1. Introduction
The identification of new plant viruses using viral metagenomics has been described in numerous publications [
1,
2]. While the wild plant species in natural settings are known to potentially harbor uncharacterized viruses, most metagenomics studies have focused on the cultivated plant species [
2]. The introduction of new crops to areas where they have never been grown before, as well as the implementation of intensive cropping systems, both of which lead to new encounters with virulent viruses infecting crops or indigenous plants, has facilitated the emergence of damaging virus epidemics around the world [
3]. In Florida, the low-chill southern highbush blueberries (SHB) are cultivated near wild plants of the same and related species. The diverse communities of native
Vaccinium spp. and the adjacent Florida’s blueberry production areas could serve as a reservoir for a diverse assemblage of viruses in these species, thus causing spillover and spillback between the wild and cultivated hosts [
4]. Another pathway that can potentially cause new emerging viruses in cultivated
Vaccinium spp. is the lack of virus screening prior to the use of native, wild blueberries in the development of new SHB cultivars.
Fifteen species of viruses have been recorded in highbush, lowbush, and rabbiteye blueberries around the world, including RNA and DNA viruses from eight known and two unassigned genera [
5]. Only two virus species have been documented in Florida: blueberry necrotic ring blotch virus (BNRBV) (genus
Blunervirus) [
6] and blueberry red ringspot virus (BRRV) (genus
Soymovirus) [
7]. Since viral diseases are not currently a major threat to the Florida blueberry industry, no survey on the status of blueberry viruses has been performed. Therefore, this study with the following objectives was carried out to (1) characterize and compare the virus population of the wild and cultivated
Vaccinium corymbosum in Florida using a metagenomic approach; and (2) identify and characterize novel and known viral sequences in the blueberry viromes. We showed that analysis of viromes determined from double-stranded RNA (dsRNA)-enriched samples using metagenomics is an excellent tool to explore viral diversity of RNA viruses of all genome types (dsRNA, positive- and negative-sense single-stranded RNA (ssRNA)). Metagenomic analysis of the RNA viromes has shown that viral diversity in the wild
V. corymbosum is found to be greater than that in the cultivated species, with considerable overlap and unique genera represented in both species. This study has also led to the discovery of two known blueberry viruses, BlLV (genus
Amalgavirus) and BlMaV (genus
Ophiovirus), that are new to Florida, as well as a tentative novel tepovirus (BlVT) that has never been reported in blueberry. In addition, this research is the first to show that BlMaV can be found in wild highbush,
V. corymbosum. 4. Discussion
Without doubt, plant viral metagenomics has contributed to our understanding of viral populations, as well as unravelling the etiology of viral diseases in various plant species. This is supported by the exponentially increased discovery of novel plant viruses that have been thoroughly described [
1,
2]. In this study, viral populations in the wild and cultivated species of
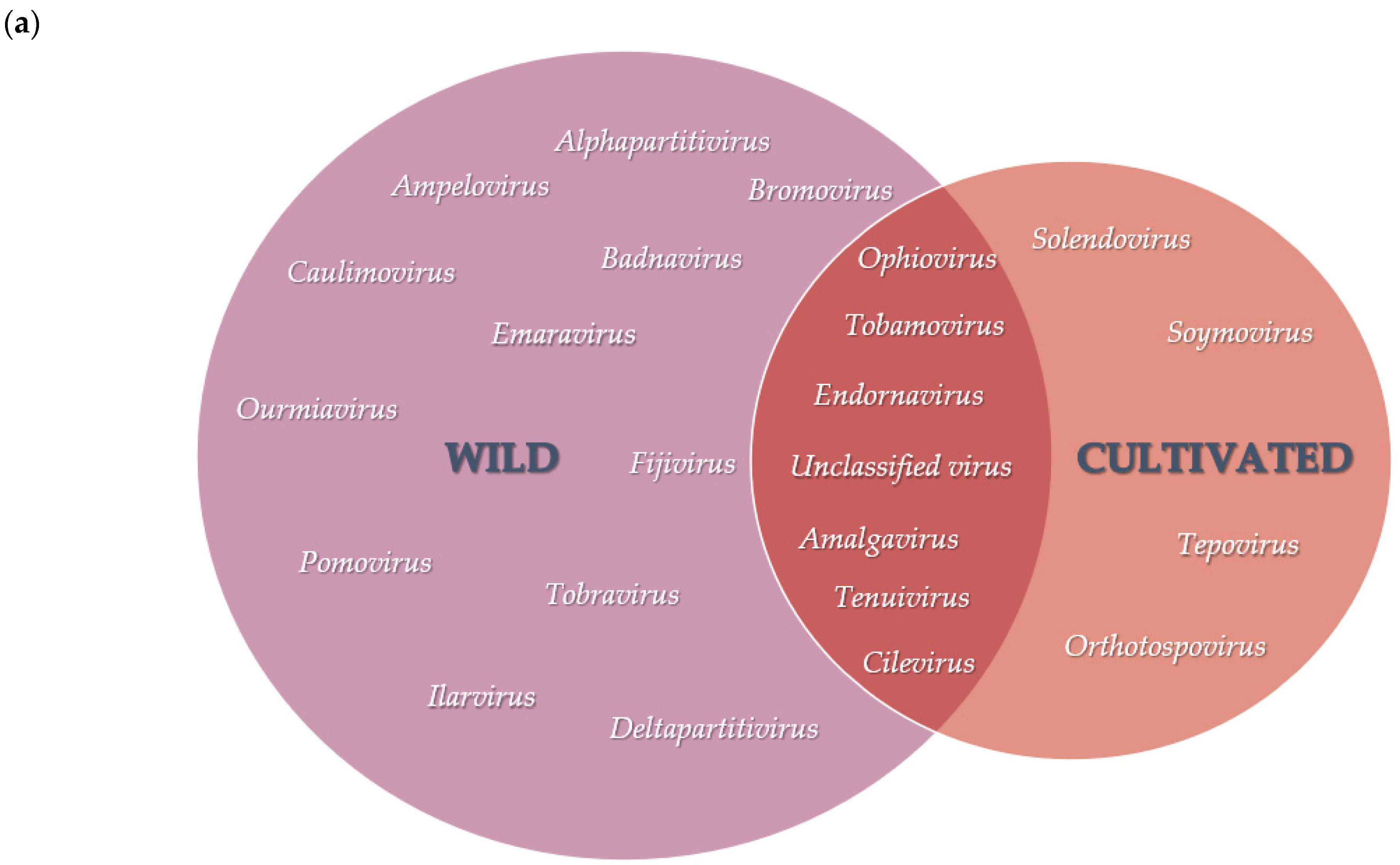
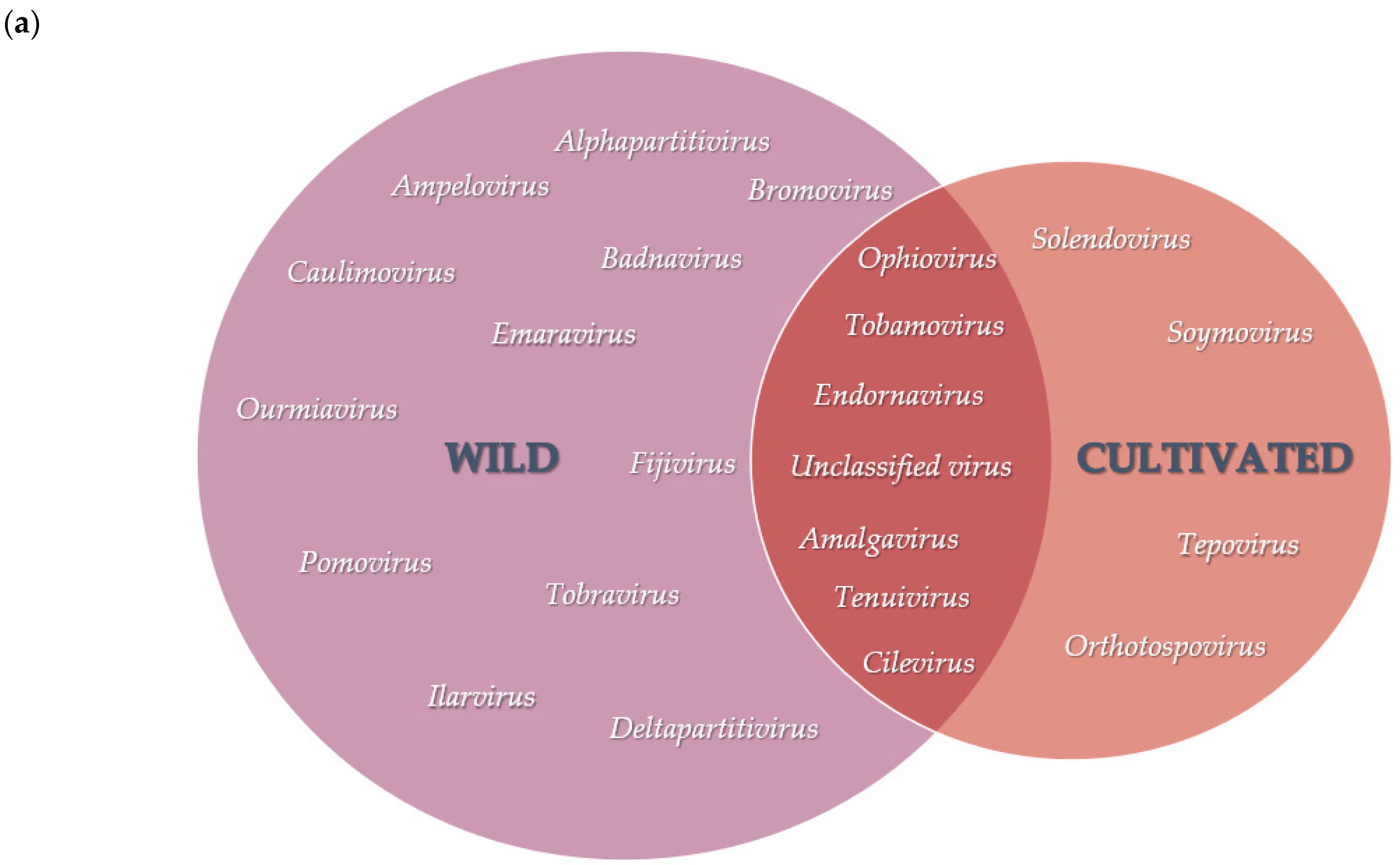
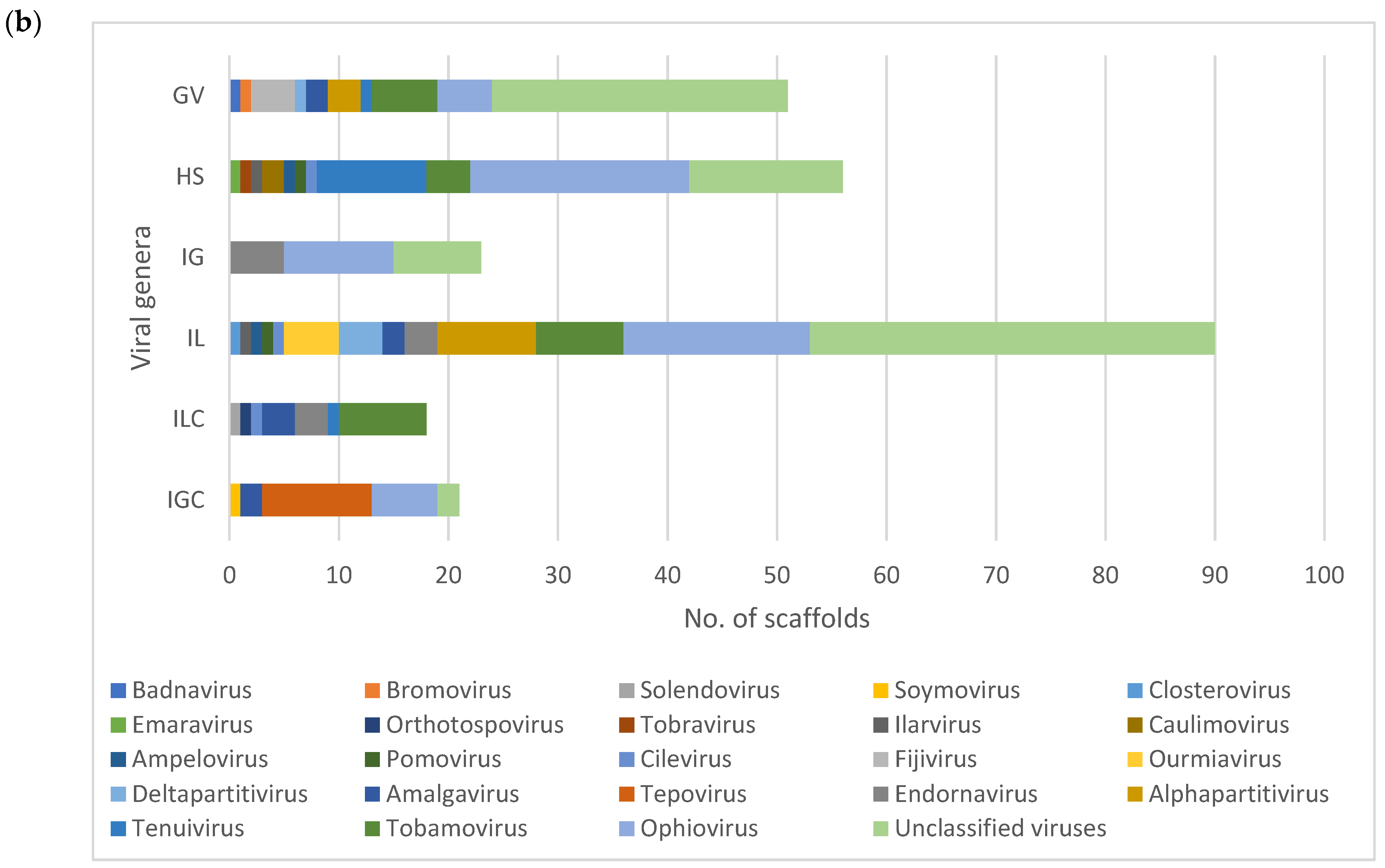
V. corymbosum, collected from different locations in Florida, were characterized using a viral metagenomic approach. Less than 2% of the de novo assembled scaffolds obtained from all the RNA viromes were considered as putative plant virus scaffolds because the majority of scaffolds produced sequence similarity to either insect or fungal viruses (data not shown). The presence of scaffolds with the highest sequence match to viruses from a total of 23 genera indicated that the population of viruses in this host are greatly diverse. This is in agreement with the findings of diverse viral species in previous studies of various wild plant species through viral metagenomics [
21,
22,
23,
24,
25]. A greater virus diversity was further observed in the viral population of wild
V. corymbosum native to Florida, as shown by the number of viral genera, which is almost twice more than those of cultivated viromes. Several conditions could contribute to the greater diversity of viruses in the viral population of the wild
V. corymbosum compared to the cultivated ones. These conditions include the presence of different wild
Vaccinium species and other plant communities within and surrounding the areas, the occurrence of vectors, and abiotic factors at these locations. In addition, the presence of diverse plant communities within and surrounding the wild
V. corymbosum may act as a virus reservoir, thus increasing the heterogeneity in the population of viruses due to their movement between these plants. The occurrence of insects as virus vectors in the wild area may be another important factor that contributes to the greater virus diversity in the wild
V. corymbosum by facilitating the movement of viruses between plants. On the other hand, the movement of viruses in the cultivated plants may be restricted due to the implementation of agricultural management practices, which could contribute to the lower number of viral genera in this host.
Metagenomic analysis of the viromes of wild and cultivated species of
V. corymbosum in Florida has enabled the reconstruction of ten consensus sequences of five virus species, including BlLV, BlMaV, BRRV, TMV, and a putative new species closely related to PrVT. Although the complete genome of TMV was detected in four out of six viromes, TMV was not included for further sequences analysis since it is well known that tobamoviruses are frequently found in various environments due to their high stability [
26]. In addition, the presence of varying frequency of SNPs from the mapping of reads to the assembled viral genomes in the corresponding viromes suggests the possible presence of virus variants in the pooled samples. Although the assembled viral sequences could represent a consensus of various genomes, this does not affect the overall findings of this study, in which the metagenomic approach has led to the detection of several known and new viruses in wild and cultivated blueberry in Florida.
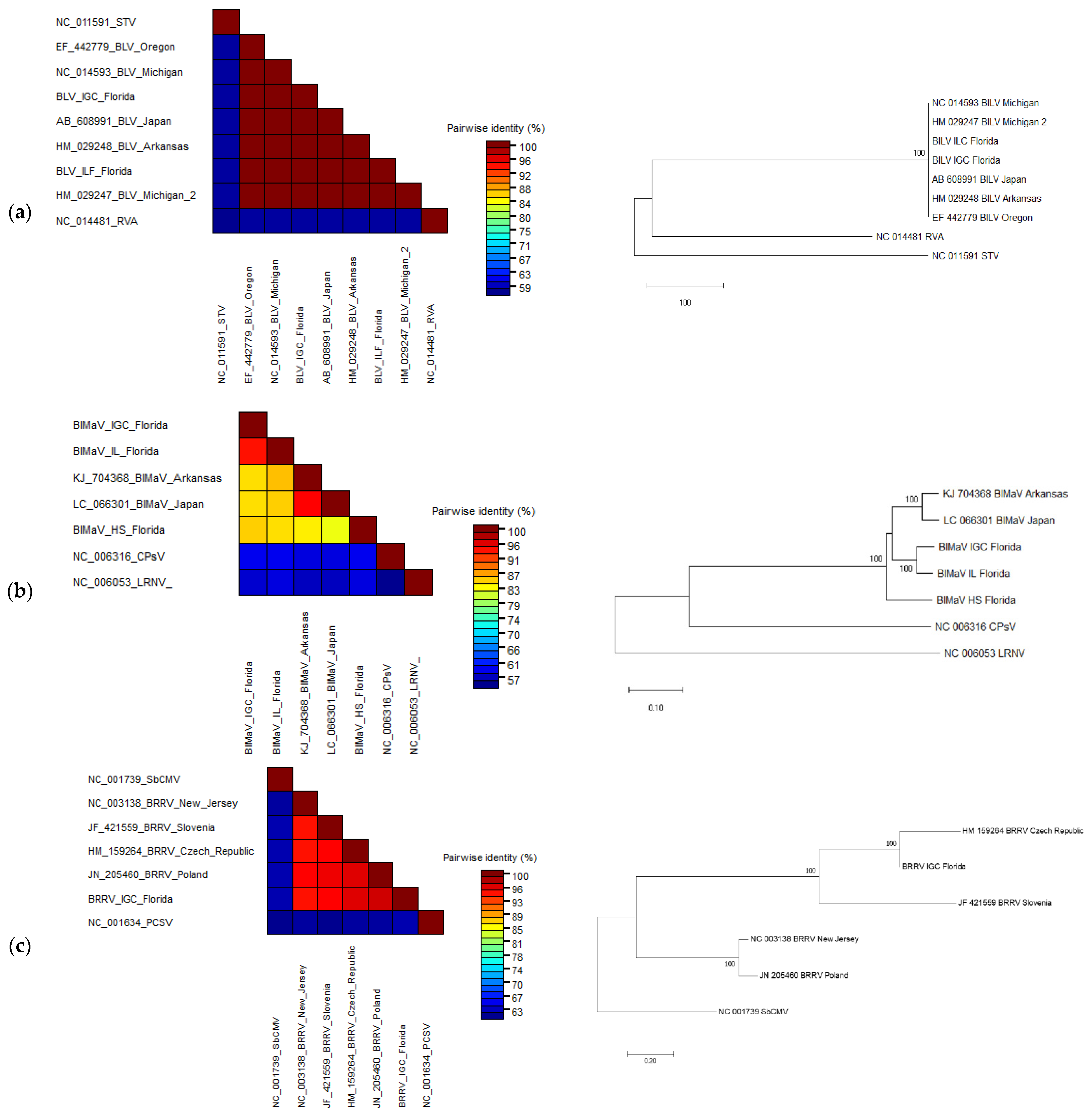
The complete genomes of BlLV, an amalgavirus, were recovered from viromes of cultivated
V. corymbosum. Pairwise sequence comparison between the whole genome of BlLV from Florida with other published isolates showed more than 99% nucleotide identity. This finding was expected since it was previously reported that BlLV has a very stable population structure, with less than 0.5% diversity among partial and complete sequences of isolates from Japan and the US [
17,
27,
28]. Phylogenetic analysis of the complete genome of BlLV isolates from Arkansas, Florida, Michigan, Oregon showed that these isolates clustered together, suggesting that the complete genome of BlLV recovered from Florida is another isolate of BlLV.
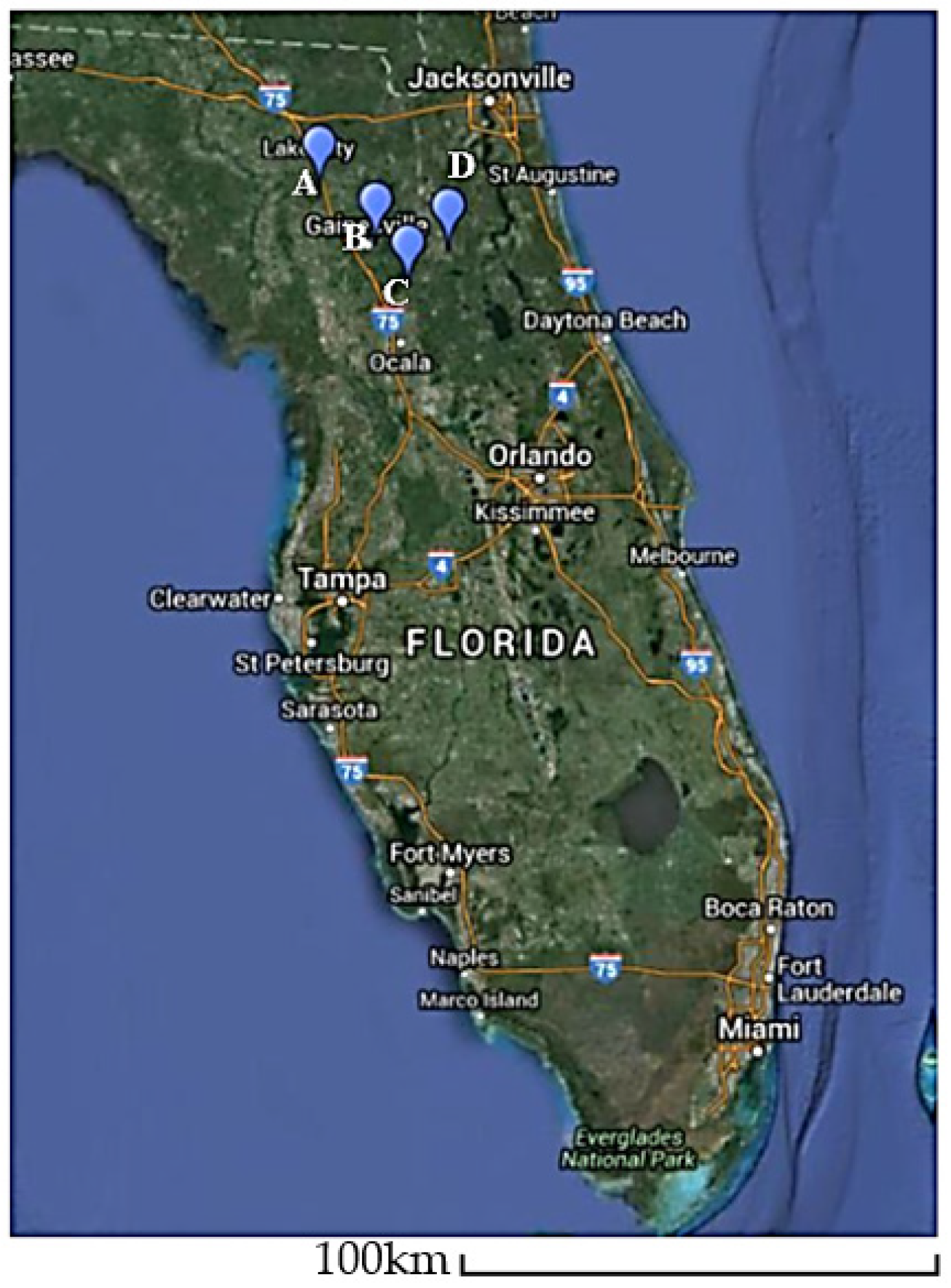
Scaffolds with high homology (96–97%) to an ophiovirus, BlMaV, were identified in all viromes except the cultivated
V. corymbosum virome at Interlachen. The presence of scaffolds closely related to BlMaV in all viromes of wild
V. corymbosum suggests that the occurrence of this virus in wild blueberry might be common. Pairwise comparison using the NP regions of BlMaV isolates assembled from this study (Island Grove, Interlachen, and High Springs) and those from Arkansas and Japan indicated that the isolate from Island Grove and Interlachen had 14% sequence divergence from the rest of the isolates. This result was supported by a previous study which found that BlMaV had low genetic diversity among isolates, as shown by 13% nt divergence in the NP regions [
29]. In contrast, the isolate from High Springs showed up to ~17% nt divergent with other isolates, thus suggesting that this sequence might have evolved from reassortment between BlMaV variants. In addition, phylogenetic analysis showed that the High Springs isolate form a separate branch from those of Island Grove and Interlachen, suggesting that BlMaV could have been established earlier in the wild
V. corymbosum at High Springs in order for diversification to occur.
The analysis of the RNA virome additionally allowed for the assembly of a complete BRRV genome, a dsDNA virus, from the virome of cultivated
V. corymbosum at Island Grove. The detection of BRRV in the RNA virome is not surprising since it produces RNA/DNA intermediate during its replication cycle, thus making it possible to form dsRNA complex at some point. The genome organization and ORFs of BRRV isolate from Island Grove are similar to the recently published BRRV genomes identified from the cultivar “Emerald” in Florida [
7]. Overall, whole-genome pairwise comparison analysis of the BRRV isolates from the Czech Republic, Island Grove, New Jersey, Poland, and Slovenia showed that these isolates had less than 10% nt divergence, implying low genetic diversity and thus suggesting that this virus has a very stable population structure. In addition, pairwise comparison and phylogenetic analysis indicated that BRRV isolate from Island Grove was closely related to an isolate from the Czech Republic, suggesting that there may be an exchange of BRRV-infected material between these regions.
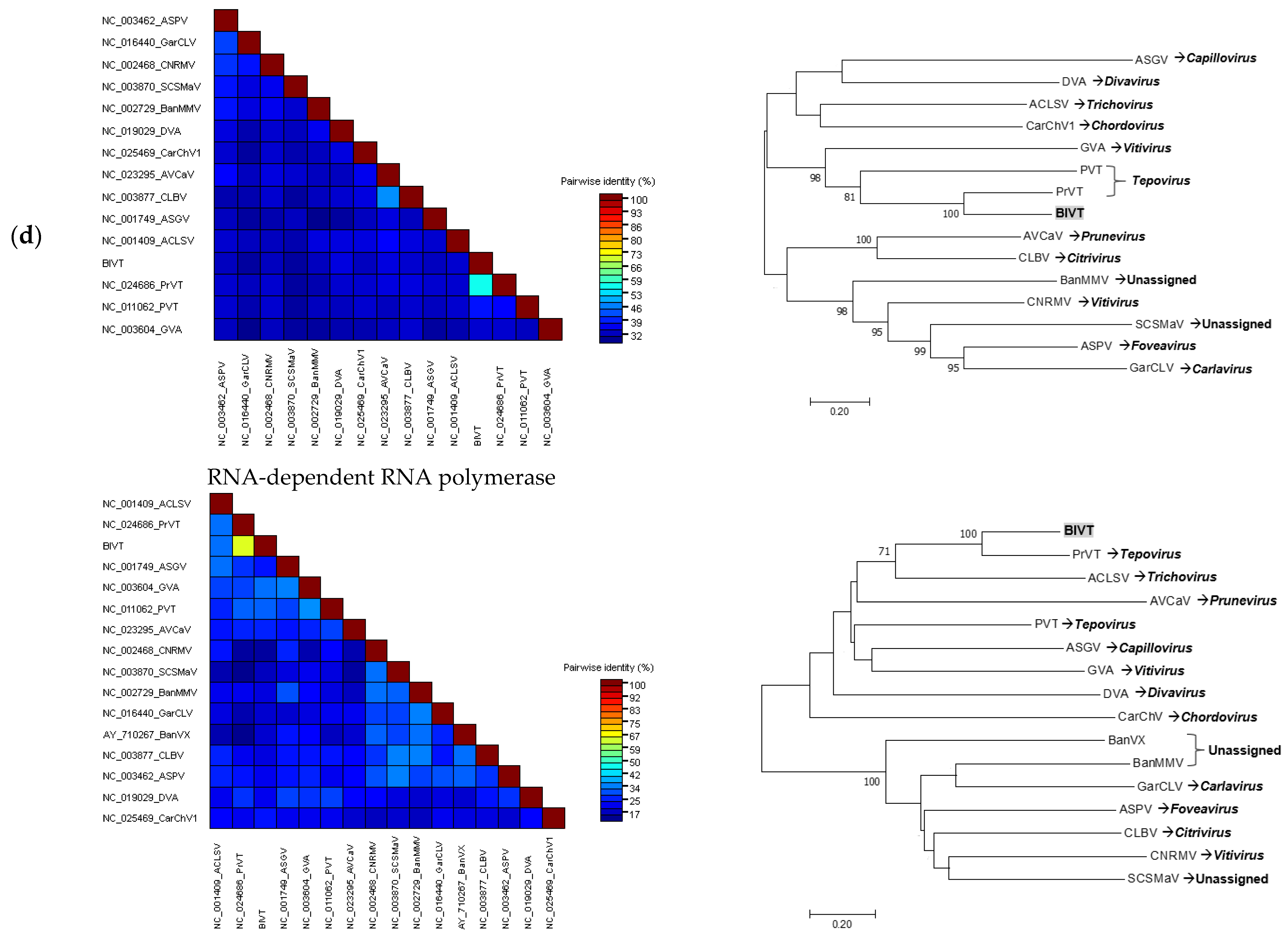
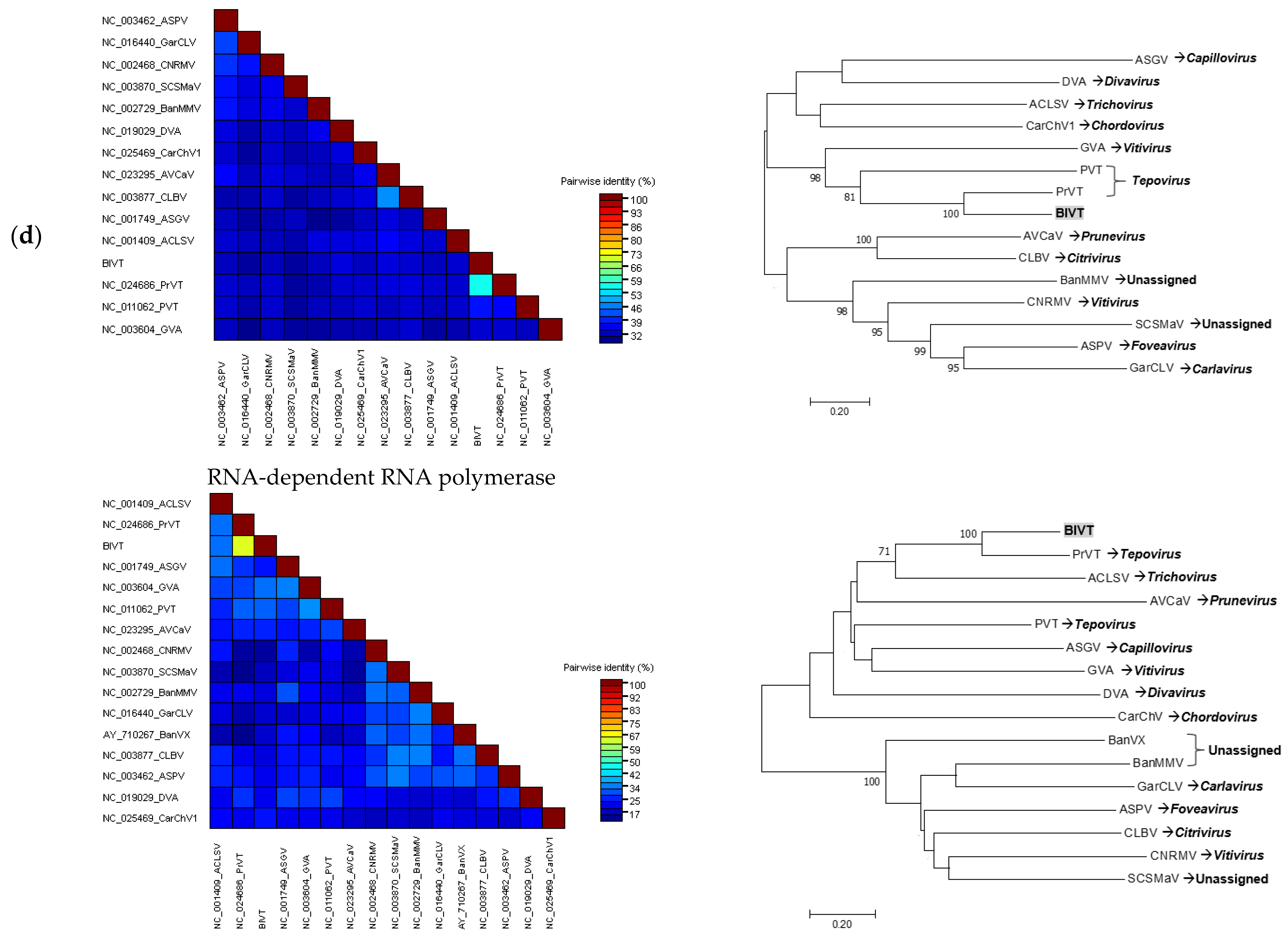
In addition to the detection of known viruses of blueberry that are new to Florida, this study also led to the discovery of a novel virus that is closely related to PrVT, a member in the genus
Tepovirus belonging to the family
Betaflexiviridae. Partial resequencing completely validated the CP region of the de novo assembled scaffold obtained by metagenome analysis. Pairwise comparison of the RdRp and CP of the putative novel virus with the corresponding proteins of selected members representing different genera in the family
Betaflexiviridae demonstrated that this novel virus was distantly related to other members of the respective viral family, though higher sequence homology was observed with PrVT. Furthermore, pairwise nucleotide comparison showed that the putative novel virus only shared 61 and 65% identity with the RdRp and CP of PrVT, respectively, which fall well below the currently accepted species demarcation criteria in the genus
Tepovirus, described as having less than 72% nt identity between the RdRp and CP genes [
30]. Phylogenetic analyses of these genes demonstrated that the novel virus is consistently grouped with PrVT. The similar size and organization of the new viral genome to other members of the family
Betaflexiviridae as well as statistically supported phylogenetic grouping further suggested that this novel virus, proposed as BlVT, should be considered as a new species in the genus
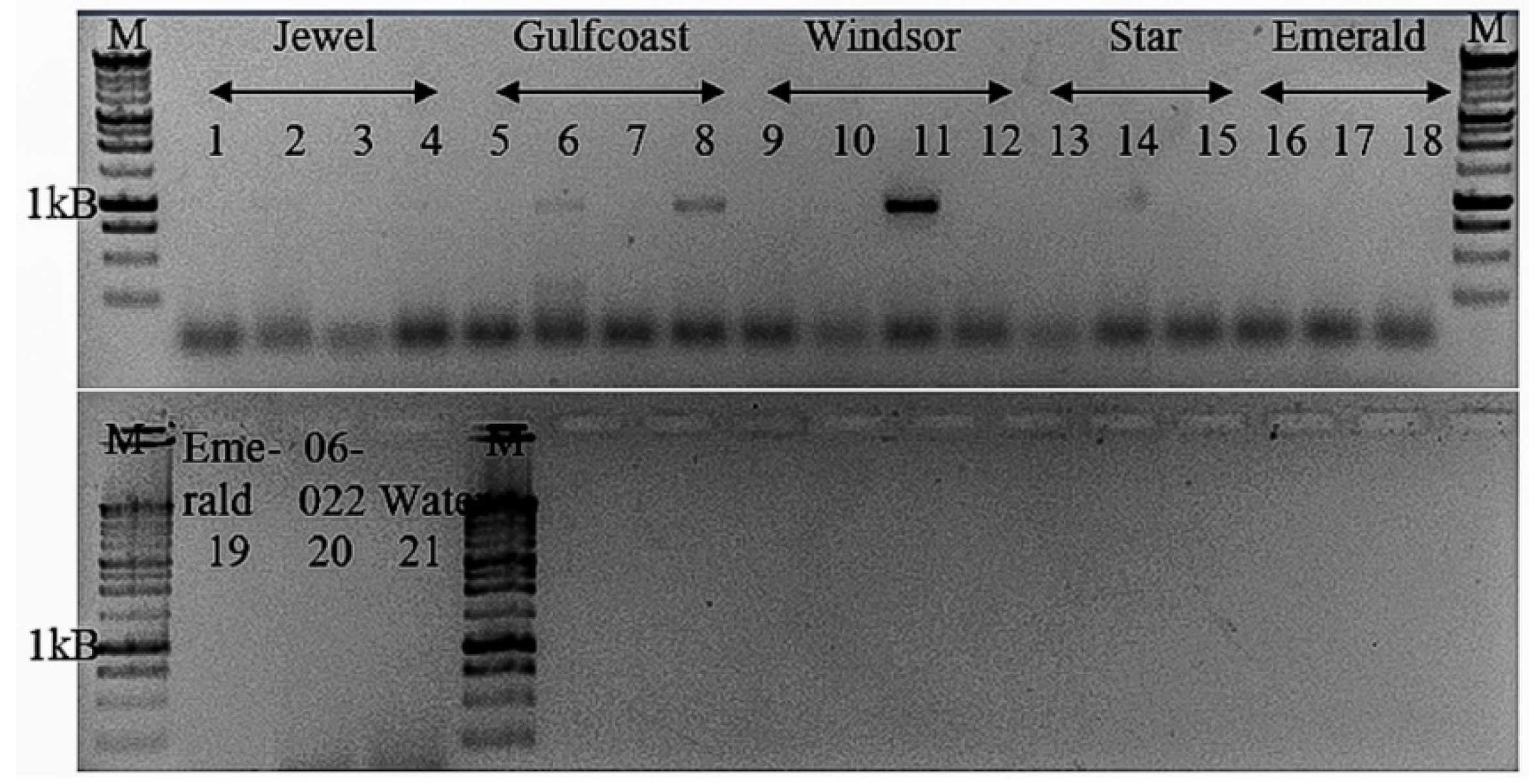
Tepovirus. The biological information regarding BlVT is still lacking due to the unknown vector as well as the limited knowledge on the spread of this virus in the field. Although molecular screening of BlVT in twenty
V. corymbosum samples collected from Island Grove, Florida, indicated a 15% virus incidence, the infection of BlVT still could not be associated with specific virus symptoms at this point due to mixed viral infections in the plants. Hence, the primers developed in this study could potentially be used for detection of BlVT in blueberry or related crops in the future.


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}