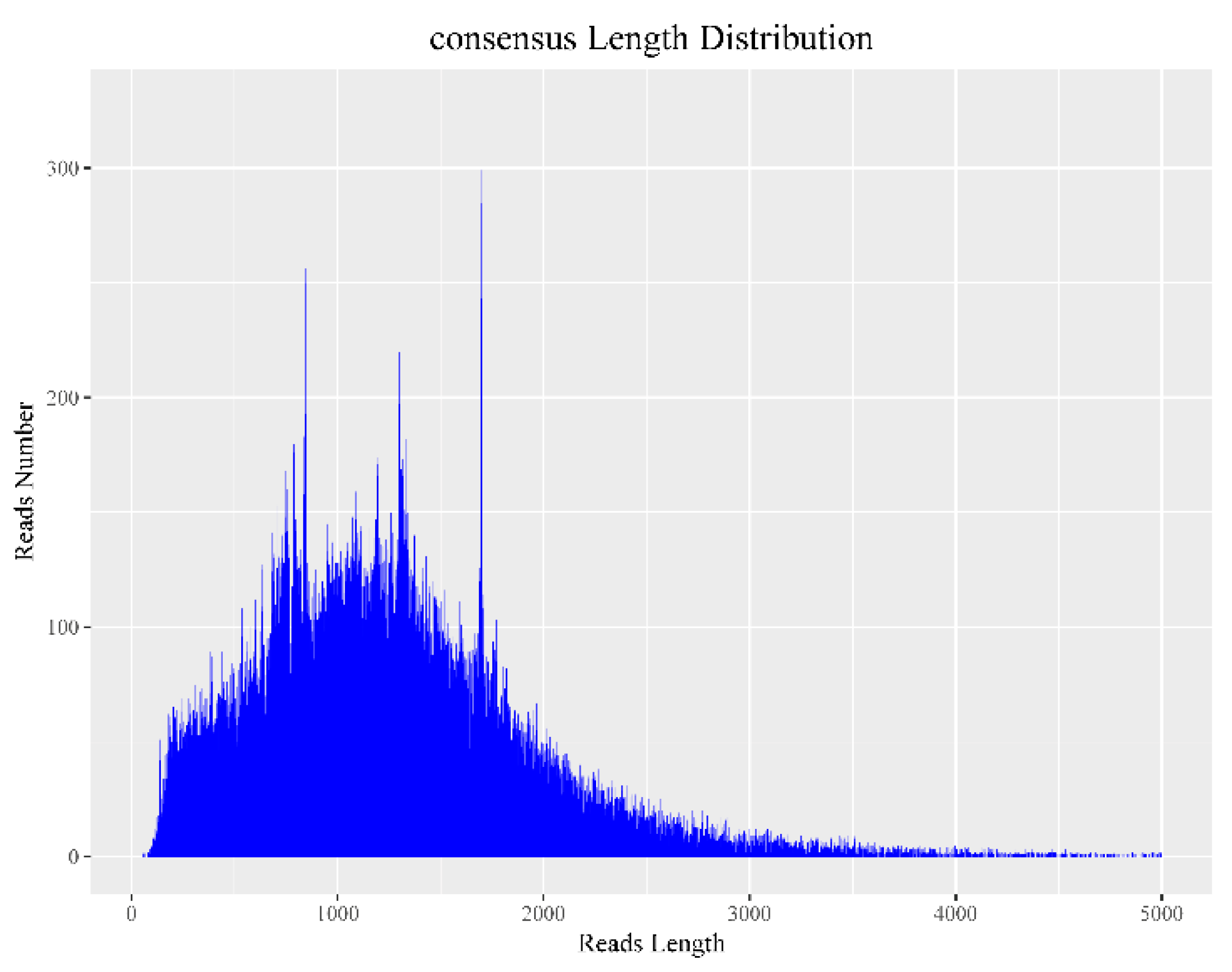
3.1. Sequencing Data Statistics
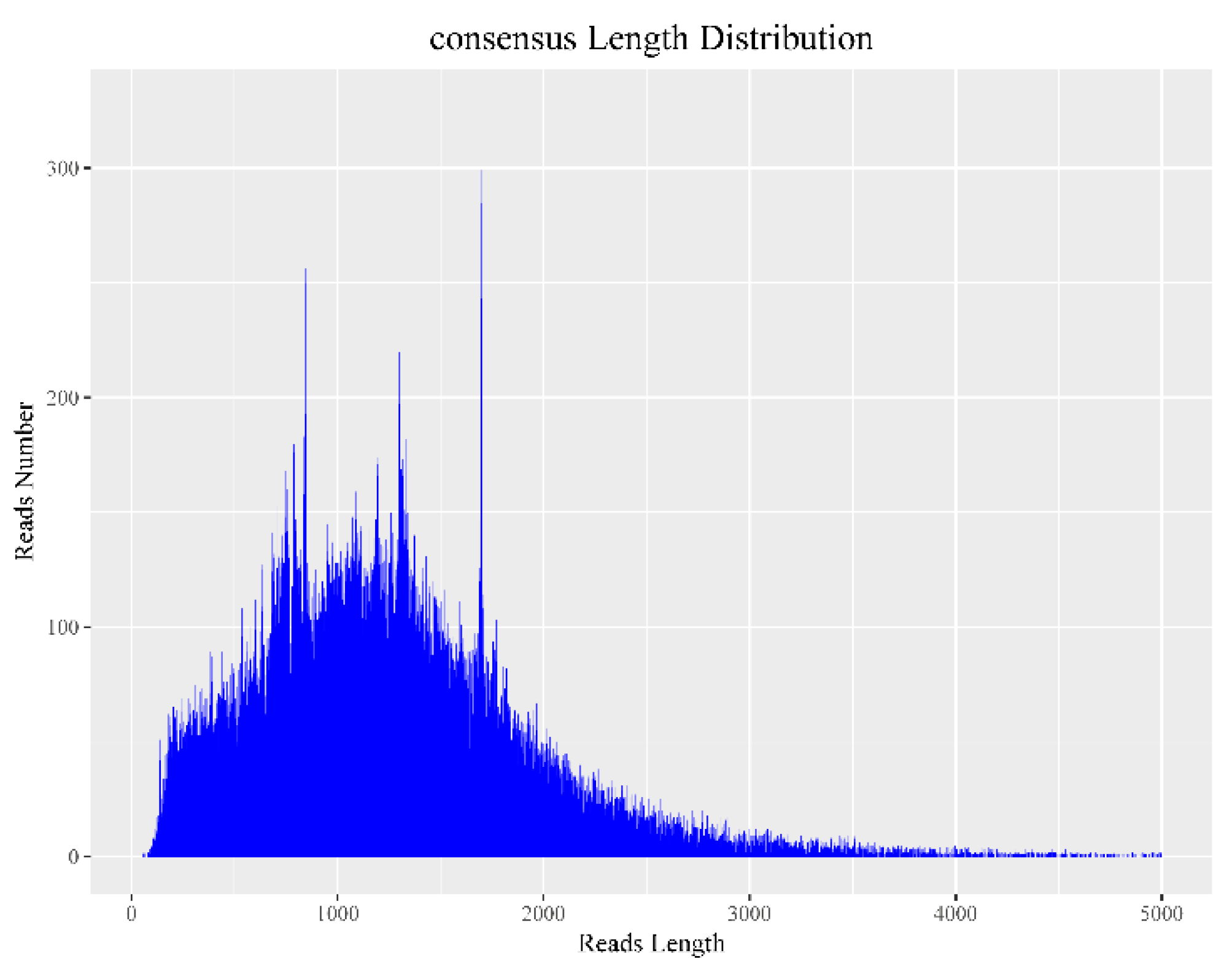
3.2. Data Correction
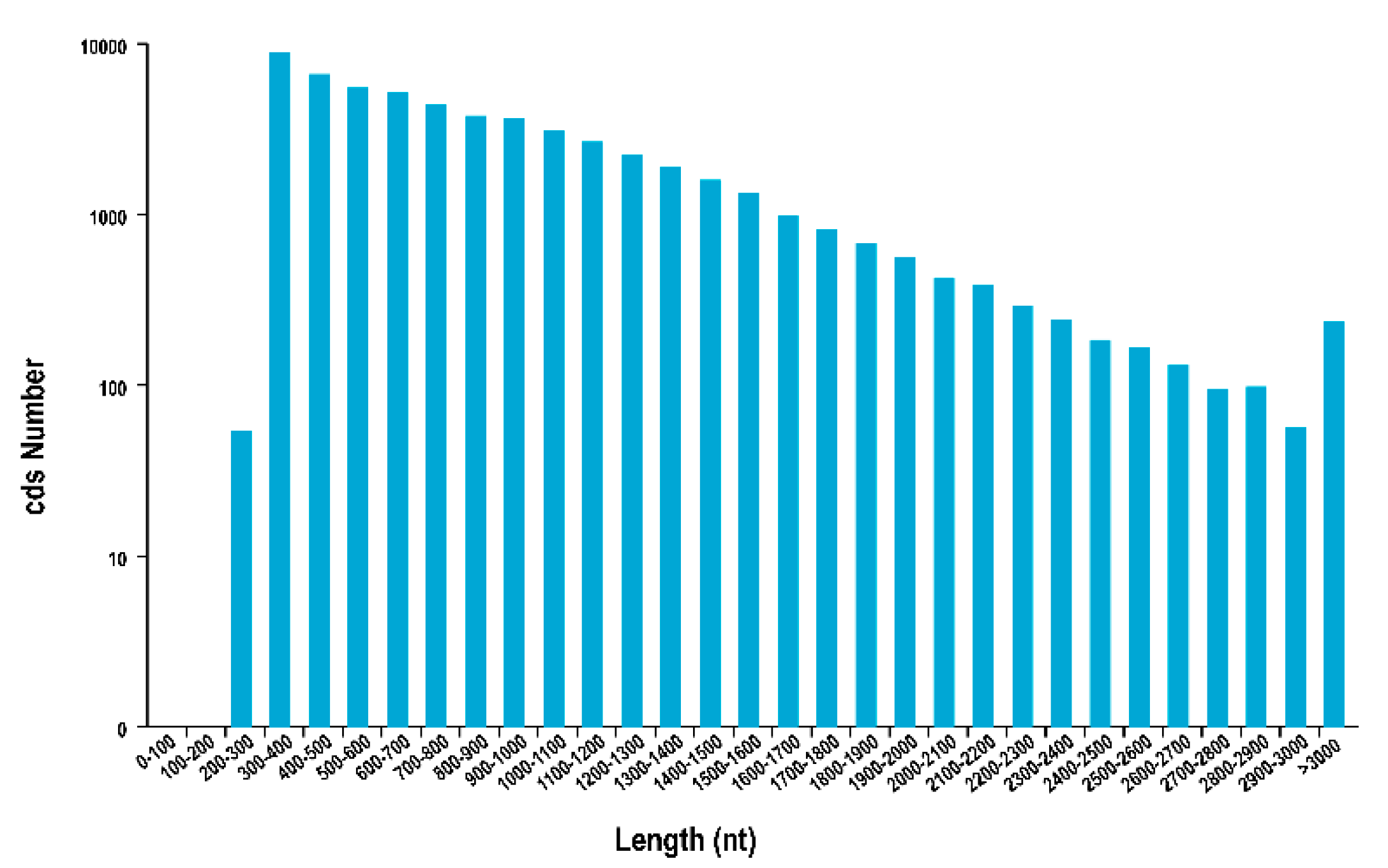
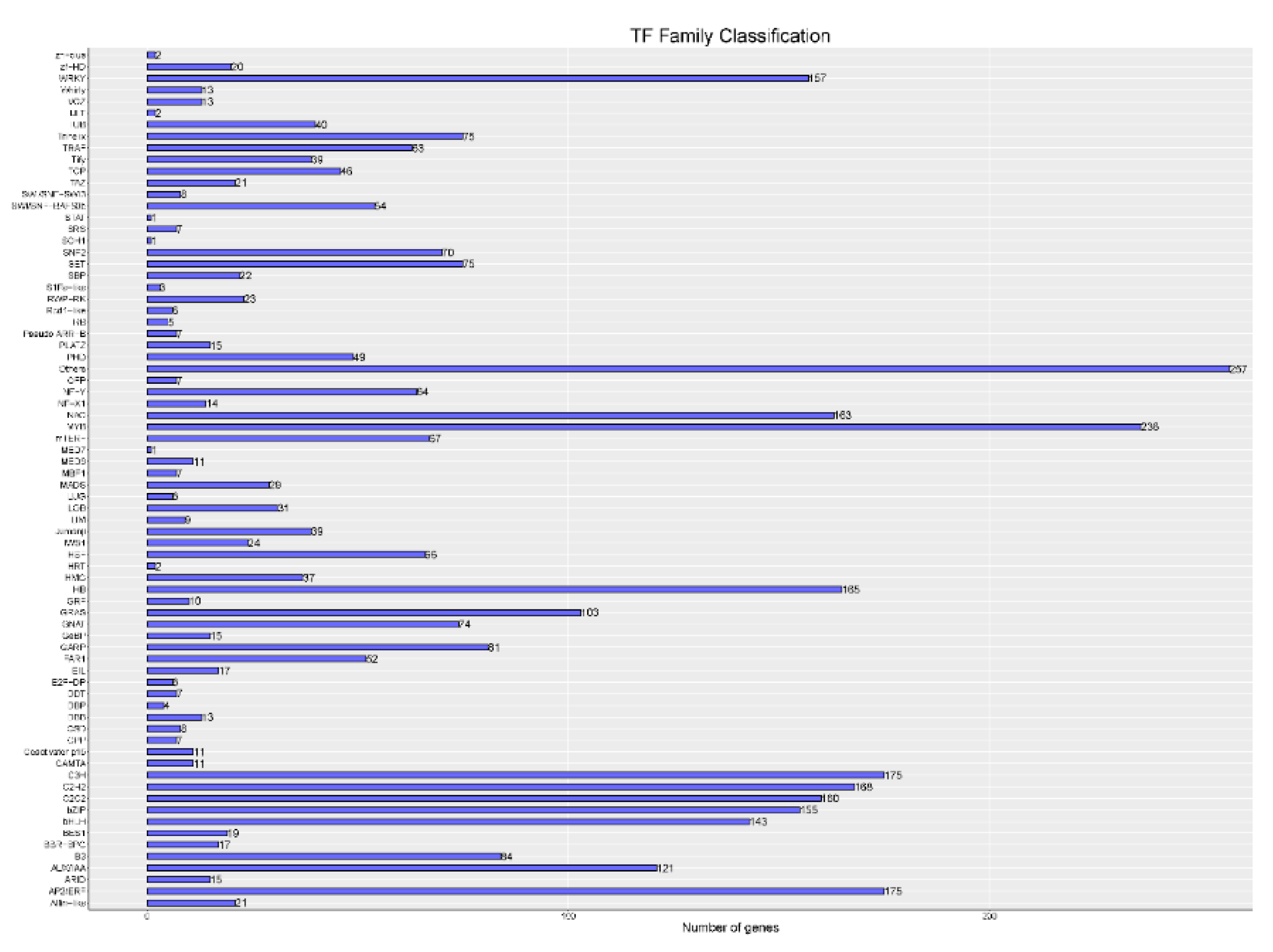
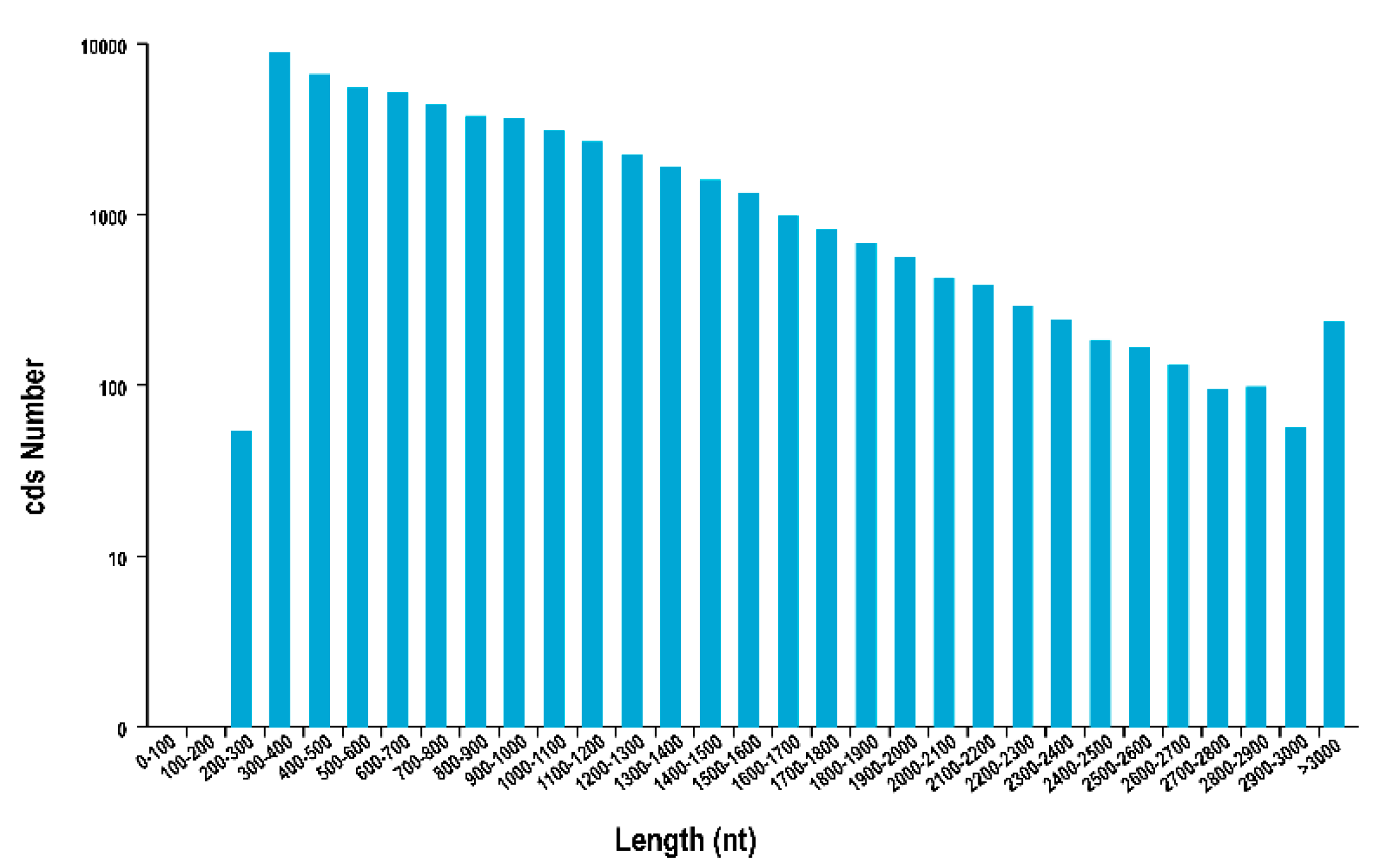
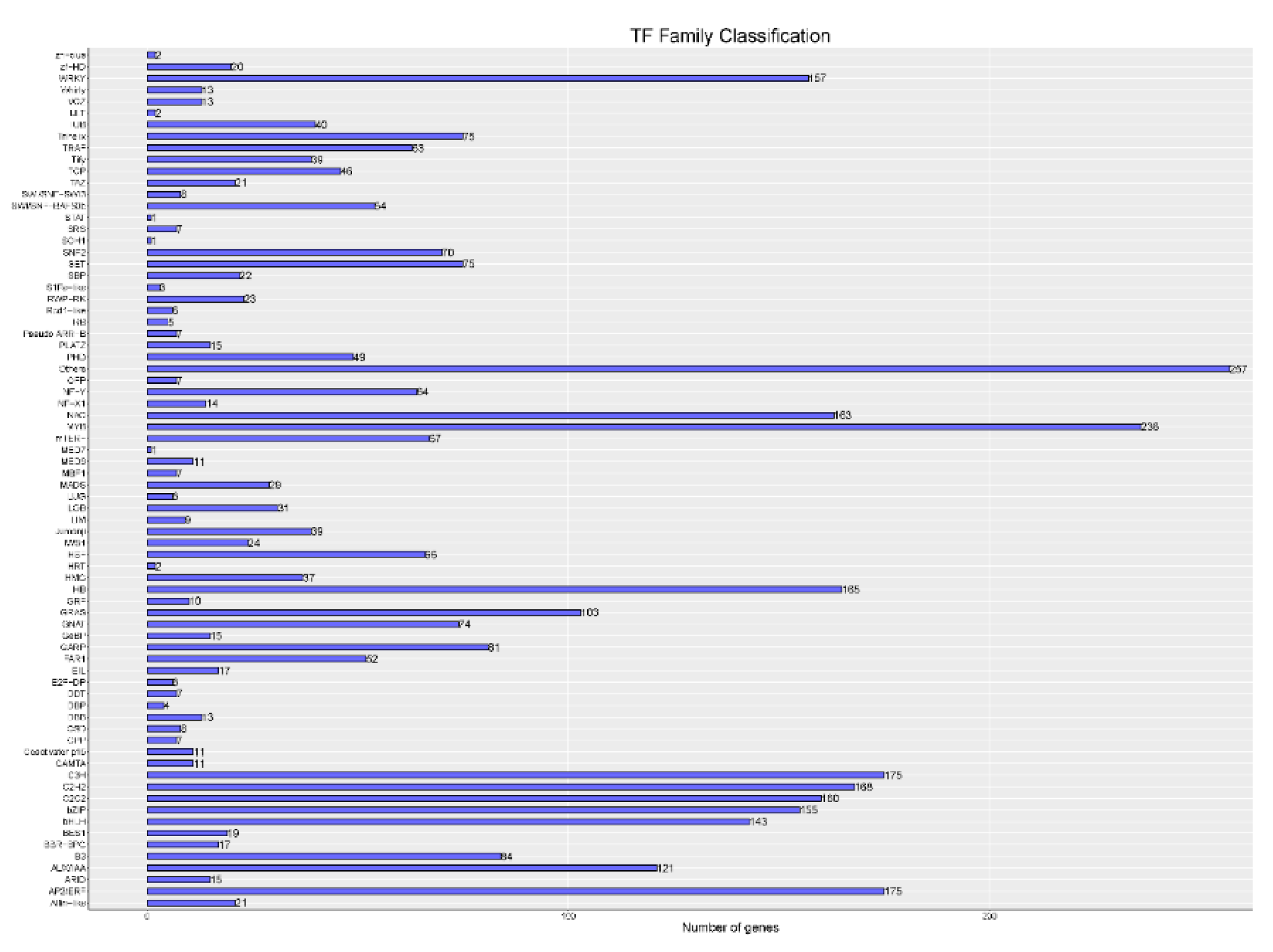
3.3. CDS, TFs, lncRNA Analysis
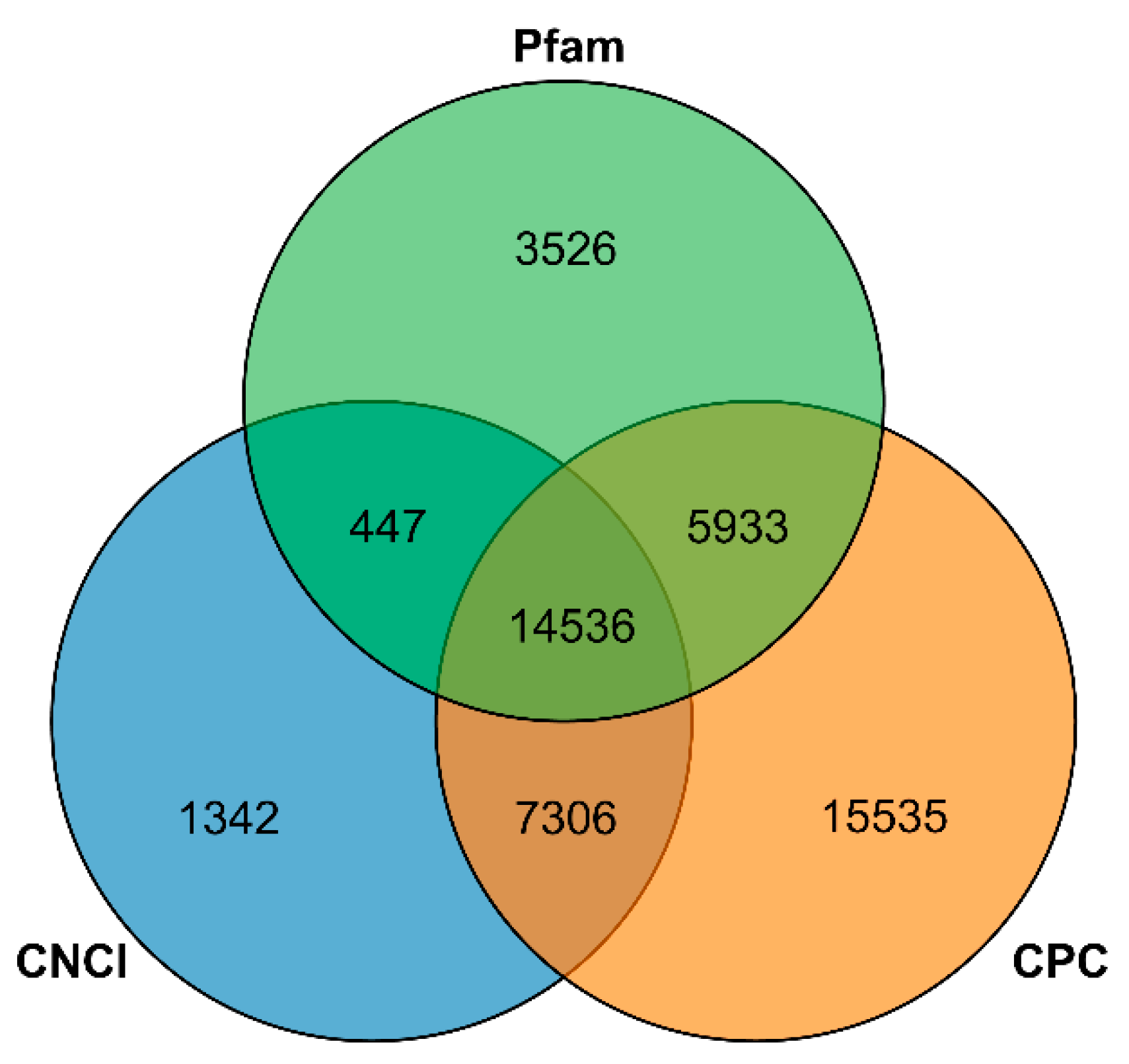
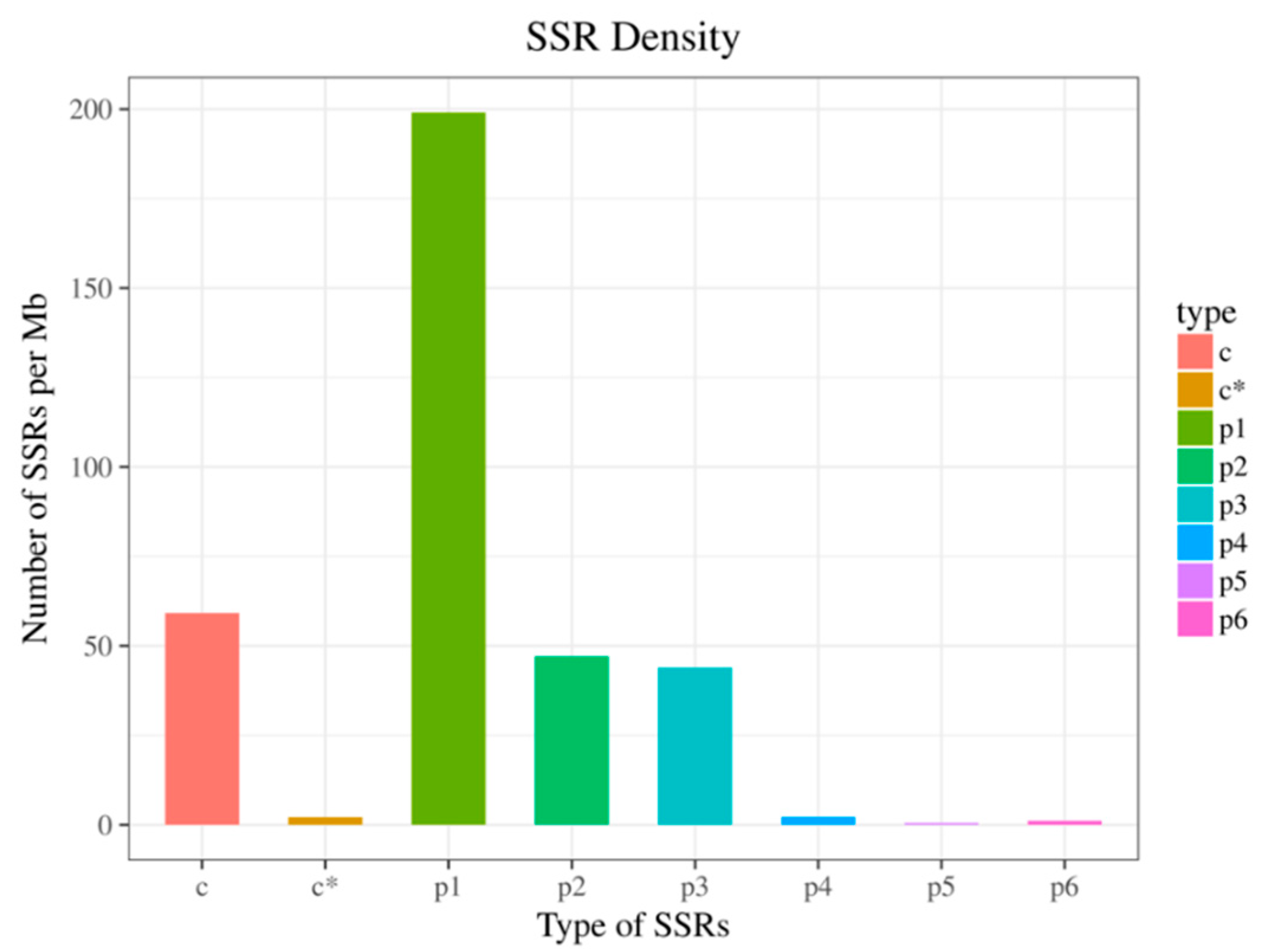
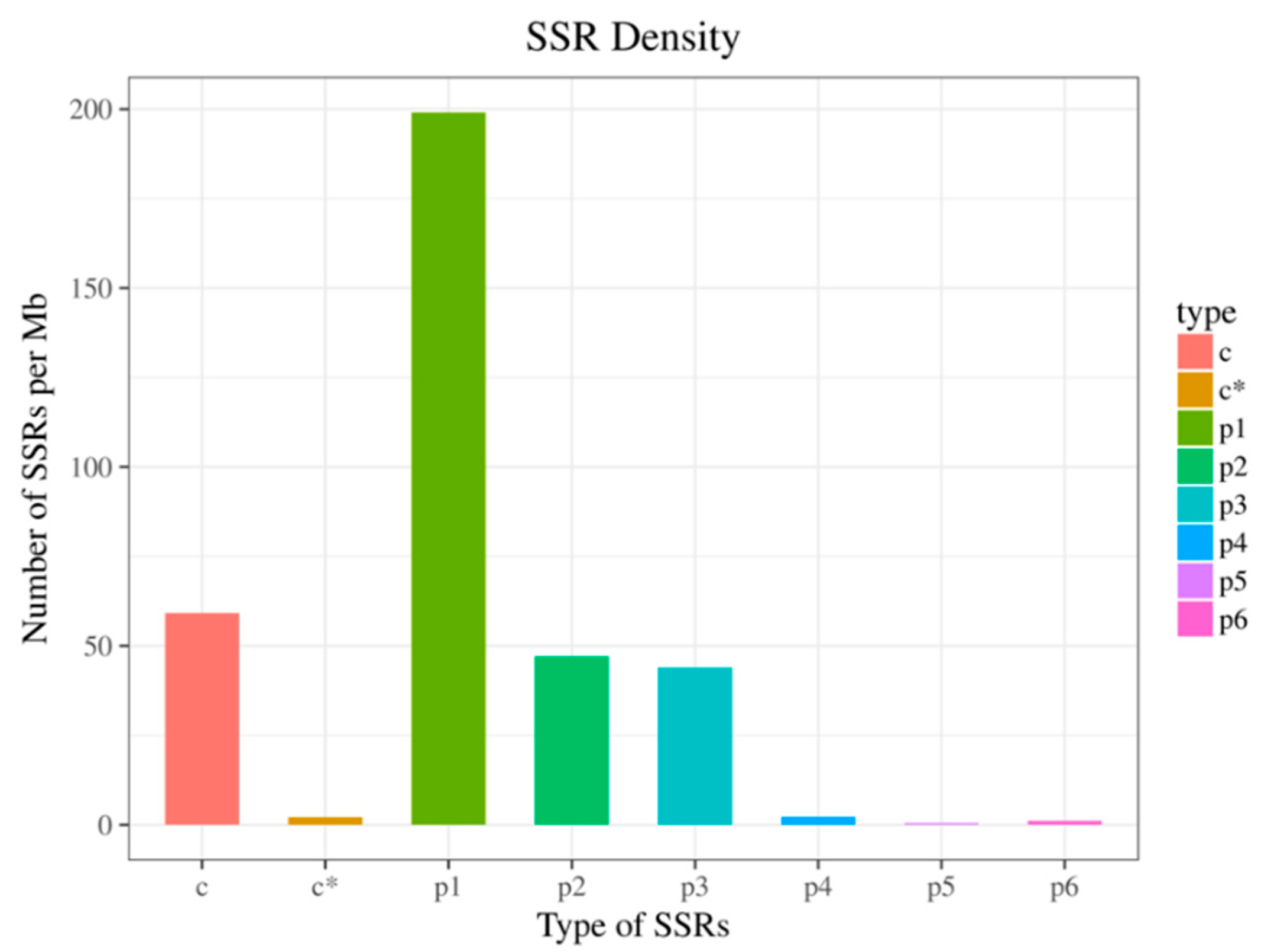
3.4. SSR Analysis
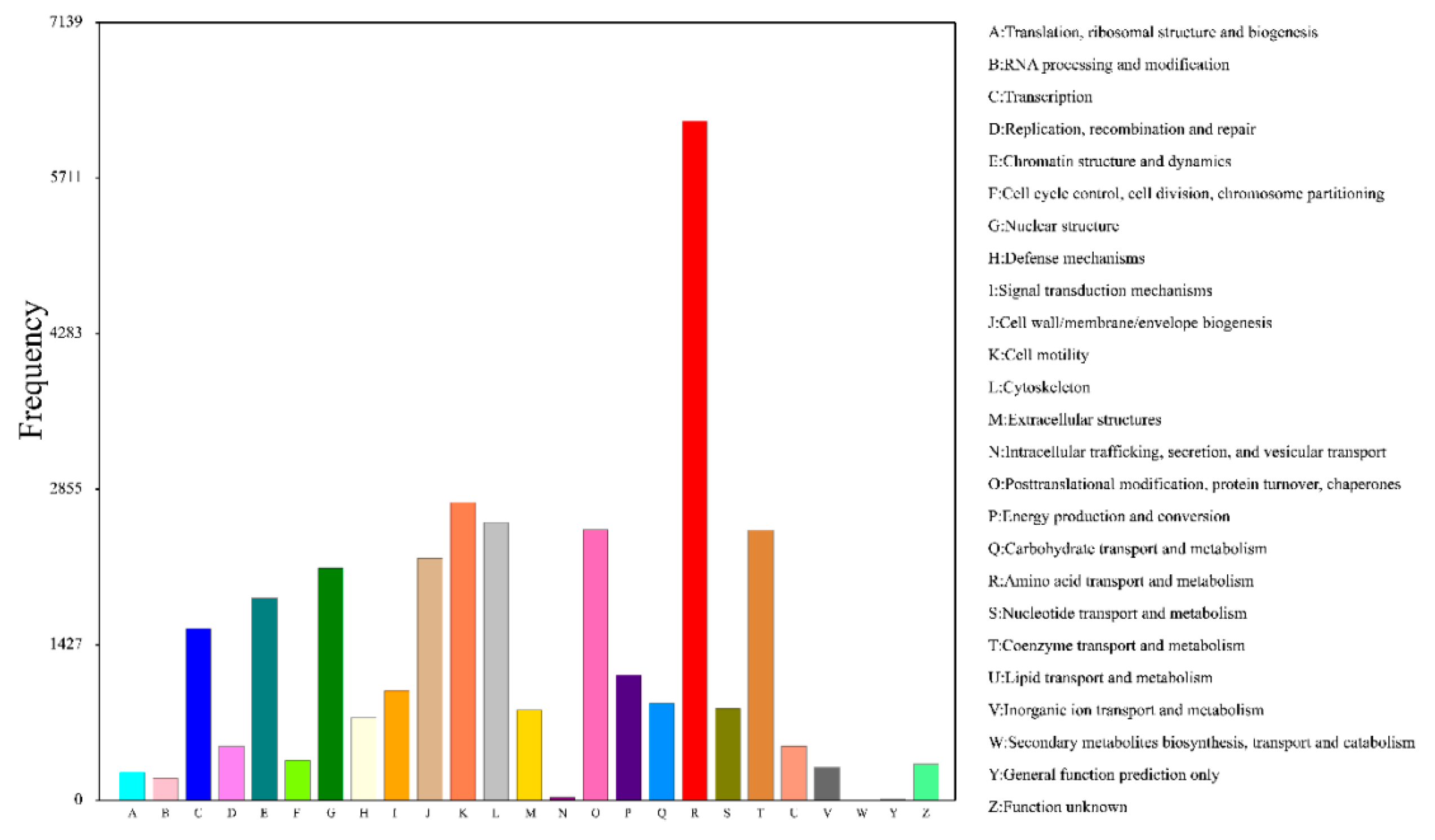
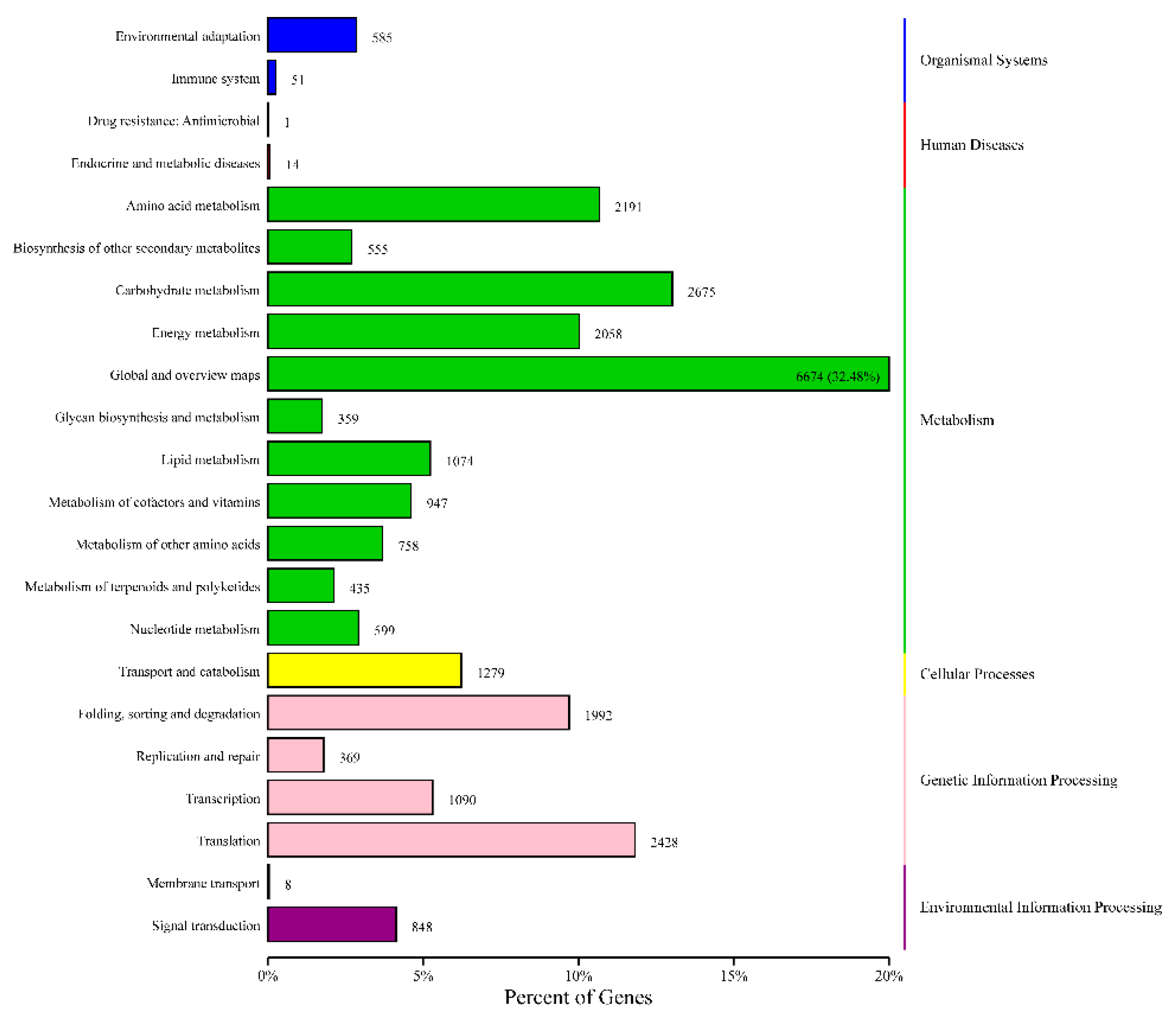
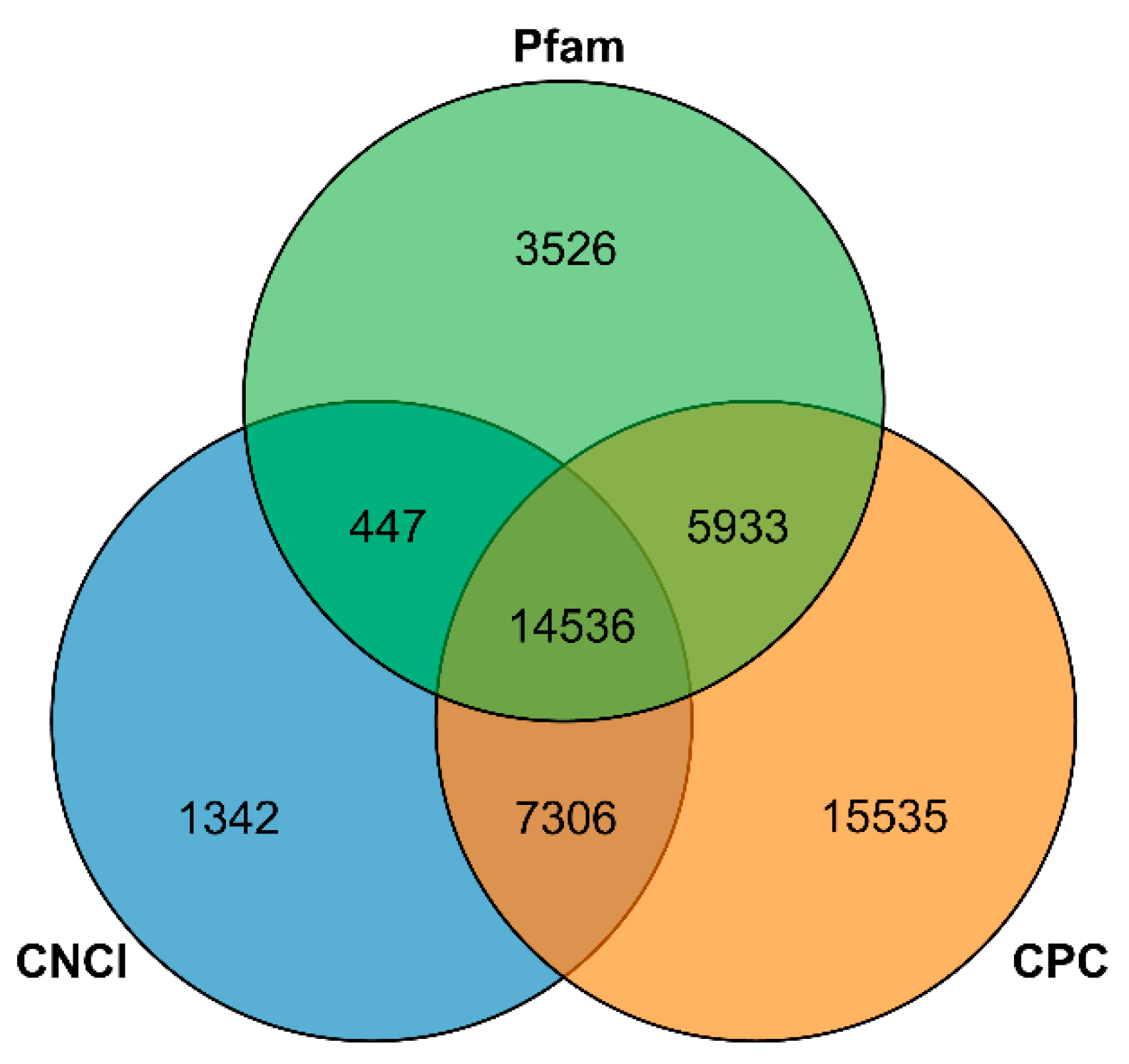
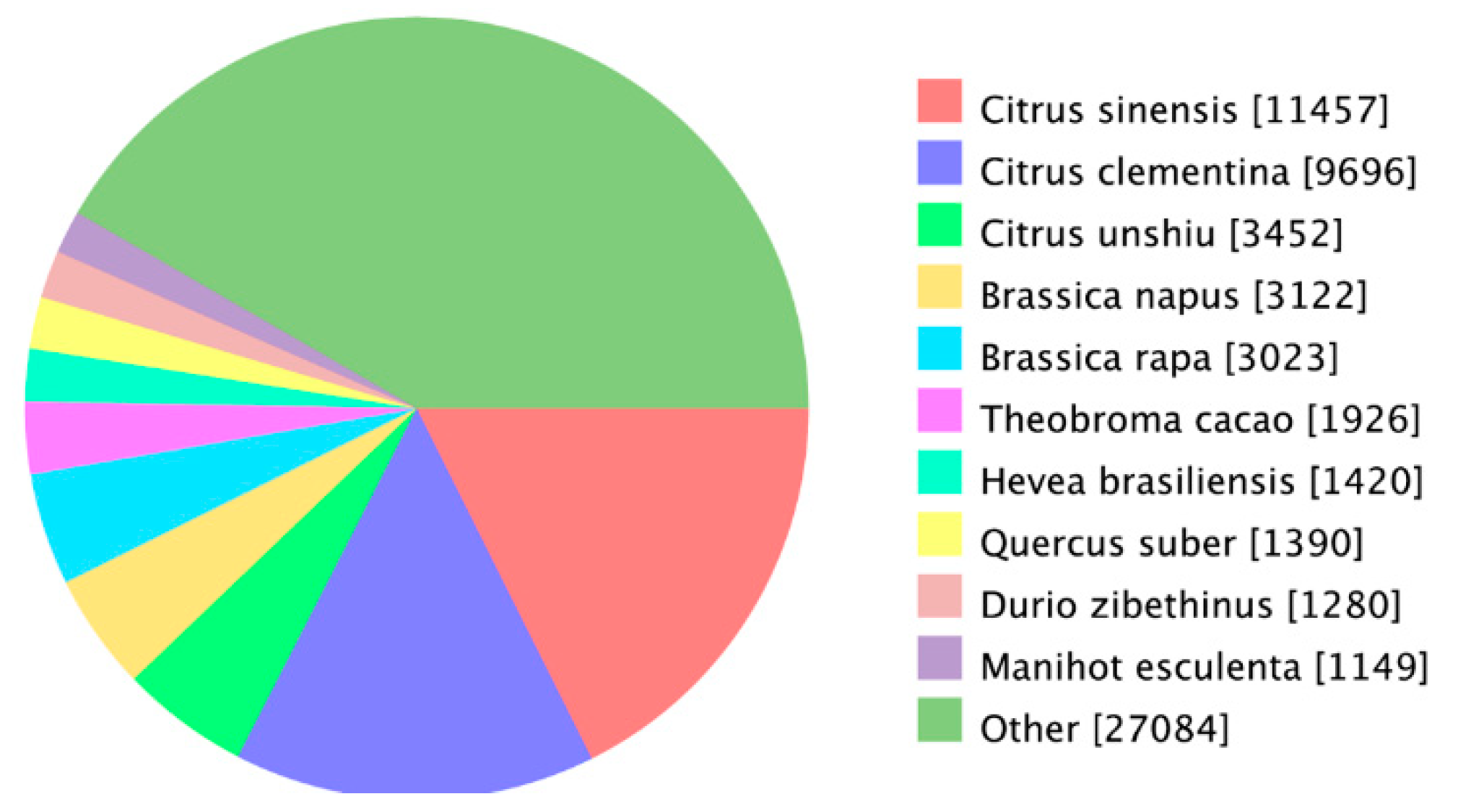
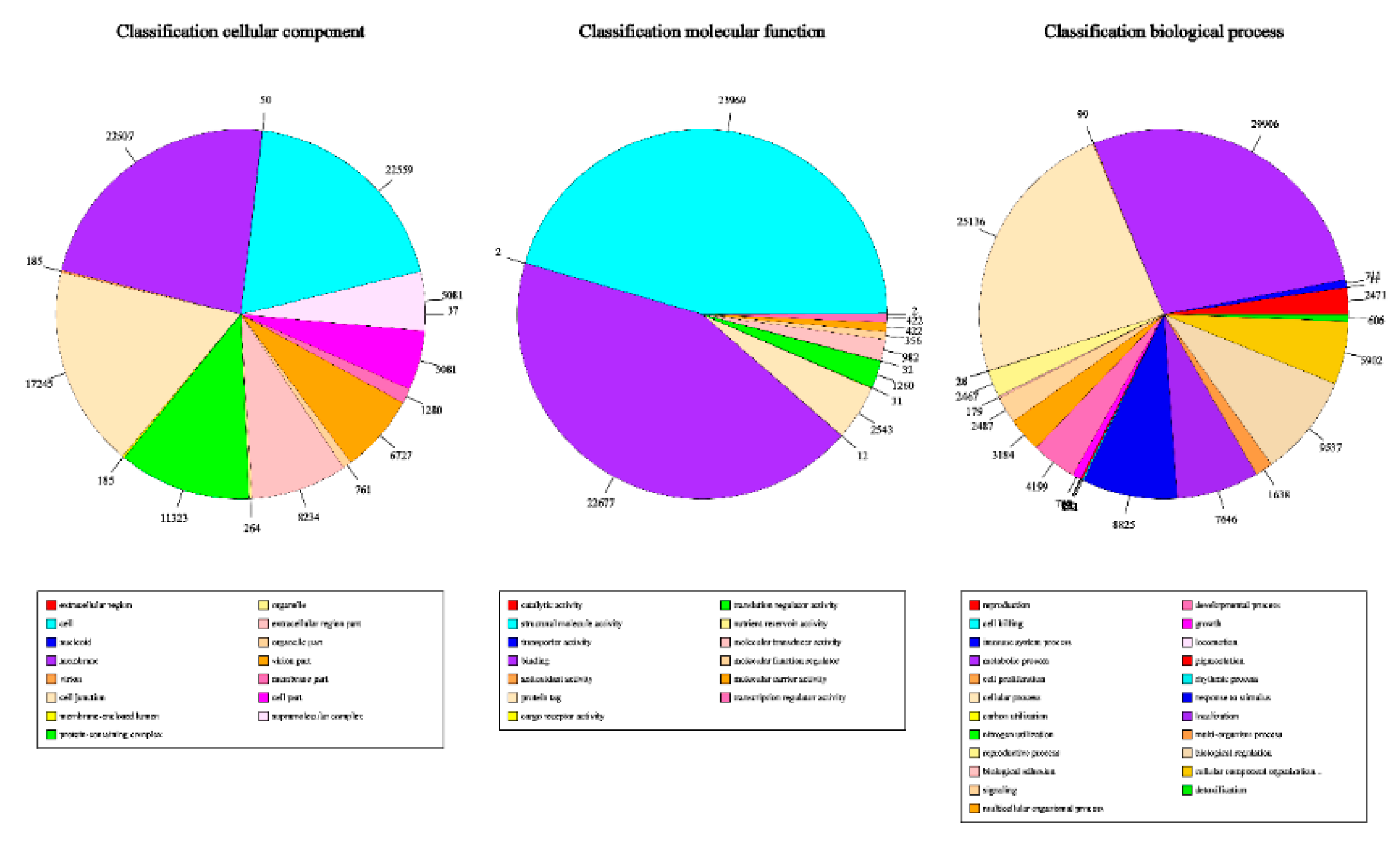
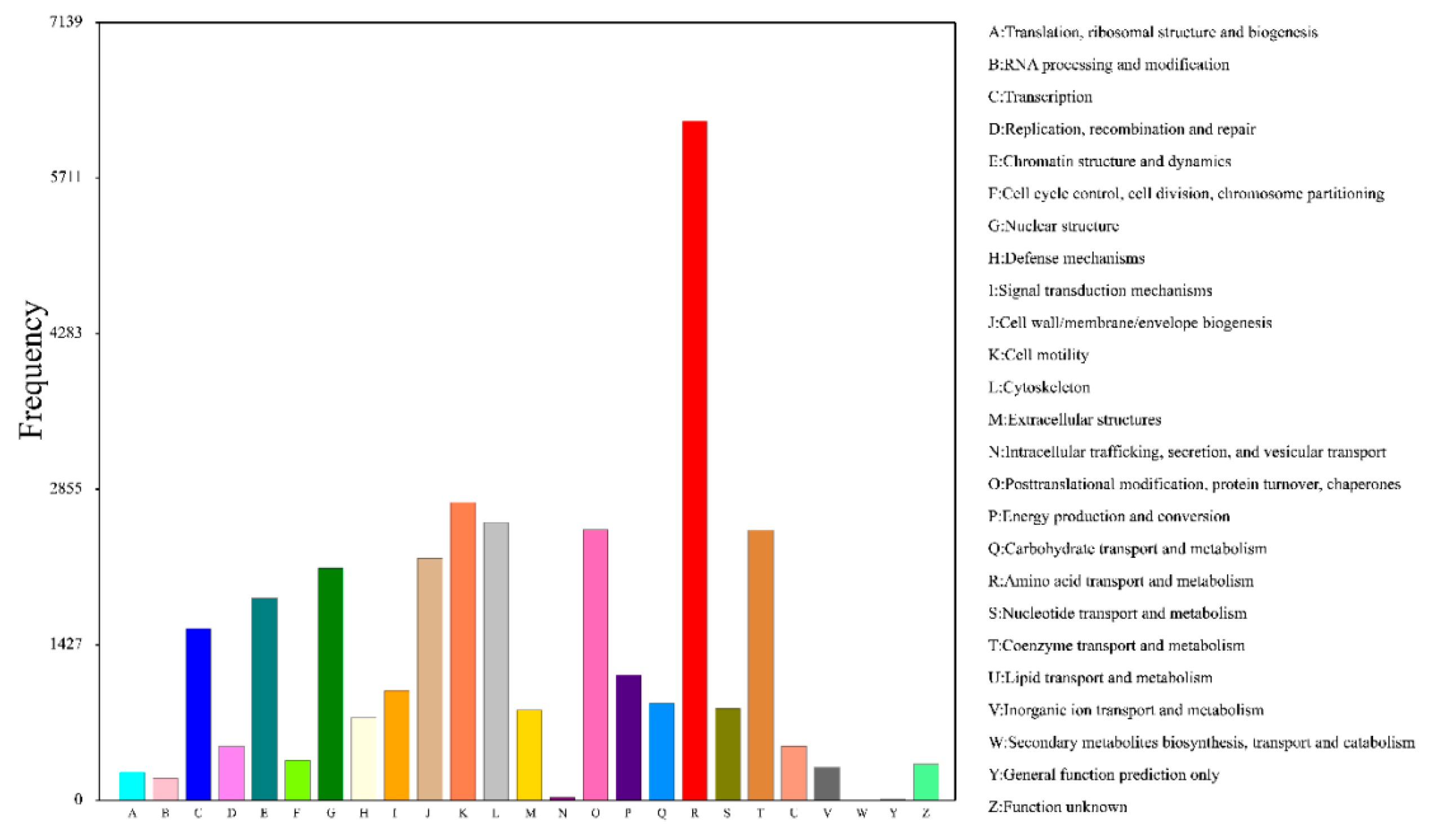
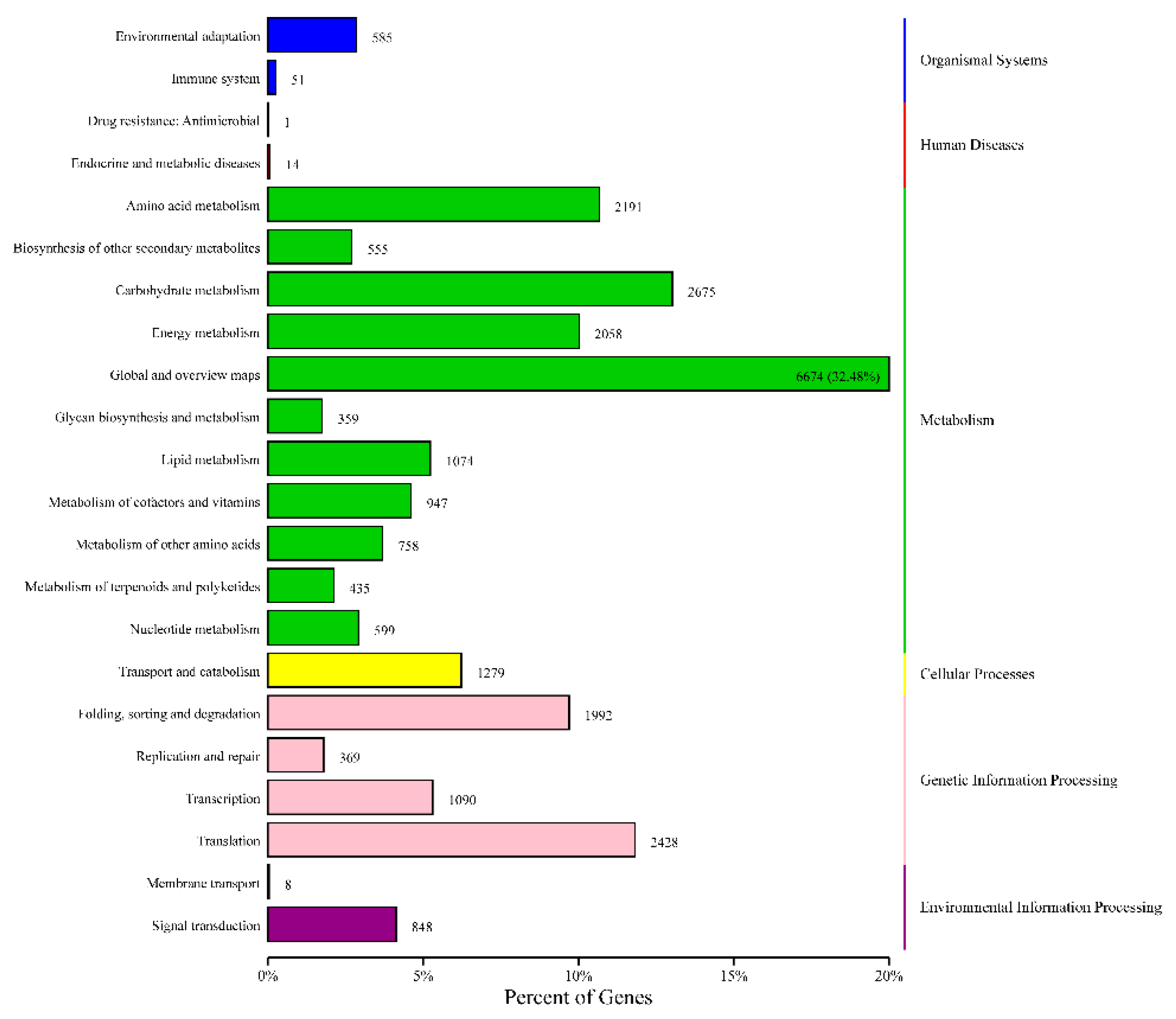
3.5. Functional Annotation
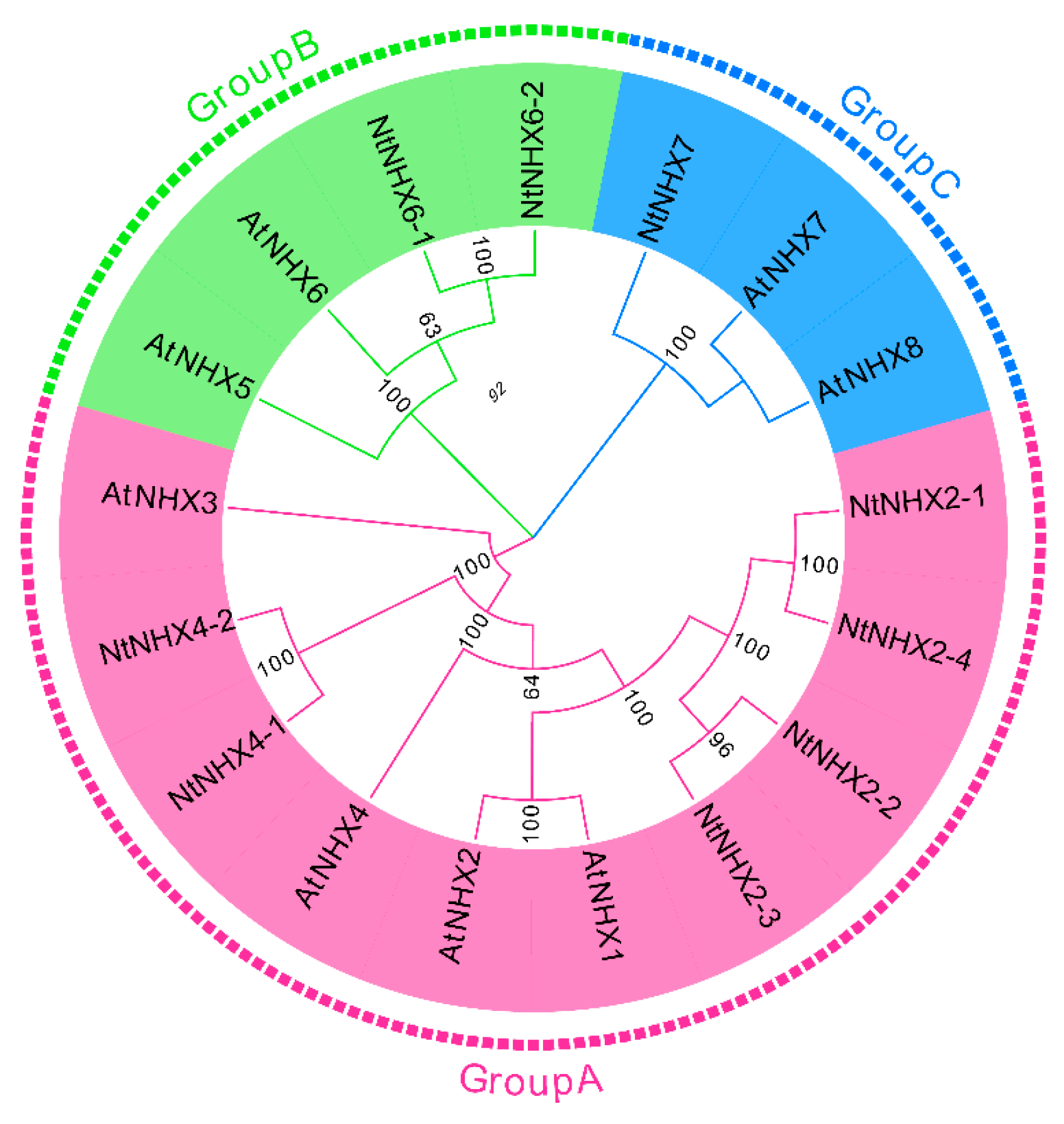
3.6. Identification of Na+/H+ Antiporter Genes in the Full-Length Transcriptome
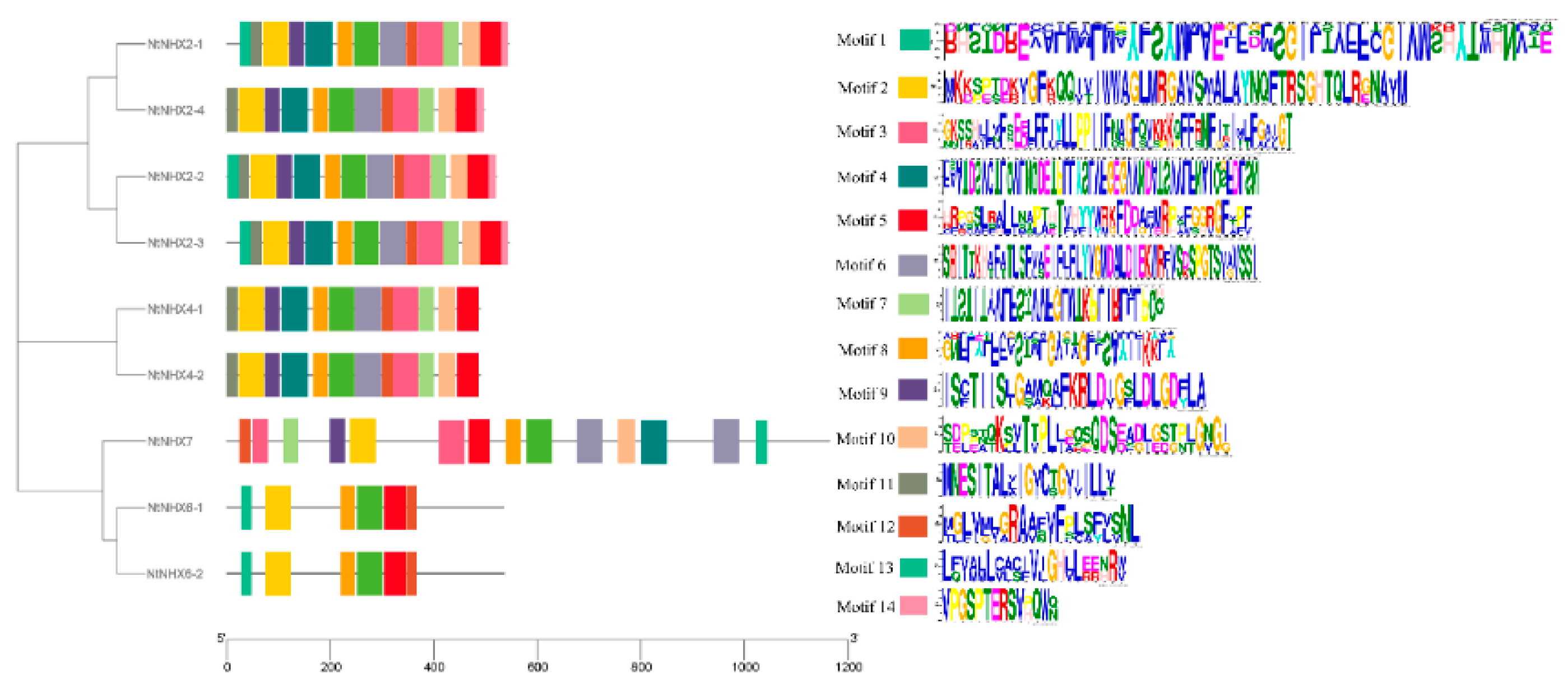
3.7. Gene Conserved Motifs Analysis of NHXs
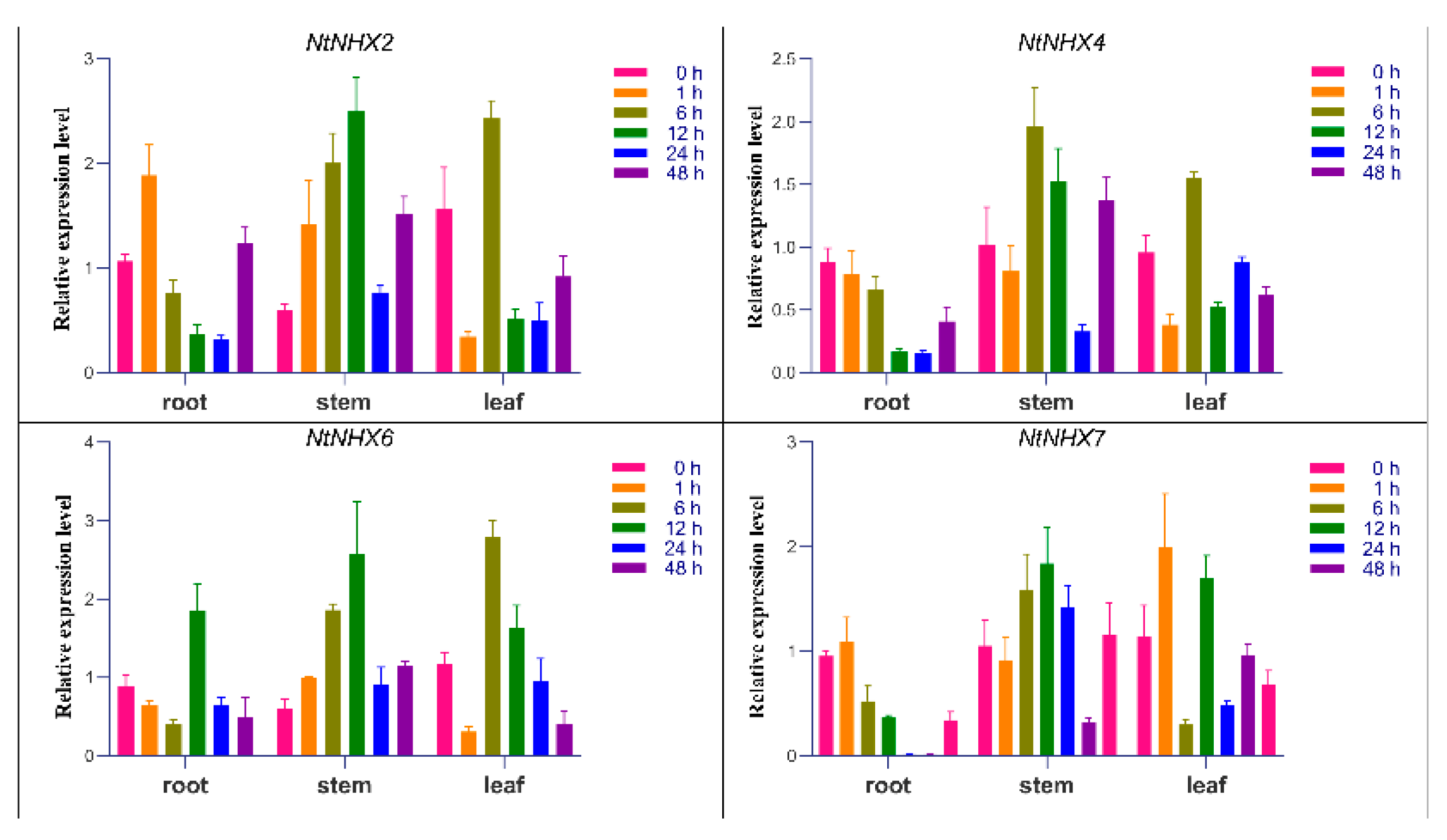
3.8. Real-Time Quantitative PCR Analysis
3.9. Clone and Subcellular Localization of NtNHX7
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NHX | Na+/H+ antiporter gene |
| SRMT | Single-molecule real-time |
| CDS | Coding sequence |
| SSR | Simple sequence repeat |
| LncRNA | Long non-coding RNA |
| CPC | Coding potential calculator |
| CNCI | Coding non-coding index |
| CPC | Coding potential calculator |
| mRNA | Messenger RNA |
| qRT-PCR | Quantitative real-time PCR |
| TF | Transcription factors |
| cDNA | Complementary DNA |
| Pfam | Protein family |
| NR | Non-redundant protein sequence database |
| Swiss-Prot | Swiss-Prot protein sequence database |
| GO | Gene Ontology Consortium |
| COG | Cluster of Orthologous Groups of proteins |
| KOG | EuKaryotic Orthologous Groups |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
References
- Du, Q.; Xin, H.; Peng, C. Pharmacology and phytochemistry of the Nitraria genus (Review). Mol. Med. Rep. 2015, 11, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Ma, J.; Bi, H.; Song, J.; Yang, H.; Xia, Z. Characterization and cardioprotective activity of anthocyanins from Nitraria tangutorum Bobr. by-products. Food Funct. 2017, 8, 2771–2782. [Google Scholar] [CrossRef]
- Zhou, H.; Zhao, W.; Luo, W.; Liu, B. Species diversity and vegetation distribution in nebkhas of Nitraria tangutorum in the Desert Steppes of China. Ecol. Res. 2015, 30, 735–744. [Google Scholar] [CrossRef]
- Du, J.; Yan, P.; Dong, Y. Phenological response of Nitraria tangutorum to climate change in Minqin County, Gansu Province, northwest China. Int. J. Biometeorol. 2010, 54, 583–593. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, F.; Li, X.; Shi, R.; Lu, J. Signal regulation of proline metabolism in callus of the halophyte Nitraria tangutorum Bobr. grown under salinity stress. Plant Cell Tissue Organ Cult. (PCTOC) 2013, 112, 33–42. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, Y.; Yang, Y.; Zeng, Y.; Wang, Q.; Shao, Y.; Mei, L.; Shi, Y.; Tao, Y. Isolation and identification of antioxidant and α-glucosidase inhibitory compounds from fruit juice of Nitraria tangutorum. Food Chem. 2017, 227, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.; Zheng, J.; Li, W.; Suo, Y. Isolation, stability, and antioxidant activity of anthocyanins from Lycium ruthenicum Murray and Nitraria tangutorum Bobr of Qinghai-Tibetan plateau. Sep. Sci. Technol. 2014, 49, 2897–2906. [Google Scholar] [CrossRef]
- Bock, C.; Farlik, M.; Sheffield, N.C. Multi-omics of single cells: Strategies and applications. Trends Biotechnol. 2016, 34, 605–608. [Google Scholar] [CrossRef] [Green Version]
- Hrdlickova, R.; Toloue, M.; Tian, B. RNA-Seq methods for transcriptome analysis. Wiley Interdiscip. Rev. RNA 2017, 8, e1364. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B. Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, P.; Liang, F.; Ye, Z.; Li, J.; Shen, C.; Pei, L.; Wang, F.; Hu, J.; Tu, L. A global survey of alternative splicing in allopolyploid cotton: Landscape, complexity and regulation. New Phytol. 2018, 217, 163–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.T.; Kim, J. Trends in next-generation sequencing and a new era for whole genome sequencing. Int. Neurourol. J. 2016, 20 (Suppl. 2), S76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wall, P.K.; Leebens-Mack, J.; Chanderbali, A.S.; Barakat, A.; Wolcott, E.; Liang, H.; Landherr, L.; Tomsho, L.P.; Hu, Y.; Carlson, J.E. Comparison of next generation sequencing technologies for transcriptome characterization. BMC Genom. 2009, 10, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Nebenführ, A. Identifying subcellular protein localization with fluorescent protein fusions after transient expression in onion epidermal cells. In Plant Cell Morphogenesis; Humana Press: Totowa, NJ, USA, 2014; pp. 77–85. [Google Scholar]
- Li, Q.; Li, Y.; Song, J.; Xu, H.; Xu, J.; Zhu, Y.; Li, X.; Gao, H.; Dong, L.; Qian, J. High-accuracy de novo assembly and SNP detection of chloroplast genomes using a SMRT circular consensus sequencing strategy. New Phytol. 2014, 204, 1041–1049. [Google Scholar] [CrossRef]
- Fukuda, A.; Nakamura, A.; Hara, N.; Toki, S.; Tanaka, Y. Molecular and functional analyses of rice NHX-type Na+/H+ antiporter genes. Planta 2011, 233, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.Y.; Zhang, H.C.; Chen, J.H.; Xia, X.L.; Yin, W.L. Molecular characterization of putative vacuolar NHX-type Na+/H+ exchanger genes from the salt-resistant tree Populus euphratica. Physiol. Plantarum. 2009, 137, 166–174. [Google Scholar] [CrossRef]
- Shi, H.; Quintero, F.J.; Pardo, J.M.; Zhu, J. The putative plasma membrane Na+/H+ antiporter SOS1 controls long-distance Na+ transport in plants. Plant Cell 2002, 14, 465–477. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, A. Third generation DNA sequencing: Pacific biosciences’ single molecule real time technology. Chem. Biol. 2010, 17, 675–676. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Ghany, S.E.; Hamilton, M.; Jacobi, J.L.; Ngam, P.; Devitt, N.; Schilkey, F.; Ben-Hur, A.; Reddy, A.S. A survey of the sorghum transcriptome using single-molecule long reads. Nat. Commun. 2016, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minoche, A.E.; Dohm, J.C.; Schneider, J.; Holtgräwe, D.; Viehöver, P.; Montfort, M.; Sörensen, T.R.; Weisshaar, B.; Himmelbauer, H. Exploiting single-molecule transcript sequencing for eukaryotic gene prediction. Genome Biol. 2015, 16, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, R.; Ye, K.; Gu, Z.; Sun, X. Diminishing returns in next-generation sequencing (NGS) transcriptome data. Gene 2015, 557, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, A.; Au, K.F. PacBio sequencing and its applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Au, K.F.; Sebastiano, V.; Afshar, P.T.; Durruthy, J.D.; Lee, L.; Williams, B.A.; van Bakel, H.; Schadt, E.E.; Reijo-Pera, R.A.; Underwood, J.G. Characterization of the human ESC transcriptome by hybrid sequencing. Proc. Natl. Acad. Sci. USA 2013, 110, E4821–E4830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Tseng, E.; Regulski, M.; Clark, T.A.; Hon, T.; Jiao, Y.; Lu, Z.; Olson, A.; Stein, J.C.; Ware, D. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat. Commun. 2016, 7, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Zang, F.; Xie, X.; Ma, Y.; Zheng, Y.; Zang, D. Full-length transcriptome sequencing analysis and development of EST-SSR markers for the endangered species Populus wulianensis. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Xu, M.; Liu, X.; Wang, J.; Teng, S.; Shi, J.; Li, Y.; Huang, M. Transcriptome sequencing and development of novel genic SSR markers for Dendrobium officinale. Mol. Breed. 2017, 37, 18. [Google Scholar] [CrossRef]
- Chen, J.; Tang, X.; Ren, C.; Wei, B.; Wu, Y.; Wu, Q.; Pei, J. Full-length transcriptome sequences and the identification of putative genes for flavonoid biosynthesis in safflower. BMC Genom. 2018, 19, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Ishitani, M.; Kim, C.; Zhu, J. The Arabidopsis thaliana salt tolerance gene SOS1 encodes a putative Na+/H+ antiporter. Proc. Natl. Acad. Sci. USA 2000, 97, 6896–6901. [Google Scholar] [CrossRef] [Green Version]
- Akram, U.; Song, Y.; Liang, C.; Abid, M.A.; Askari, M.; Myat, A.A.; Abbas, M.; Malik, W.; Ali, Z.; Guo, S. Genome-wide characterization and expression analysis of NHX gene family under salinity stress in Gossypium barbadense and its comparison with Gossypium Hirsutum. Genes 2020, 11, 803. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, W.; Yang, Q.; Lin, J.; Chang, Y. Isolation and comparative analysis of two Na+/H+ antiporter NHX2 genes from Pyrus betulaefolia. Plant Mol. Biol. Rep. 2018, 36, 439–450. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Subreads Base (G) | Subreads Number | Average Length of Subreads | N30 | N50 | N90 |
|---|---|---|---|---|---|---|
| N. tangutorum | 21.83 | 17,951, 056 | 1216 | 1788 | 1427 | 744 |
| Sample | CCS | Nfl-Reads | Flnc-Reads | Mean-Flnc | Consensus Reads |
|---|---|---|---|---|---|
| N. tangutorum | 21.83 | 179,510, 56 | 1216 | 1788 | 1427 |
| Feature | Number |
|---|---|
| Total number of sequences examined | 71,089 |
| Total size of examined sequences (bp) | 111,431,918 |
| Identified SSRs | 51,875 |
| SSR containing sequences | 29,249 |
| Sequences containing more than 1 SSR | 11,082 |
| SSRs present in compound formation | 12,283 |
| Mononucleotides | 37,182 |
| Dinucleotides | 7804 |
| Trinucleotides | 6294 |
| Tetranucleotides | 330 |
| Pentanucleotides | 89 |
| Hexanucleotides | 176 |
| Annotated Databases | Number of Unigenes | 300–1000 bp | ≥1000 bp |
|---|---|---|---|
| COG | 26,526 | 3596 | 22,903 |
| GO | 45,222 | 7626 | 37,501 |
| KEGG | 20,548 | 3528 | 16,970 |
| KOG | 40,450 | 6479 | 33,887 |
| Pfam | 47,073 | 6746 | 40,303 |
| Swiss-Prot | 51,635 | 8760 | 42,752 |
| Nr | 65,055 | 12,596 | 52,211 |
| All | 65,361 | 12,728 | 52,376 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, L.; Lu, L.; Yang, L.; Hao, Z.; Chen, J.; Cheng, T. The Full-Length Transcriptome Sequencing and Identification of Na+/H+ Antiporter Genes in Halophyte Nitraria tangutorum Bobrov. Genes 2021, 12, 836. https://doi.org/10.3390/genes12060836
Zhu L, Lu L, Yang L, Hao Z, Chen J, Cheng T. The Full-Length Transcriptome Sequencing and Identification of Na+/H+ Antiporter Genes in Halophyte Nitraria tangutorum Bobrov. Genes. 2021; 12(6):836. https://doi.org/10.3390/genes12060836
Chicago/Turabian StyleZhu, Liming, Lu Lu, Liming Yang, Zhaodong Hao, Jinhui Chen, and Tielong Cheng. 2021. "The Full-Length Transcriptome Sequencing and Identification of Na+/H+ Antiporter Genes in Halophyte Nitraria tangutorum Bobrov" Genes 12, no. 6: 836. https://doi.org/10.3390/genes12060836