
Synthesis, Properties, and Electrochemistry of bis(iminophosphorane)pyridine Iron(II) Pincer Complexes

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
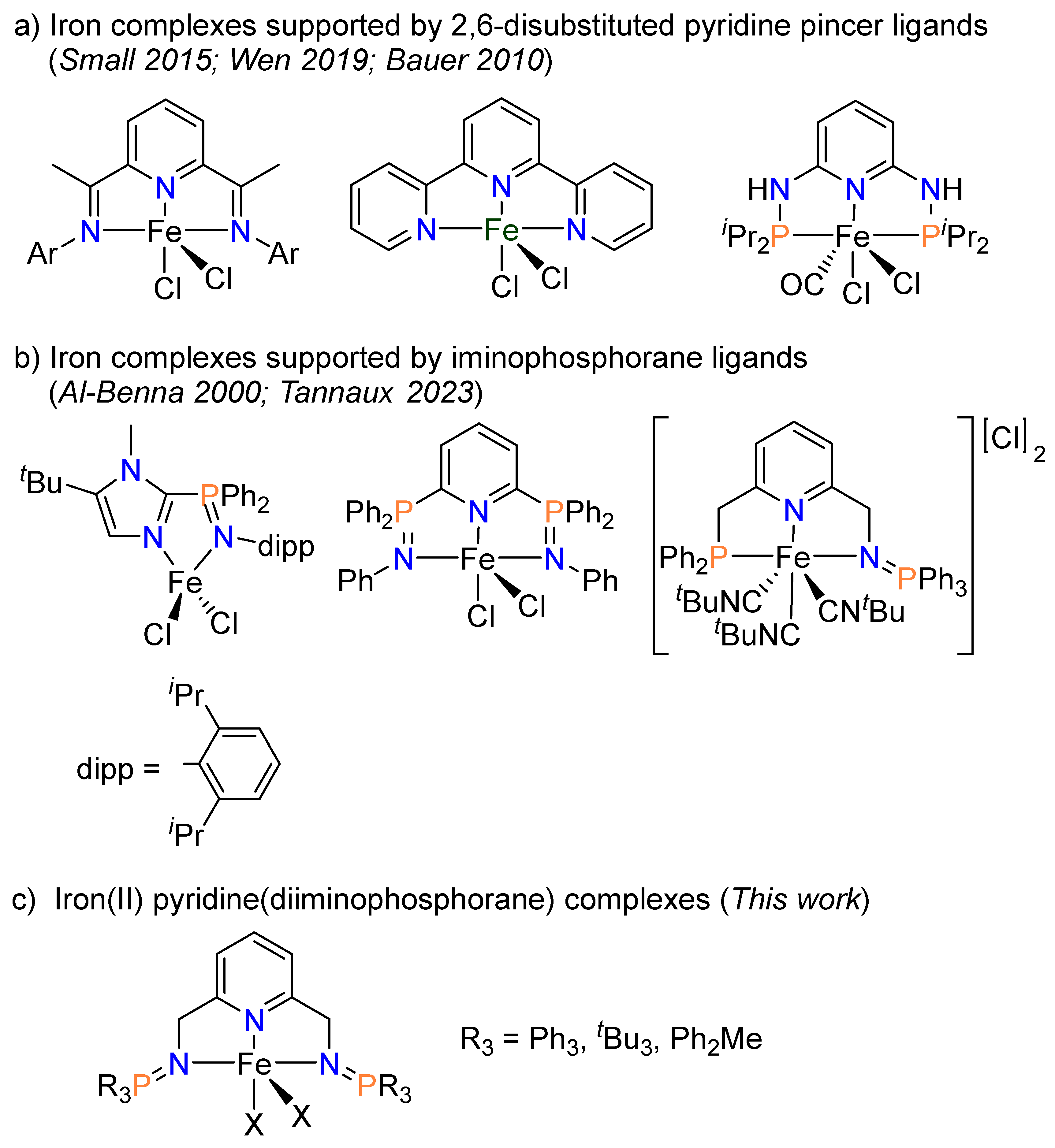
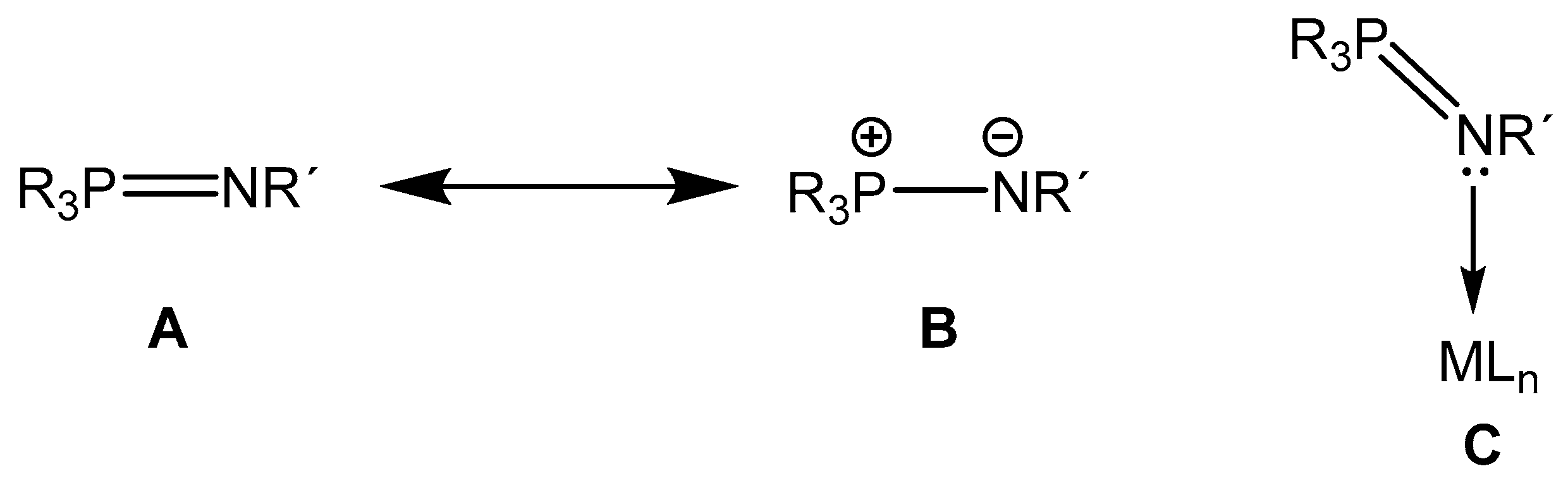
1. Introduction
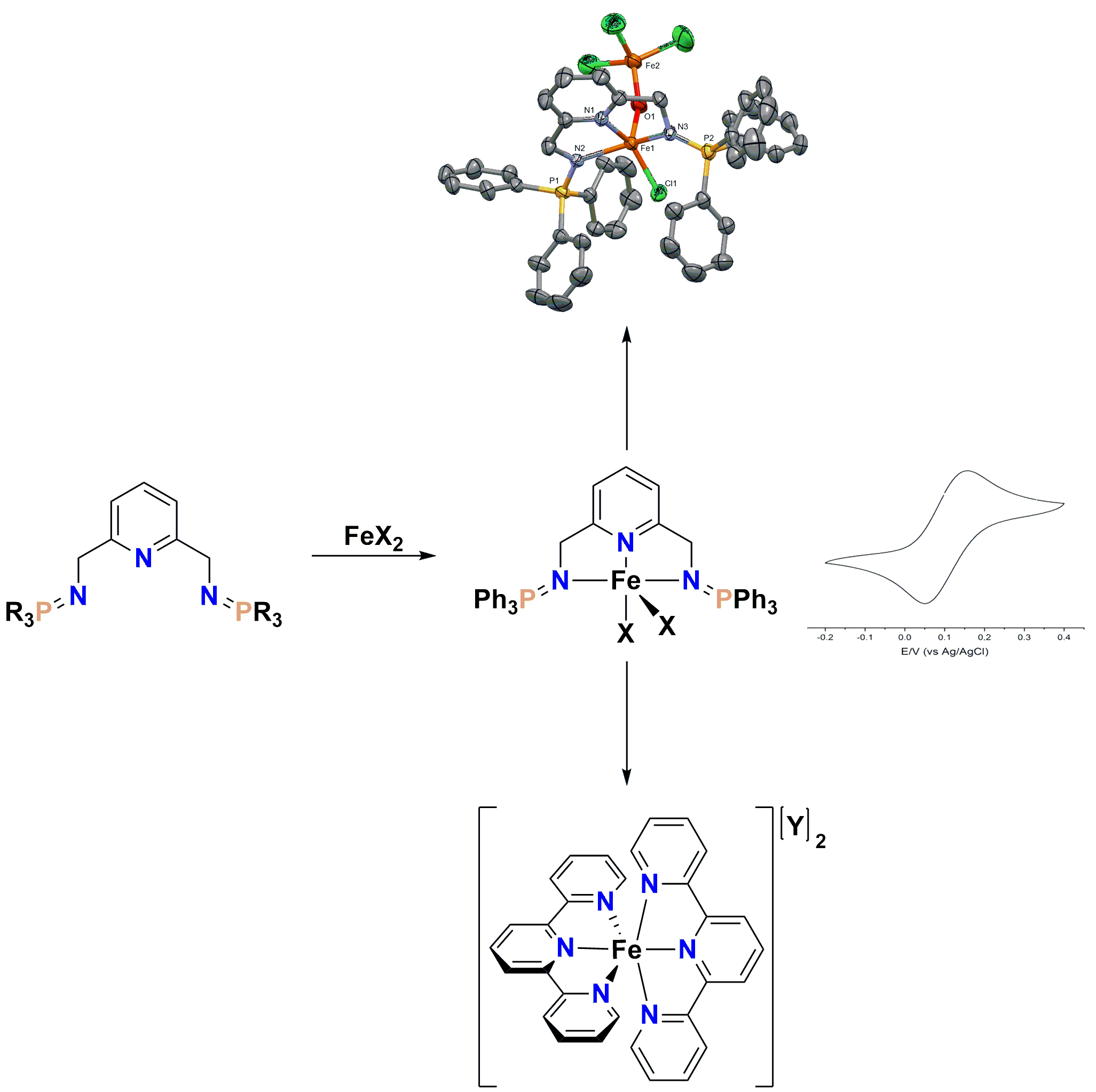
2. Results and Discussion
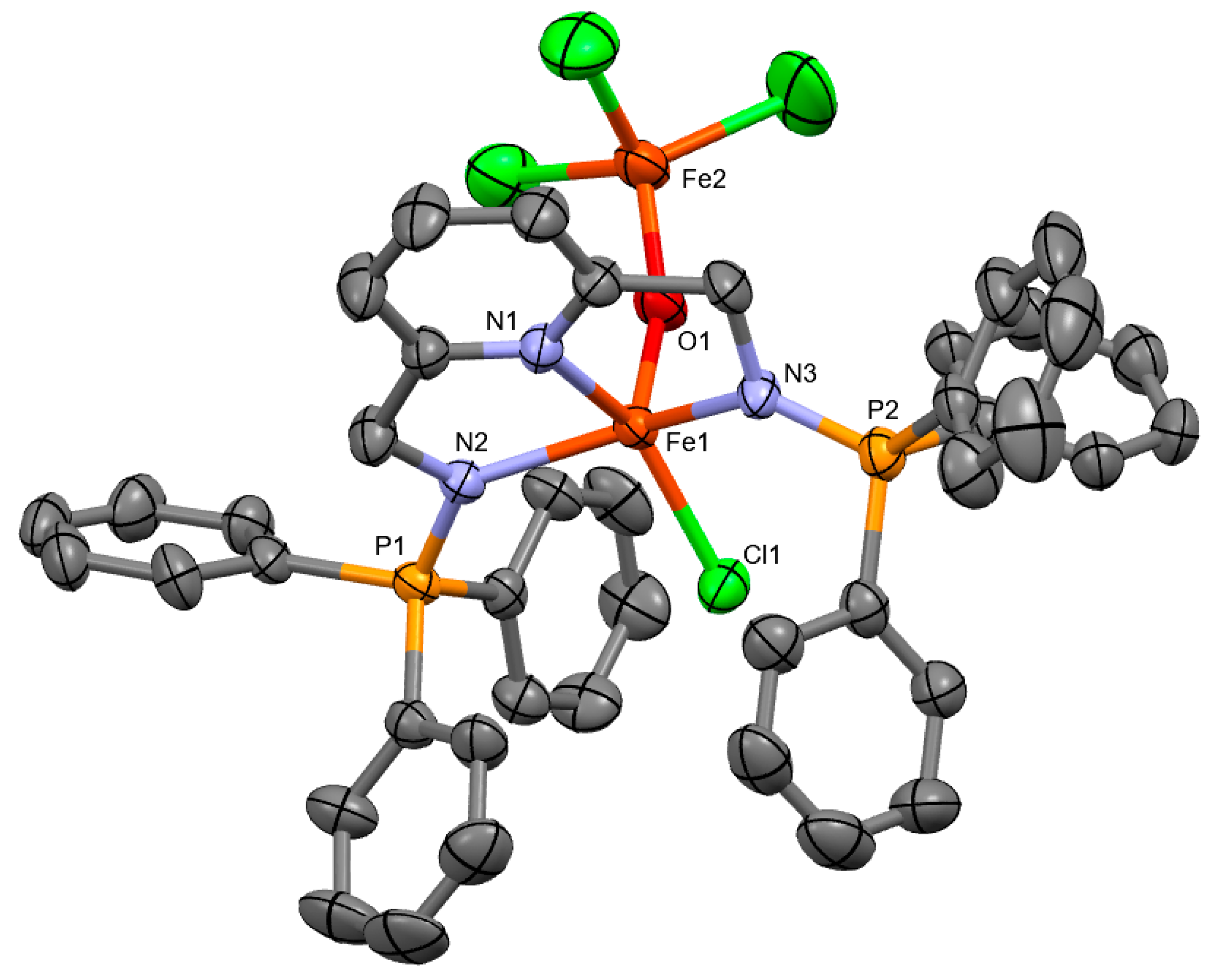
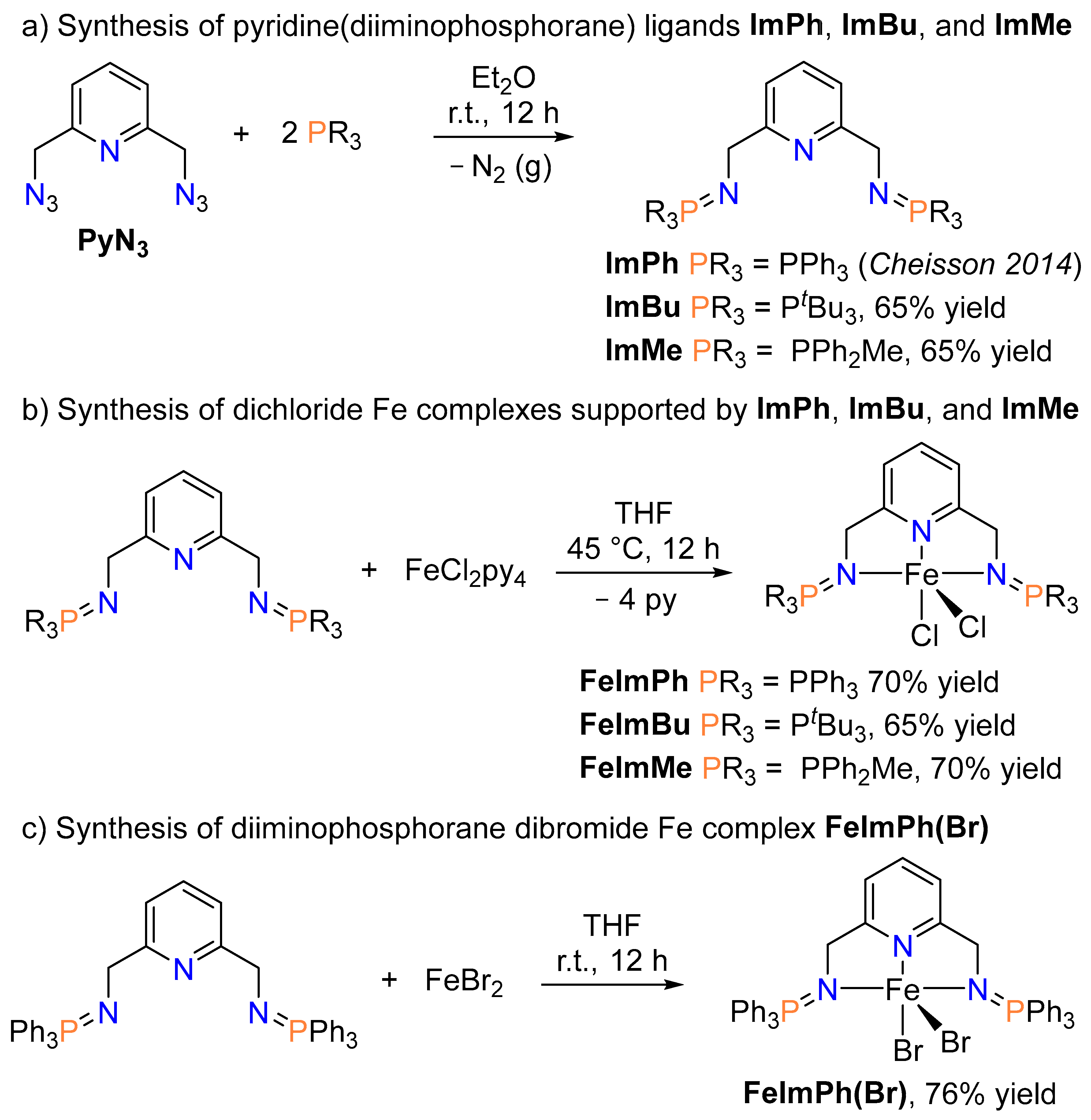
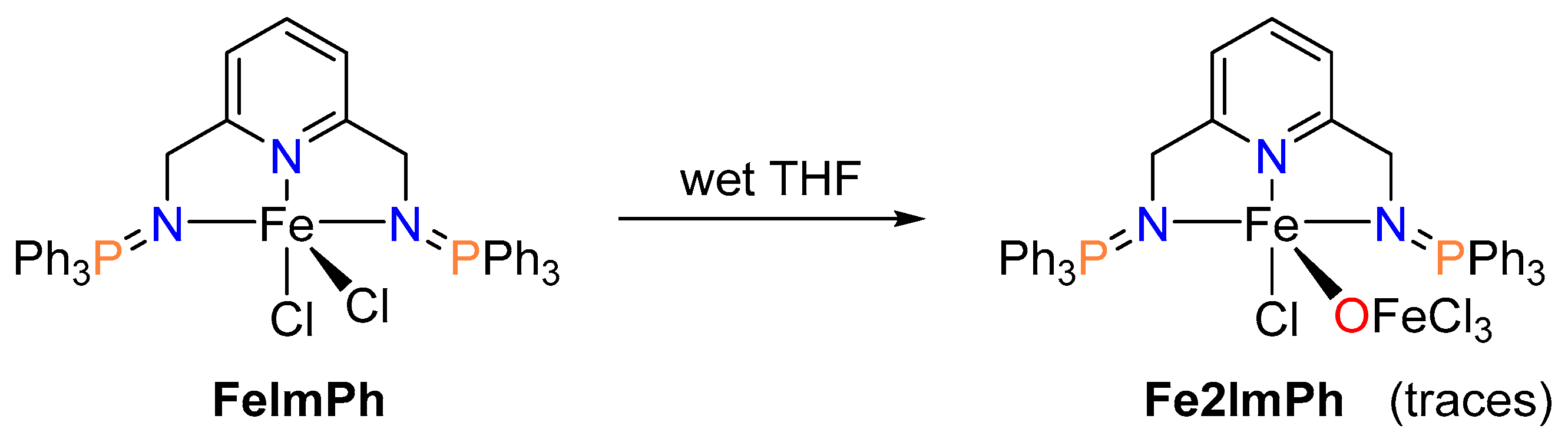
2.1. Synthesis of Ligands and Iron Complexes
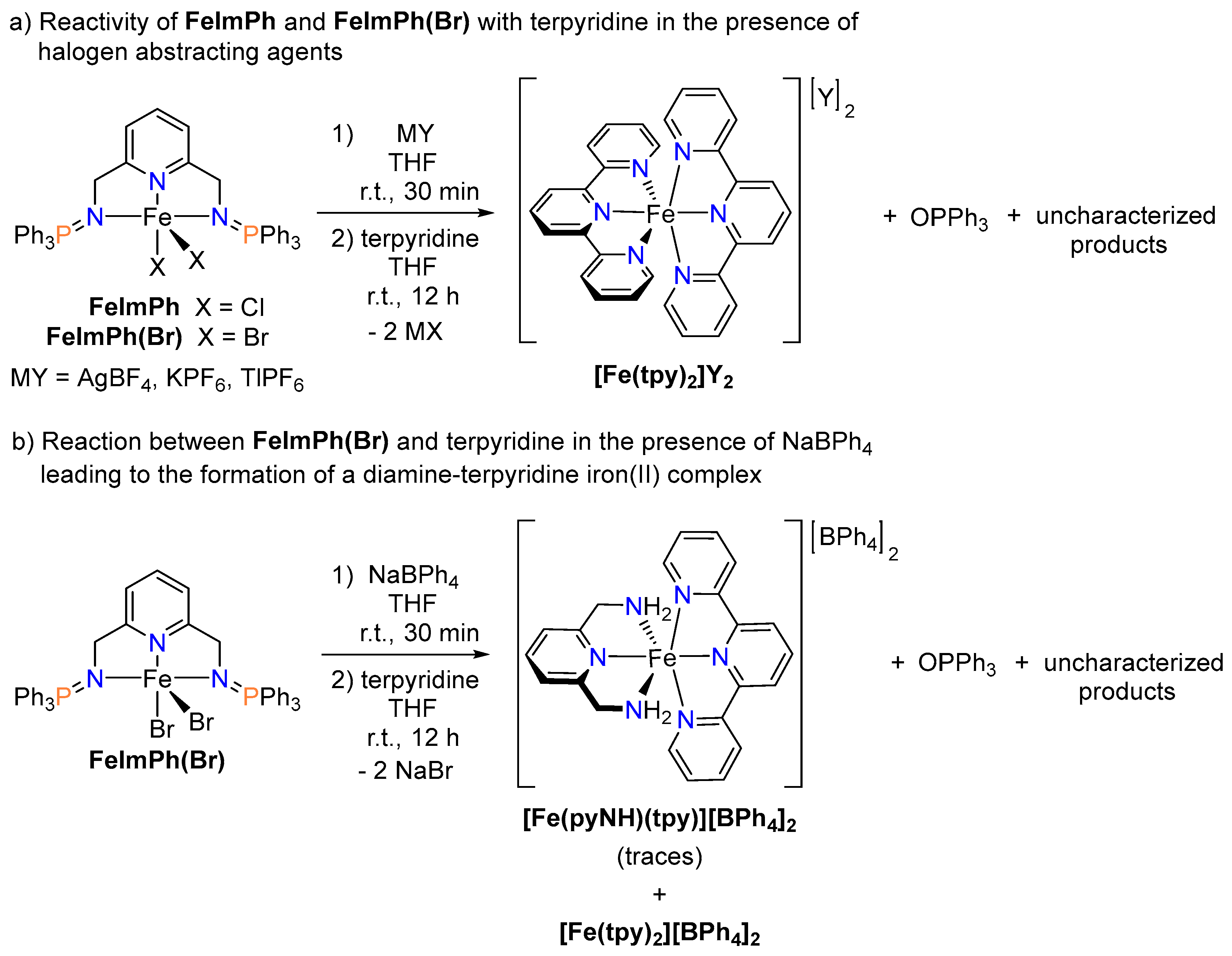
2.2. Efforts toward the Synthesis of Heteroleptic Diiminophosphorane–Terpyridine Iron(II) Complexes
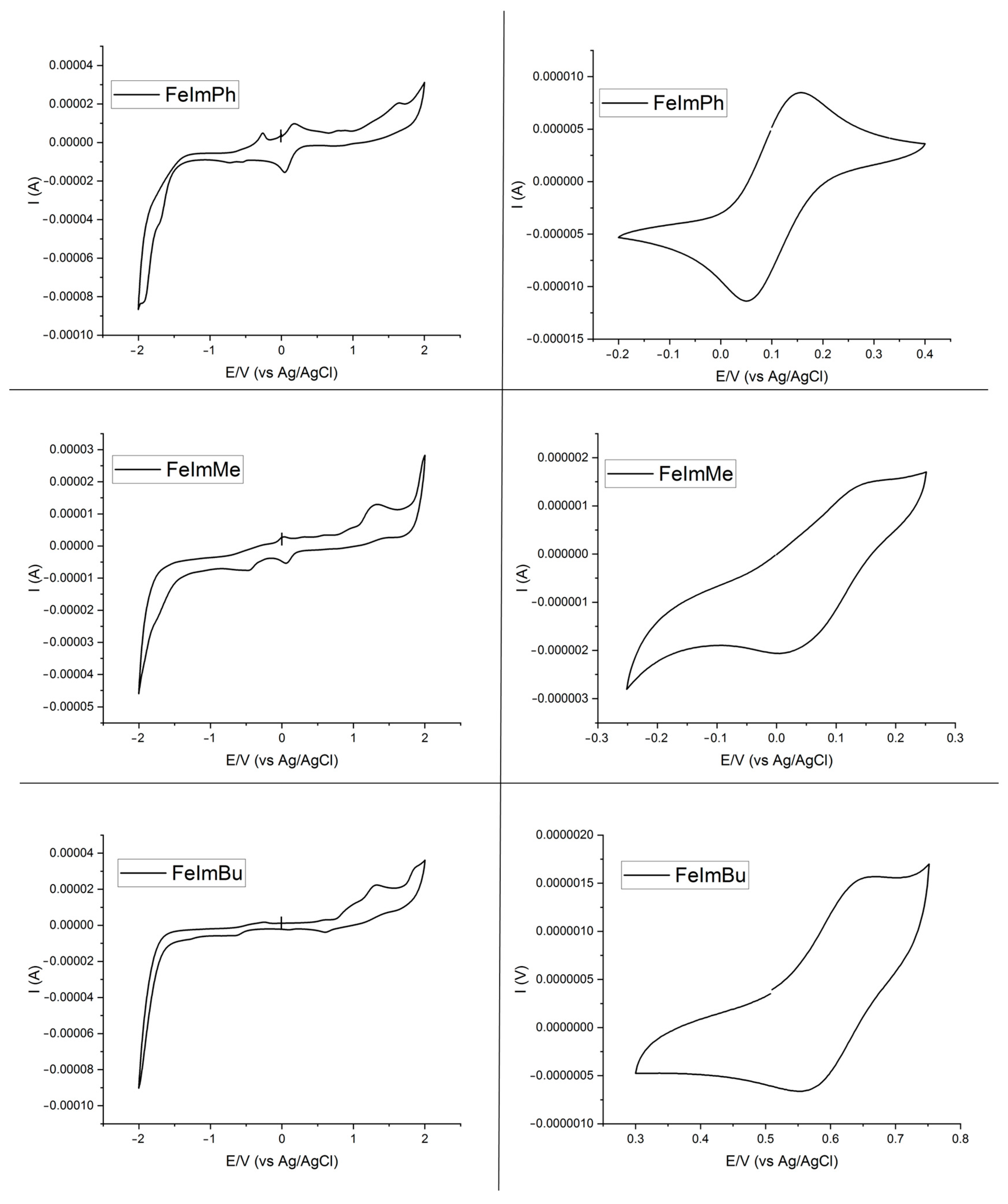
2.3. Cyclic Voltammetry Studies
3. Materials and Methods
3.1. Materials and Reagents
3.2. Synthesis of ImBu
3.3. Synthesis of ImMe
3.4. Synthesis of FeImPh
3.5. Synthesis of FeImBu
3.6. Synthesis of FeImMe
3.7. Synthesis of FeImPh(Br)
3.8. Reaction between FeImPh and terpyridine
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Small, B.L. Discovery and Development of Pyridine-bis(imine) and Related Catalysts for Olefin Polymerization and Oligomerization. Acc. Chem. Res. 2015, 48, 2599–2611. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Liu, G.; Huang, Z. Recent advances in tridentate iron and cobalt complexes for alkene and alkyne hydrofunctionalizations. Coord. Chem. Rev. 2019, 386, 138–153. [Google Scholar] [CrossRef]
- Bauer, G.; Hu, X. Recent developments of iron pincer complexes for catalytic applications. Inorg. Chem. Front. 2016, 3, 741–765. [Google Scholar] [CrossRef]
- Tondreau, A.M.; Milsmann, C.; Patrick, A.D.; Hoyt, H.M.; Lobkovsky, E.; Wieghardt, K.; Chirik, P.J. Synthesis and Electronic Structure of Cationic, Neutral, and Anionic Bis(imino)pyridine Iron Alkyl Complexes: Evaluation of Redox Activity in Single-Component Ethylene Polymerization Catalysts. J. Am. Chem. Soc. 2010, 132, 15046–15059. [Google Scholar] [CrossRef] [PubMed]
- Arevalo, R.; Chirik, P.J. Enabling Two-Electron Pathways with Iron and Cobalt: From Ligand Design to Catalytic Applications. J. Am. Chem. Soc. 2019, 141, 9106–9123. [Google Scholar] [CrossRef] [PubMed]
- García-Álvarez, J.; García-Garrido, S.E.; Cadierno, V. Iminophosphorane–phosphines: Versatile ligands for homogeneous catalysis. J. Organomet. Chem. 2014, 751, 792–808. [Google Scholar] [CrossRef]
- Al-Benna, S.; Sarsfield, M.J.; Thornton-Pett, M.; Ormsby, D.L.; Maddox, P.J.; Brès, P.; Bochmann, M. Sterically hindered iminophosphorane complexes of vanadium, iron, cobalt and nickel: A synthetic, structural and catalytic study †. J. Chem. Soc. Dalton Trans. 2000, 4247–4257. [Google Scholar] [CrossRef]
- Tannoux, T.; Mazaud, L.; Cheisson, T.; Casaretto, N.; Auffrant, A. Fe(II) complexes supported by an iminophosphorane ligand: Synthesis and reactivity. Dalton Trans. 2023, 52, 12010–12019. [Google Scholar] [CrossRef] [PubMed]
- Sudhakar, P.V.; Lammertsma, K. Nature of bonding in phosphazoylides. A comparative study of N2H4, NPH4, and P2H4. J. Am. Chem. Soc. 2002, 113, 1899–1906. [Google Scholar] [CrossRef]
- Molina, P.; Alajarin, M.; Lopez Leonardo, C.; Claramunt, R.M.; Foces-Foces, M.d.l.C.; Hernandez Cano, F.; Catalan, J.; De Paz, J.L.G.; Elguero, J. Experimental and theoretical study of the R3P+-X- bond. Case of betaines derived from N-iminophosphoranes and alkyl isocyanates. J. Am. Chem. Soc. 2002, 111, 355–363. [Google Scholar] [CrossRef]
- Tannoux, T.; Auffrant, A. Complexes featuring tridentate iminophosphorane ligands: Synthesis, reactivity, and catalysis. Coord. Chem. Rev. 2023, 474, 214845. [Google Scholar] [CrossRef]
- Chaplin, A.B.; Harrison, J.A.; Dyson, P.J. Revisiting the Electronic Structure of Phosphazenes. Inorg. Chem. 2005, 44, 8407–8417. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.E.; Schleyer, P.v.R. Chemical bonding in hypervalent molecules. The dominance of ionic bonding and negative hyperconjugation over d-orbital participation. J. Am. Chem. Soc. 2002, 112, 1434–1445. [Google Scholar] [CrossRef]
- Winslow, C.; Lee, H.B.; Field, M.J.; Teat, S.J.; Rittle, J. Structure and Reactivity of a High-Spin, Nonheme Iron(III)- Superoxo Complex Supported by Phosphinimide Ligands. J. Am. Chem. Soc. 2021, 143, 13686–13693. [Google Scholar] [CrossRef] [PubMed]
- Hein, N.M.; Pick, F.S.; Fryzuk, M.D. Synthesis and Reactivity of a Low-Coordinate Iron(II) Hydride Complex: Applications in Catalytic Hydrodefluorination. Inorg. Chem. 2017, 56, 14513–14523. [Google Scholar] [CrossRef] [PubMed]
- Spentzos, A.Z.; May, S.R.; Confer, A.M.; Gau, M.R.; Carroll, P.J.; Goldberg, D.P.; Tomson, N.C. Investigating Metal-Metal Bond Polarization in a Heteroleptic Tris-Ylide Diiron System. Inorg. Chem. 2023, 62, 11487–11499. [Google Scholar] [CrossRef] [PubMed]
- Spencer, L.P.; Altwer, R.; Wei, P.; Gelmini, L.; Gauld, J.; Stephan, D.W. Pyridine− and Imidazole−Phosphinimine Bidentate Ligand Complexes: Considerations for Ethylene Oligomerization Catalysts. Organometallics 2003, 22, 3841–3854. [Google Scholar] [CrossRef]
- Oheix, E.; Herrero, C.; Moutet, J.; Rebilly, J.N.; Cordier, M.; Guillot, R.; Bourcier, S.; Banse, F.; Senechal-David, K.; Auffrant, A. Fe(III) and Fe(II) Phosphasalen Complexes: Synthesis, Characterization, and Catalytic Application for 2-Naphthol Oxidative Coupling. Chemistry 2020, 26, 13634–13643. [Google Scholar] [CrossRef]
- Peris, E.; Crabtree, R.H. Key factors in pincer ligand design. Chem. Soc. Rev. 2018, 47, 1959–1968. [Google Scholar] [CrossRef]
- Cheisson, T.; Auffrant, A. Versatile coordination chemistry of a bis(methyliminophosphoranyl)pyridine ligand on copper centres. Dalton Trans. 2014, 43, 13399–13409. [Google Scholar] [CrossRef]
- Cheisson, T.; Ricard, L.; Heinemann, F.W.; Meyer, K.; Auffrant, A.; Nocton, G. Synthesis and Reactivity of Low-Valent f-Element Iodide Complexes with Neutral Iminophosphorane Ligands. Inorg. Chem. 2018, 57, 9230–9240. [Google Scholar] [CrossRef]
- Cheisson, T.; Auffrant, A.; Nocton, G. η5–η1 Switch in Divalent Phosphaytterbocene Complexes with Neutral Iminophosphoranyl Pincer Ligands: Solid-State Structures and Solution NMR 1JYb–P Coupling Constants. Organometallics 2015, 34, 5470–5478. [Google Scholar] [CrossRef]
- Staudinger, H.; Meyer, J. Über neue organische phosphorverbindungen III. Phosphinmethylenderivate und phosphinimine. Helv. Chim. Acta 1919, 2, 635–646. [Google Scholar] [CrossRef]
- Gorenstein, D.G. Conformation and Dynamics of DNA and Protein-DNA Complexes by 31P NMR. Chem. Rev. 1994, 94, 1315–1338. [Google Scholar] [CrossRef]
- Kuveke, R.E.H.; Barwise, L.; van Ingen, Y.; Vashisth, K.; Roberts, N.; Chitnis, S.S.; Dutton, J.L.; Martin, C.D.; Melen, R.L. An International Study Evaluating Elemental Analysis. ACS Cent. Sci. 2022, 8, 855–863. [Google Scholar] [CrossRef]
- Small, B.L.; Brookhart, M. Polymerization of propylene by a new generation of iron catalysts: Mechanisms of chain initiation, propagation, and termination. Macromolecules 1999, 32, 2120–2130. [Google Scholar] [CrossRef]
- Bocian, A.; Napierała, S.; Gorczyński, A.; Kubicki, M.; Wałęsa-Chorab, M.; Patroniak, V. The first example of an asymmetrical μ-oxo bridged dinuclear iron complex with a terpyridine ligand. New J. Chem. 2019, 43, 12650–12656. [Google Scholar] [CrossRef]
- Addison, A.W.; Rao, T.N.; Reedijk, J.; van Rijn, J.; Verschoor, G.C. Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen–sulphur donor ligands; the crystal and molecular structure of aqua [1,7-bis(N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane]copper(II) perchlorate. J. Chem. Soc. Dalton Trans. 1984, 7, 1349–1356. [Google Scholar] [CrossRef]
- Hagfeldt, A.; Boschloo, G.; Sun, L.; Kloo, L.; Pettersson, H. Dye-sensitized solar cells. Chem. Rev. 2010, 110, 6595–6663. [Google Scholar] [CrossRef]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W. Visible light photoredox catalysis with transition metal complexes: Applications in organic synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef]
- Lebon, E.; Bastin, S.; Sutra, P.; Vendier, L.; Piau, R.E.; Dixon, I.M.; Boggio-Pasqua, M.; Alary, F.; Heully, J.-L.; Igau, A. Can a functionalized phosphine ligand promote room temperature luminescence of the [Ru (bpy)(tpy)] 2+ core? Chem. Commun. 2012, 48, 741–743. [Google Scholar] [CrossRef]
- Liu, Y.; Persson, P.; Sundstrom, V.; Warnmark, K. Fe N-heterocyclic carbene complexes as promising photosensitizers. Acc. Chem. Res. 2016, 49, 1477–1485. [Google Scholar] [CrossRef]
- Fukui, M.; Itoh, K.; Ishii, Y. Iminophosphorane Complexes of Palladium(II). Bull. Chem. Soc. Jpn. 1975, 48, 2044–2046. [Google Scholar] [CrossRef]
- Martin, D.J.; McCarthy, B.D.; Piro, N.A.; Dempsey, J.L. Synthesis and electrochemical characterization of a tridentate Schiff-base ligated Fe(II) complex. Polyhedron 2016, 114, 200–204. [Google Scholar] [CrossRef]
- Wu, J.Y.; Stanzl, B.N.; Ritter, T. A strategy for the synthesis of well-defined iron catalysts and application to regioselective diene hydrosilylation. J. Am. Chem. Soc. 2010, 132, 13214–13216. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez López, N.; Nuñez Bahena, E.; Ryabov, A.D.; Sutra, P.; Igau, A.; Le Lagadec, R. Synthesis, Properties, and Electrochemistry of bis(iminophosphorane)pyridine Iron(II) Pincer Complexes. Inorganics 2024, 12, 115. https://doi.org/10.3390/inorganics12040115
Sánchez López N, Nuñez Bahena E, Ryabov AD, Sutra P, Igau A, Le Lagadec R. Synthesis, Properties, and Electrochemistry of bis(iminophosphorane)pyridine Iron(II) Pincer Complexes. Inorganics. 2024; 12(4):115. https://doi.org/10.3390/inorganics12040115
Chicago/Turabian StyleSánchez López, Nicolás, Erick Nuñez Bahena, Alexander D. Ryabov, Pierre Sutra, Alain Igau, and Ronan Le Lagadec. 2024. "Synthesis, Properties, and Electrochemistry of bis(iminophosphorane)pyridine Iron(II) Pincer Complexes" Inorganics 12, no. 4: 115. https://doi.org/10.3390/inorganics12040115