3. Experimental
3.1. General Procedures
NMR spectra were recorded on a 200/50 MHz Bruker DPX-200, 250/62.5 MHz Bruker DPX-250, 400/100 MHz Bruker DPX-400, 500/125 MHz Bruker Avance-500, 400/100 MHz Varian 400-Mr, 300/75 MHz Varian Unity-300 spectrometer. The spectra were performed in DMSO-d6 and CDCl3 with TMS as internal standard. Chemical shifts (δ) are given in ppm and coupling constants (J) in Hertz. Melting points were determined with a Quimis Q340.23 apparatus and are uncorrected. Elemental analyses were carried out on a Thermo Scientific Flash EA 1112 Series CHN-Analyzer. Thin layer chromatography was performed on Merck Kieselgel 60 HF 254 plates and detection took place using UV (254 and 365 nm). All reagents and solvents were purchased from commercial suppliers and used without further purification.
3.2. Ethyl 4-methyl-2-phenylpyrimidine-5-carboxylate (4a)
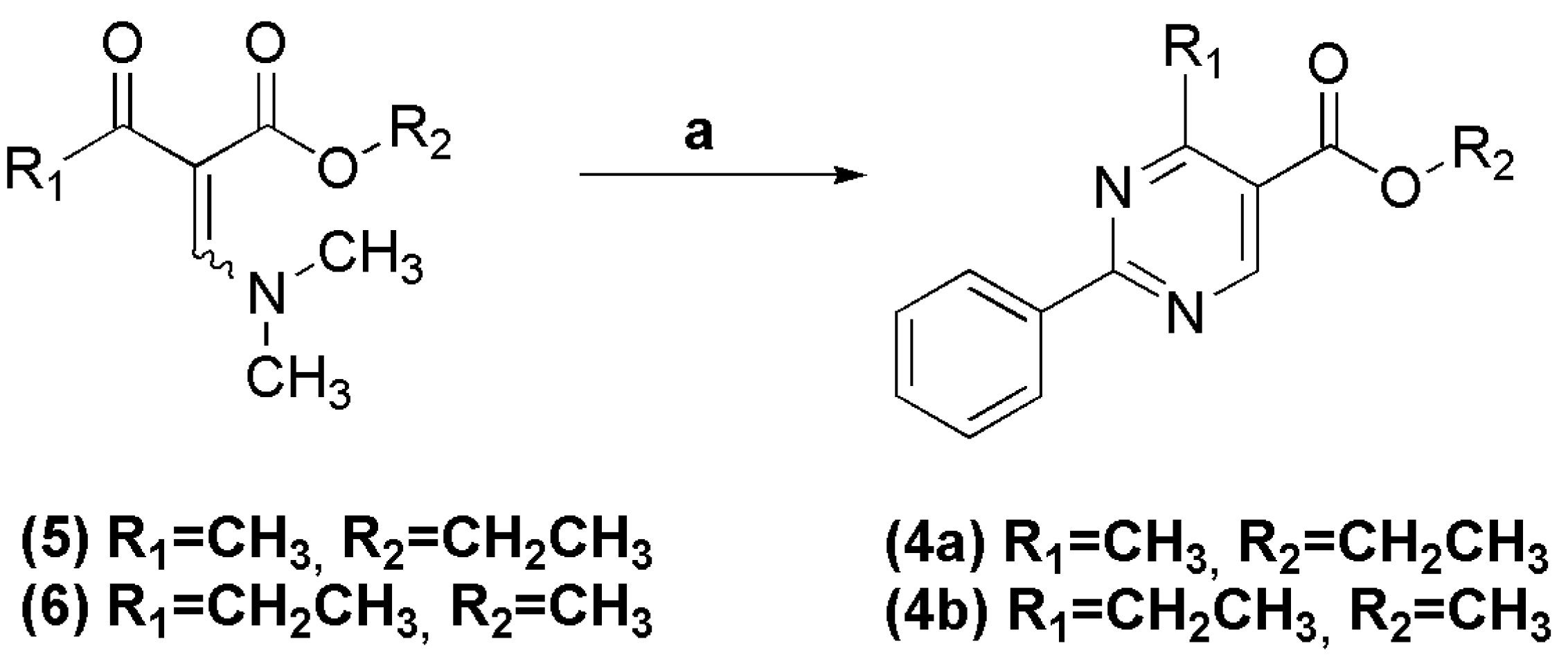
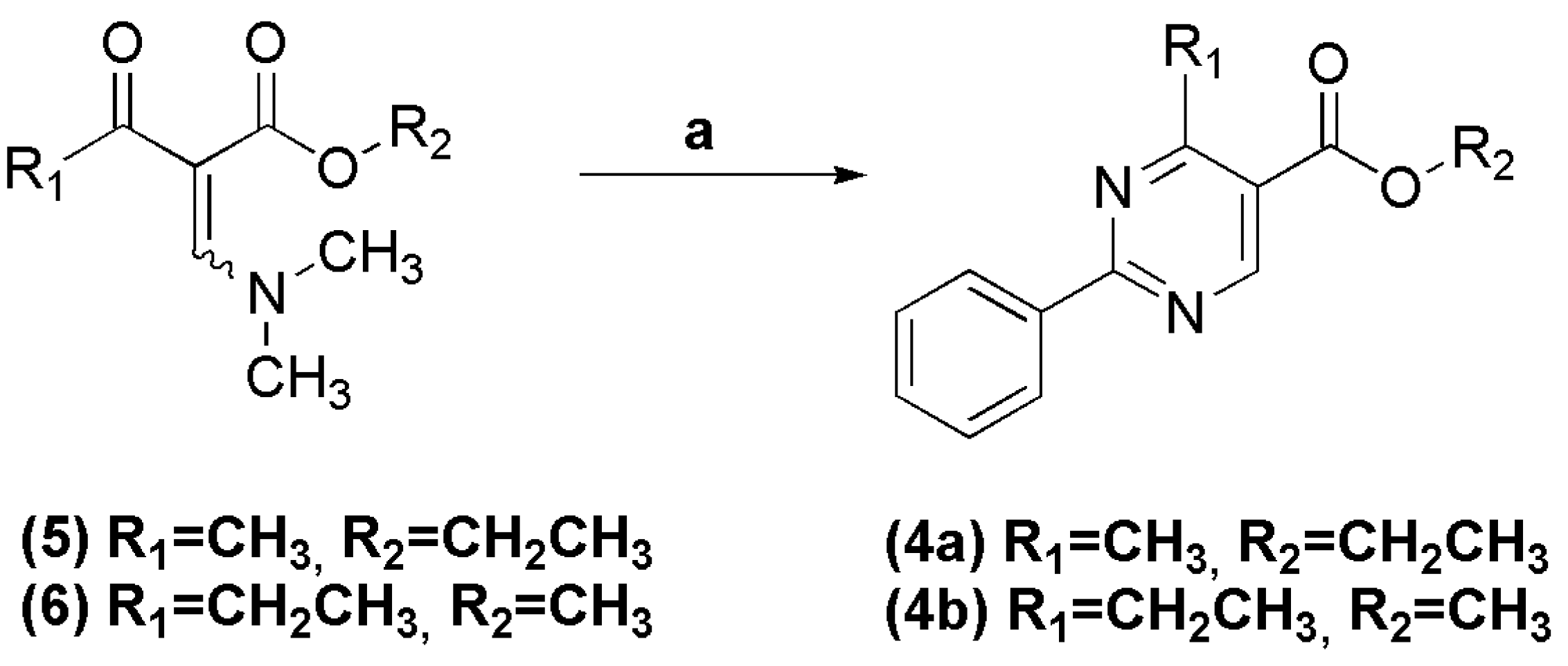
In a round bottomed flask under argon atmosphere were successively added anhydrous ethanol (20 mL), sodium metal (0.745 g, 32.4 mmol), benzamidine hydrochloride (5.07 g, 32.4 mmol) and (E/Z)-ethyl 2-((dimethylamino)methylene)-3-oxobutanoate (6.00 g, 32.4 mmol). The reaction mixture was stirred at room temperature for 2 h. The solvent was evaporated under vacuum and the resulting residue was filtered and washed with cold water. The title compound was obtained as a white crystalline solid in 80% yield. 1H-NMR (200 MHz, DMSO-d6) δ 9.17 (s, 1H, H6het); 8.46–8.44 (m, 2H, H2′Ph, H6′Ph); 7.58–7.55 (m, 3H, H3′Ph, H4′Ph, H5′Ph); 4.36 (q, 2H, J = 8.0 Hz, OCH2); 2.80 (s, 3H, CH3het); 1.35 (t, 3H, J = 8.0 Hz, CH3Et). 13C-NMR (50 MHz, DMSO-d6) δ 168.13 (C=O); 164.50 (C2het); 164.31 (C4het); 158.90 (C6het); 136.11 (C1′Ph); 131.76 (C4′Ph); 128.84 (C3′Ph, C5′Ph); 128.37 (C2′Ph, C6′Ph); 121.30 (C5het); 61.30 (OCH2); 24.28 (CH3het); 14.01 (CH3Et).
3.3. Methyl 4-Ethyl-2-Phenylpyrimidine-5-Carboxylate (4b)
To a solution of benzamidine hydrochloride (0.085 g, 0.540 mmol) and sodium hydroxide (0.022 g, 0.550 mmol) in methanol (2 mL), was added (E/Z)-methyl 2-((dimethylamino)methylene)-3-oxopentanoate (0.10 g, 0.540 mmol). The reaction mixture was stirred at room temperature for 1 h, and then concentrated under reduced pressure. The precipitate was filtered and washed with cold water, affording the title compound as a white amorphous solid in 73% yield. 1H-NMR (400 MHz, DMSO-d6) δ 9.12 (s, 1H, H6het); 9.48–9.46 (m, 2H, H2′Ph, H6′Ph); 7.46–7.41 (m, 3H, H3′Ph, H4′Ph, H5′Ph); 3.89 (s, 3H, CH3het); 3.18 (q, 2H, J = 7.5 Hz, OCH2); 1.32 (t, 3H, J = 7.5 Hz, CH3Et). 13C-NMR (100 MHz, DMSO-d6) δ 172.98 (C=O); 165.42 (C4het); 159.19 (C6het); 136.88 (C1′Ph); 132.50 (C4′Ph); 128.88 (C3′Ph, C5′Ph); 128.50 (C2′Ph, C6′Ph); 120.16 (C5het); 52.14 (OCH3); 29.84 (CH2); 12.36 (CH3).
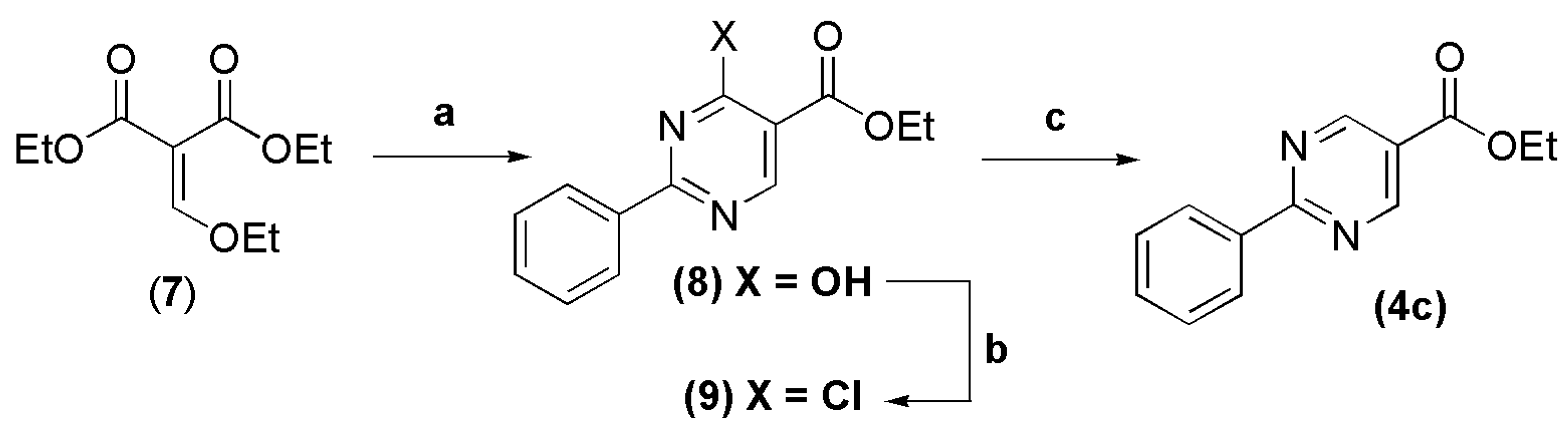
3.4. Ethyl 2-phenylpyrimidine-5-carboxylate (4c)
In a round bottomed flask under argon atmosphere were successively added anhydrous ethanol (5 mL), sodium metal (0.426 g, 18.5 mmol), benzamidine hydrochloride (2.90 g, 18.5 mmol) and diethyl 2-(ethoxymethylene)malonate (4.0 g, 18.5 mmol). The reaction mixture was stirred at room temperature for 9 h, and then poured into ice. The precipitate was filtered, washed with coldwater and recrystallized in EtOH-MeOH-CHCl3 (1:1:1) (45 mL), giving the ethyl 4-hydroxy-2-phenylpyrimidine-5-carboxylate (8) as a white crystalline solid in 50% yield. The derivative 8 (1.2 g, 4.91 mmol) and POCl3 (9.8 g, 63.9 mmol) were refluxed at 100oC for 1 h. The excess of POCl3 was removed under vacuum, ice was added on the resulting solid into the reaction flask, followed by filtration and washing with cold water. The chloride compound 9, obtained as a white amorphous solid in 98% yield, was successively dehalogenated with zinc powder (0.176 g, 2.69 mmol, 4 equiv) in anhydrous THF (2 mL). The reaction mixture was stirred at 60 °C for 1 h, and then 5 drops of acetic acid were added to the reaction vessel. After stirring at 60 °C for 23 h, the reaction mixture was cooled to room temperature, followed by addition of CH2Cl2 (3 mL), filtration and evaporation of solvent. Purification by silica gel column chromatography (n-hexane-EtOAc 0.6%) afforded the title compound as a white crystalline solid in 50% yield. Compound 8: 1H-NMR (200 MHz, DMSO-d6/TMS) δ 12.22 (s, 1H); 8.64 (s, 1H); 8.16 (d, 2H, J = 6.0 Hz); 7.68–7.51 (m, 3H); 4.25 (q, 2H, J = 6.0 Hz); 1.28 (t, 3H, J = 6.0 Hz). 13C-NMR (62.5 MHz, DMSO-d6) δ 163.9; 161.3; 160.1; 159.0; 133.0; 131.9; 129.1; 128.8; 115.0; 60.8; 14.5. Compound 9: 1H-NMR (200 MHz, CDCl3/TMS) δ 9.19 (s, 1H); 8.50 (dd, 2H, J = 2.2 Hz, 8.0 Hz); 7.61–7.46 (m, 3H); 4.46 (q, 2H, J = 7.0 Hz); 1.45 (t, 3H, J = 7.0 Hz). 13C-NMR (50 MHz, CDCl3/TMS) δ 166.57; 162.94; 160.61; 160.52; 135.32; 132.48; 129.29; 128.85; 121.61; 62.28; 14.23. Compound 4c: 1H-NMR (250 MHz, DMSO-d6/TMS) δ 9.27 (s, 2H, H4het, H6het); 8.45–8.41 (m, 2H, H2′Ph, H6′Ph); 7.59–7.52 (m, 3H, H3′Ph, H4′Ph, H5′Ph); 4.36 (q, 2H, J = 7.5 Hz, OCH2); 1.33 (t, 3H, J = 7.5 Hz, CH3). 13C-NMR (63 MHz, DMSO-d6/TMS) δ 166.30 (C=O); 163.84 (C2het); 158.67 (C4het, C6het); 136.49 (C1′Ph); 132.45 (C4′Ph); 129.37 (C3′Ph, C5′Ph); 128.90 (C2′Ph, C6′Ph); 122.28 (C5het); 61.87 (OCH2); 14.47 (CH3).
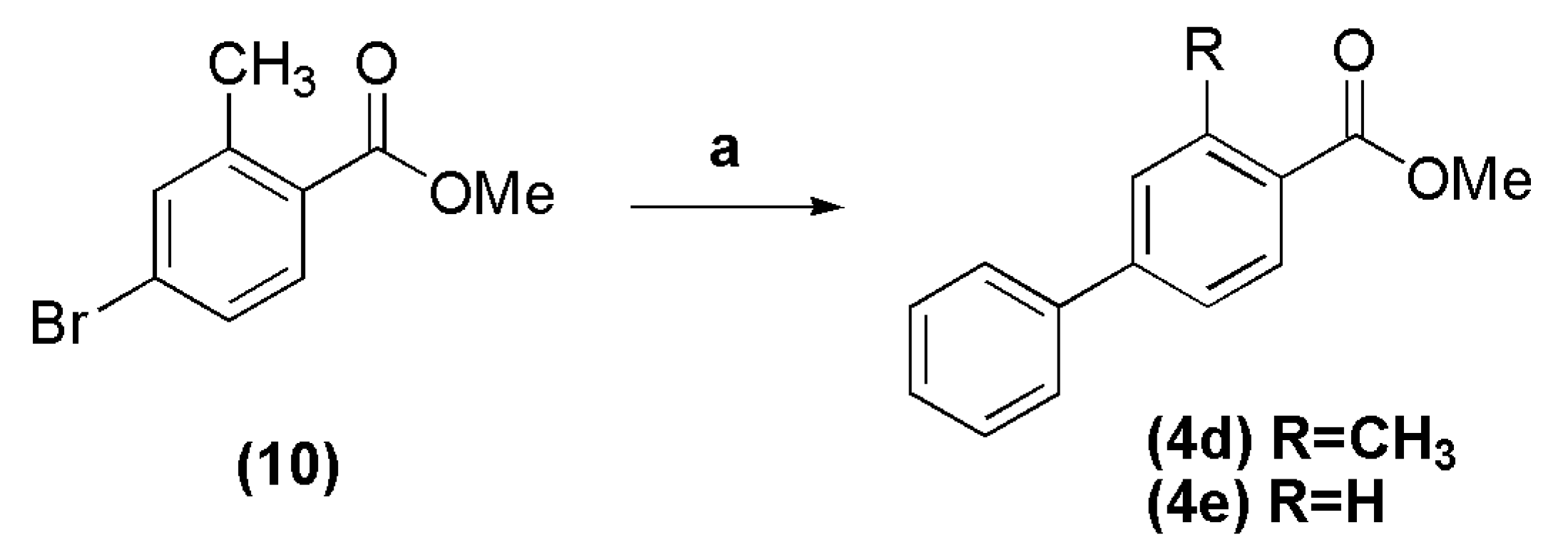
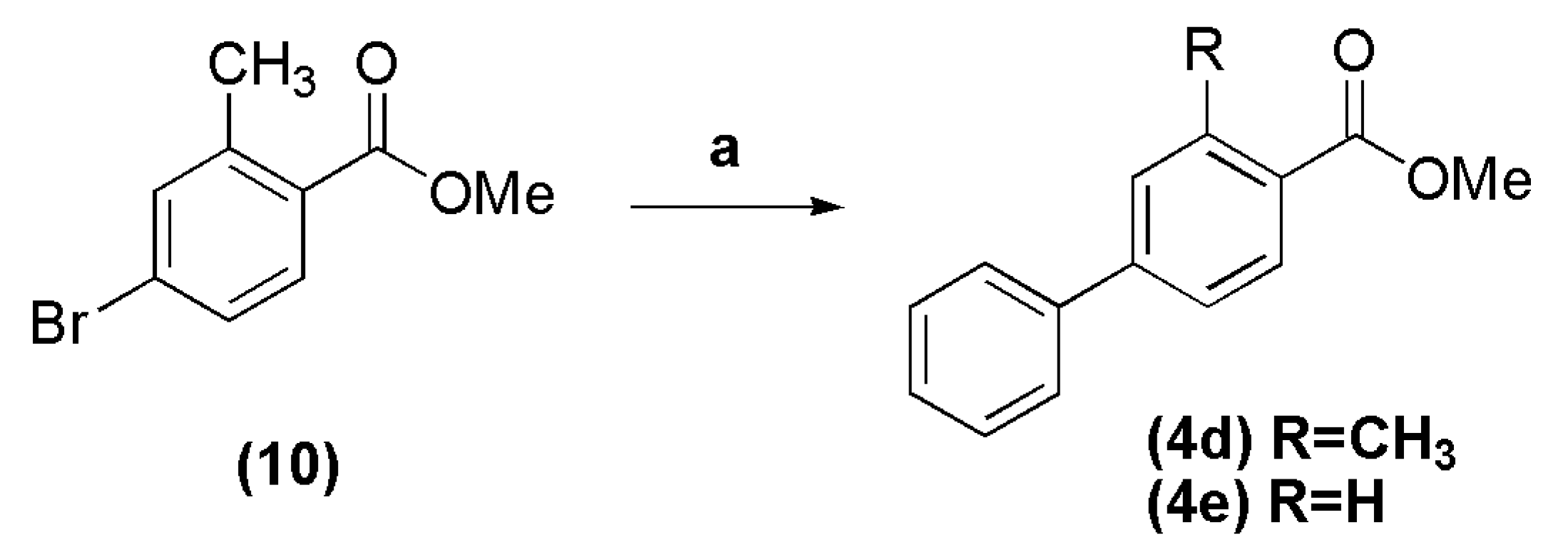
3.5. Methyl 3-methyl-biphenyl-4-carboxylate (4d)
In a round bottomed flask into ice-bath, were added methanol (15 mL), acetyl chloride (8.72 g, 111 mmol) and 4-bromo-2-methylbenzoic acid (1.0 g, 4.65 mmol). The reaction mixture was stirred at room temperature for 24 h, and then concentrated under reduced pressure. The resulting residue was extracted with CH2Cl2. The organic layer was washed with 10% sodium carbonate solution, dried over anhydrous Na2SO4, filtered and evaporated, giving the methyl 4-bromo-2-methylbenzoate 10 as a colorless oil in 86%, which was directly used in cross coupling reaction. The compound 10 (0.30 g, 1.31 mmol) was transferred to a round bottomed flask with a mixture of toluene-methanol (9:1, 4.5–0.5 mL), followed by addition of phenylboronic acid (0.192 g, 1.57 mmol), PdCl2(PPh3)2 (0.037 g, 4 mol %) and potassium carbonate (0.543 g, 3.93 mmol). The reaction mixture was stirred at 60 °C for 3 h, and after cooling to room temperature, was filtered over Celite. The resulting oil was purified by silica gel column chromatography (n-hexane-EtOAc 0%–1%), affording the title compound as a white crystalline solid in 91% yield, after evaporation under vacuum. Compound 10: 1H-NMR (200 MHz, DMSO-d6/TMS) δ 7.73 (d, 1H, J = 8.0 Hz); 7.58 (s, 1H); 7.51 (d, 1H, J = 8.0 Hz); 3.82 (s, 3H); 2.49 (s, 3H). 13C-NMR (50 MHz, DMSO-d6/TMS) δ 167.18; 142.31; 134.65; 132.51; 129.54; 129.16; 126.33; 52.58; 21.18. Compound 4d: 1H-NMR (200 MHz, CDCl3/TMS) δ 7.92 (d, 1H, J = 8.0 Hz, H5Ph); 7.55–7.51 (m, 2H, H2Ph, H6Ph); 7.40–7.25 (m, 5H, H2′Ph-H6′Ph); 3.83 (s, 3H, OCH3); 2.60 (s, 3H, CH3). 13C-NMR (50 MHz, CDCl3/TMS) δ 167.97 (C=O); 144.75 (C1Ph); 140.91 (C1′Ph); 140.14 (C3Ph); 131.36 (C5Ph); 130.49 (C2Ph); 128.95 (C3′Ph, C5′Ph); 128.30 (C4Ph); 128.09 C4′Ph); 127.33 (C2′Ph, C6′Ph); 124.47 (C6Ph); 51.89 (OCH3); 22.08 (CH3).
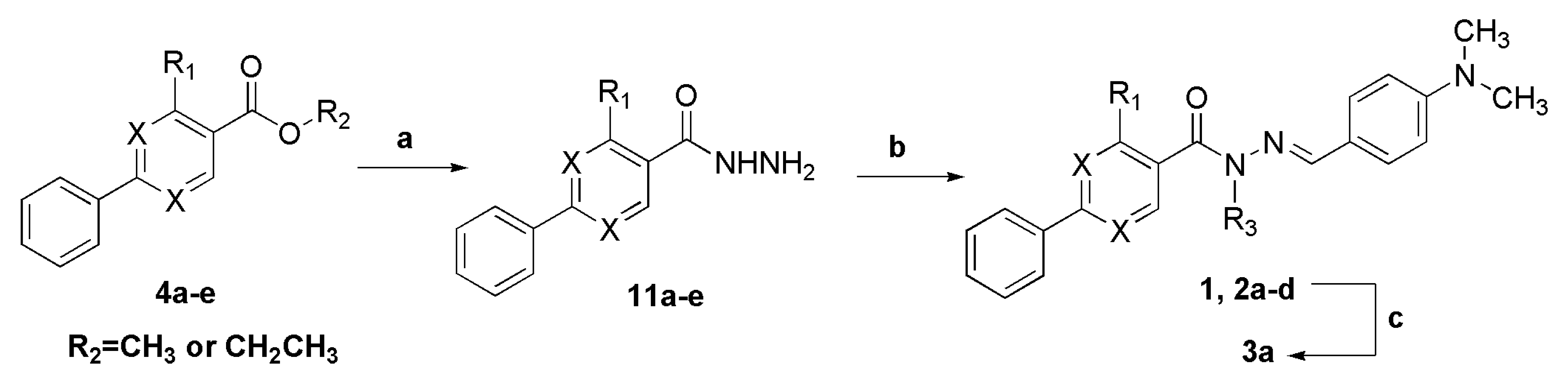
3.6. 4-Methyl-2-phenylpyrimidine-5-carbohydrazide (11a)
To a solution of ethyl 4-methyl-2-phenylpyrimidine-5-carboxylate (4a, 0.710 g, 2.92 mmol) in ethanol (5 mL) was added hydrazine monohydrate 100% (2.93 g, 2.84 mL, 58.4 mmol). The reaction mixture was stirred at 55 °C for 4 h. After cooling the reaction to room temperature, the round bottomed flask was placed in an ice-bath and the solid, filtered and washed with cold water. The title compound was obtained as a white crystalline solid in 83% yield. 1H-NMR (200 MHz, DMSO-d6) δ 9.80 (s, 1H, NH); 8.76 (s, 1H, H6het); 8.44–8.39 (m, 2H, H2′Ph, H6′Ph); 7.56–7.52 (m, 3H, H3′Ph-H5′Ph); 4.62 (s, 2H, NH2); 2.63 (s, 3H, CH3). 13C-NMR (50 MHz, DMSO-d6) δ 165.43 (C=O); 164.80 (C2het); 163.02 (C4het); 155.56 (C6het); 136.54 (C1′Ph); 131.16 (C4′Ph); 128.72 (C3′Ph, C5′Ph); 127.92 (C2′Ph, C6′Ph); 126.32 (C5het); 22.63 (CH3).
3.7. 4-Ethyl-2-phenylpyrimidine-5-carbohydrazide (11b)
To a solution of 4b (0.191 g, 0.788 mmol) in ethanol (3 mL) was added hydrazine monohydrate 100% (0.789 g, 15.8 mmol). The reaction mixture was stirred at 50 °C for 5 h, cooled to room temperature and poured into ice. The solid was filtered and washed with cold water, giving the title compound as a white amorphous solid in 79% yield. 1H-NMR (400 MHz, DMSO-d6) δ 9.77 (s, 1H, NH); 8.72 (s, 1H, H6het); 8.42–8.40 (m, 2H, H2′Ph, H6′Ph); 7.54–7.51 (m, 3H, H3′Ph, H5′Ph); 4.59 (s, 2H, NH2); 2.90 (q, 2H, J = 8.0 Hz, CH2); 1.27 (t, 3H, J = 8.0 Hz, CH3). 13C-NMR (125 MHz, DMSO-d6) δ 170.12 (C=O); 165.40 (C4het); 163.66 (C2het); 156.09 (C6het); 137.23 (C1′Ph); 131.71 (C4′Ph); 129.31 (C3′Ph, C5′Ph); 128.44 (C2′Ph, C6′Ph); 126.68 (C5het); 28.62 (CH2); 13.14 (CH3).
3.8. 2-Phenylpyrimidine-5-carbohydrazide (11c)
To a solution of ethyl 2-phenylpyrimidine-5-carboxylate (4c) (0.080 g, 0.350 mmol) in ethanol (2 mL) was added hydrazine monohydrate 100% (0.351 g, 7.01 mmol). The reaction mixture was stirred at room temperature for 2 h. Ice was added to the reaction flask and the solid, filtered and washed with cold water. The title compound was obtained as a white amorphous solid in 85% yield. 1H-NMR (250 MHz, DMSO-d6/TMS) δ 10.12 (s, 1H, NH); 9.20 (s, 2H, H4het, H6het); 8.43–8.39 (m, 2H, H2′Ph, H6′Ph); 7.55–7.52 (m, 3H, H3′Ph-H5′Ph); 4.64 (s, 2H, NH2). 13C-NMR (63 MHz, DMSO-d6/TMS) δ 165.04 (C=O); 162.88 (C2het); 156.71 (C4het, C6het); 136.78 (C1′Ph); 131.99 (C4′Ph); 129.31 (C3′Ph, C5′Ph); 128.56 (C2′Ph, C6′Ph); 125.03 (C5het).
3.9. 3-Methyl-biphenyl-4-carbohydrazide (11d)
To a solution of methyl ester (4d) (0.60 g, 2.65 mmol) in ethanol (5 mL) was added hydrazine monohydrate 100% (3.98 g, 79.5 mmol). The reaction mixture was stirred at 50 °C for 15 h, and then concentrated under reduced pressure. Ice was added to the reaction flask and the solid, filtered and washed with cold water. The title compound was obtained as a white amorphous solid in 95% yield. 1H-NMR (200 MHz, DMSO-d6/TMS) δ 9.46 (s, 1H, NH); 7.69–7.66 (m, 2H, H2Ph, H5Ph); 7.54–7.37 (m, 6H, H6Ph, H2′Ph-H6′Ph); 4.49 (s, 2H, NH2); 2.42 (s, 3H, CH3). 13C-NMR (50 MHz, DMSO-d6/TMS) δ 168.85 (C=O); 141.68 (C1Ph); 140.04 (C1′Ph); 136.98 (C3Ph); 135.06 (C4Ph); 129.50 (C3′Ph, C5′Ph); 129.28 (C5Ph); 128.54 (C2Ph); 128.28 (C4′Ph); 127.26 (C2′Ph, C6′Ph); 124.25 (C6Ph); 20.08 (CH3).
3.10. Biphenyl-4-carbohydrazide (11e)
To a solution of methyl biphenyl-4-carboxylate (4e, 0.50 g, 2.36 mmol) in ethanol (5 mL) was added hydrazine monohydrate 100% (2.36 g, 47.1 mmol). The reaction mixture was stirred at 80 °C for 8 h, and then concentrated under reduced pressure. Ice was added to the reaction flask and the solid, filtered and washed with cold water. The title compound was obtained as a pale amorphous solid in 85% yield. 1H-NMR (200 MHz, DMSO-d6/TMS) δ 9.85 (s, 1H, NH); 7.93 (d, 2H, J = 8.0 Hz, H3Ph, H5Ph); 7.77–7.69 (m, 4H, H2Ph, H6Ph, H2′Ph, H6′Ph); 7.52–7.36 (m, 3H, H3′Ph-H5′Ph); 4.55 (s, 2H, NH2). 13C-NMR (50 MHz, DMSO-d6/TMS) δ 166.13 (C=O); 143.17 (C1Ph); 139.74 (C1′Ph); 132.66 (C4Ph); 129.57 (C3Ph, C5Ph); 128.55 (C4′Ph); 128.17 (C3′Ph, C5′Ph); 127.37 (C2Ph, C6Ph); 127.08 (C2′Ph, C6′Ph).
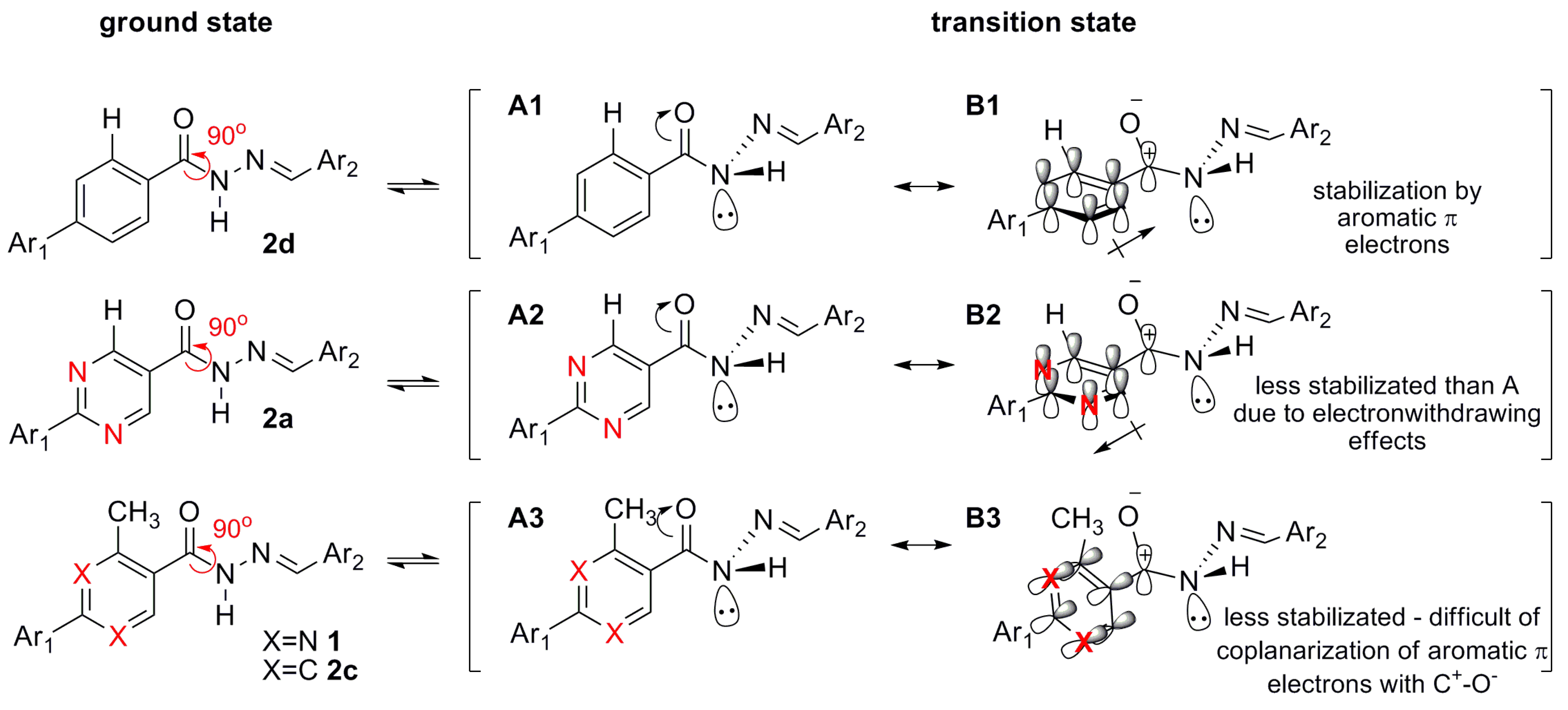
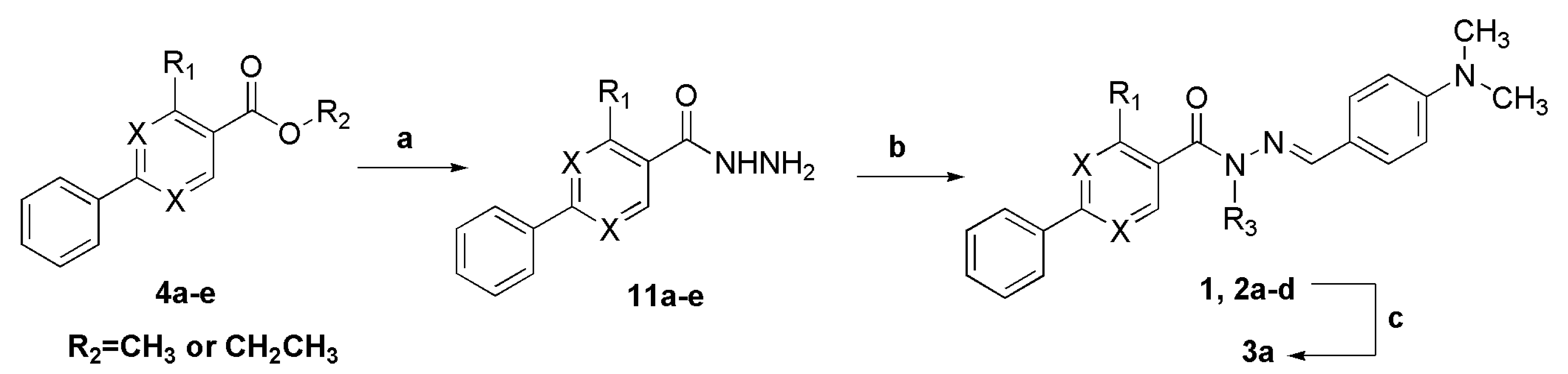
3.11. General Procedure to Synthesize the N-Acylhydrazones 1, 2a–d
To a solution of an aromatic cabohydrazide (1.10 mmol) in ethanol (5 mL) were added HCl catalytic (2 drops) and the appropriate aromatic aldehyde. The reaction mixture was stirring at room temperature for ca 30 min, and then poured into ice. The precipitate was filtered, washed with cold water, petroleum ether and recrystallized in EtOH.
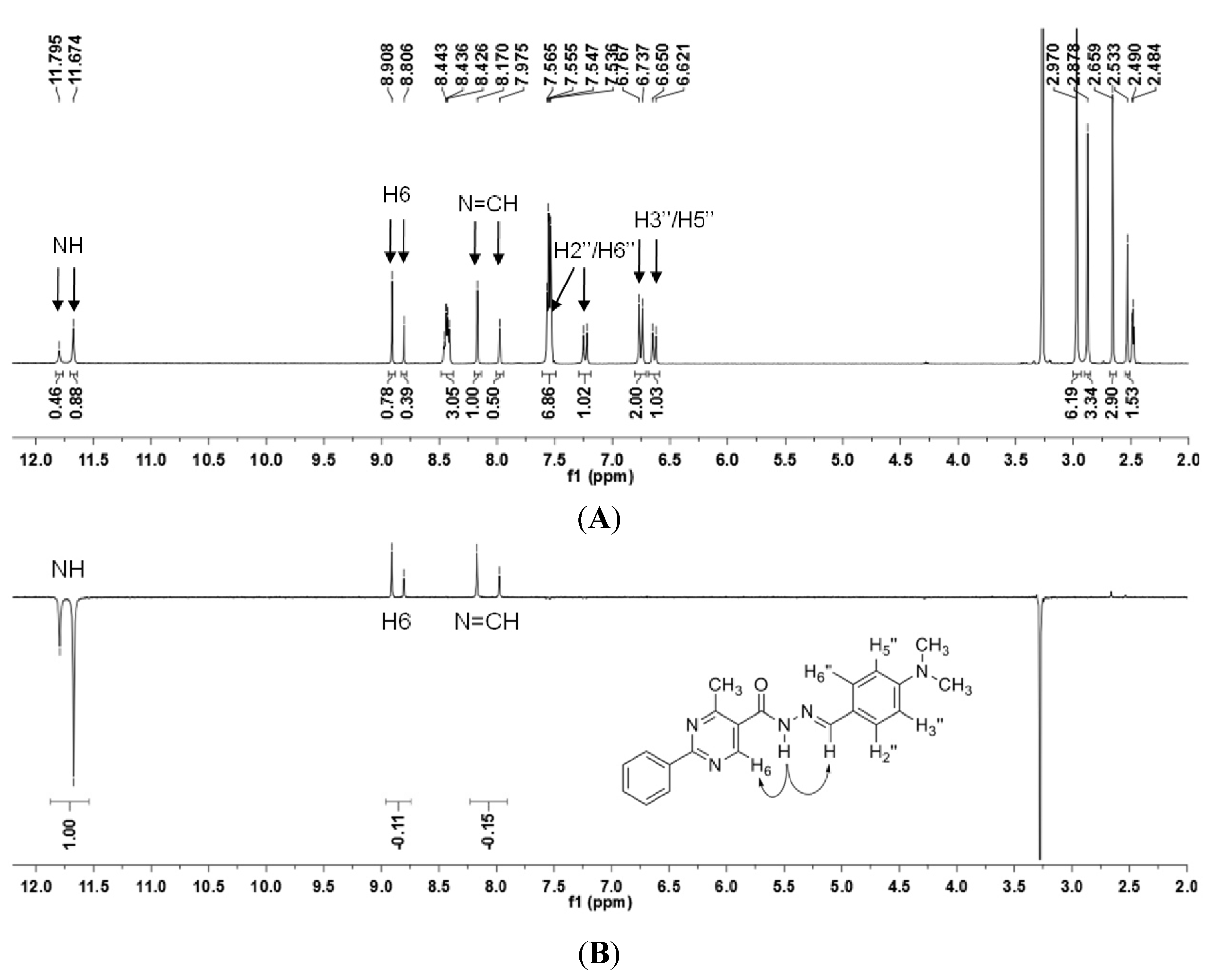
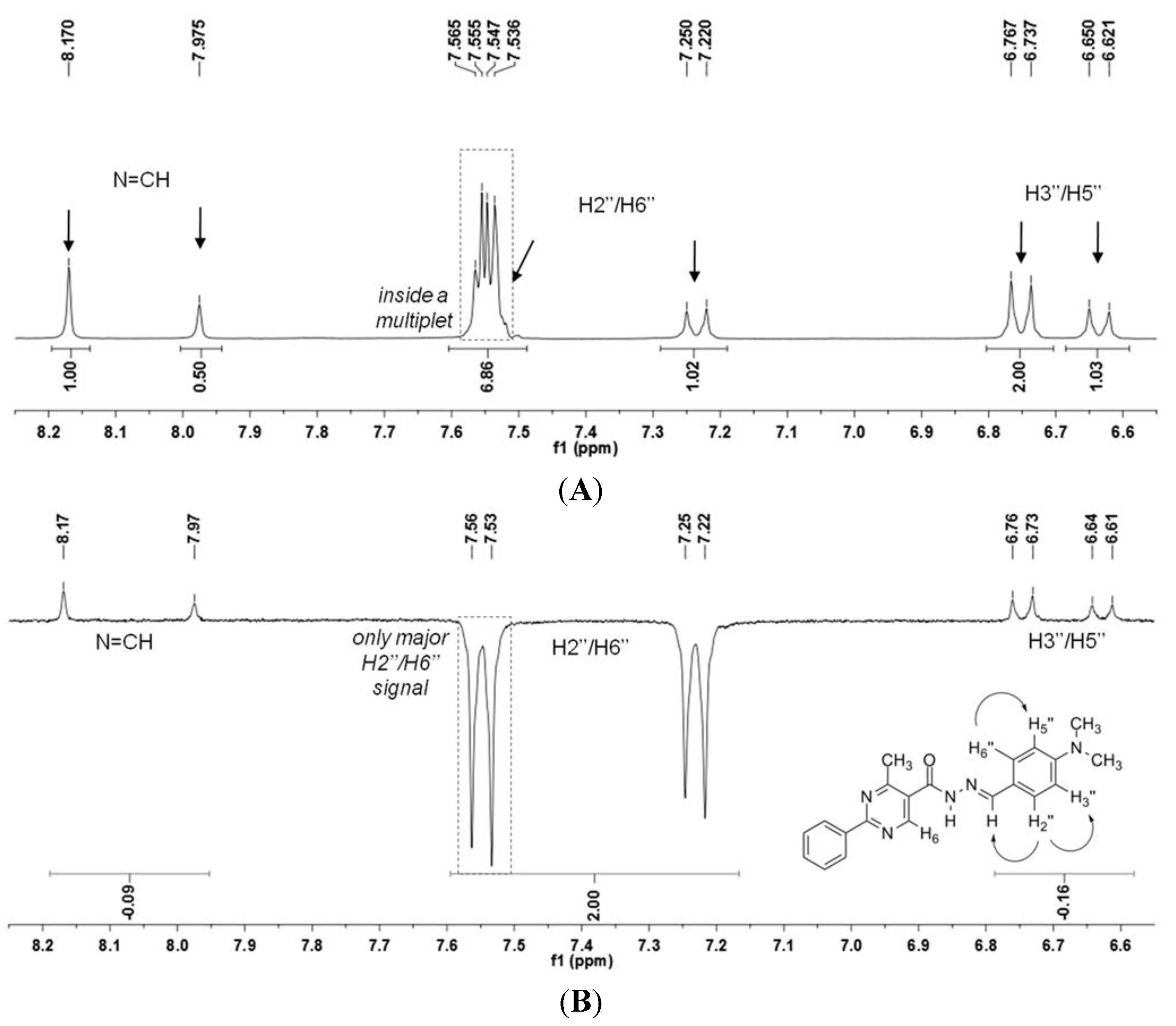
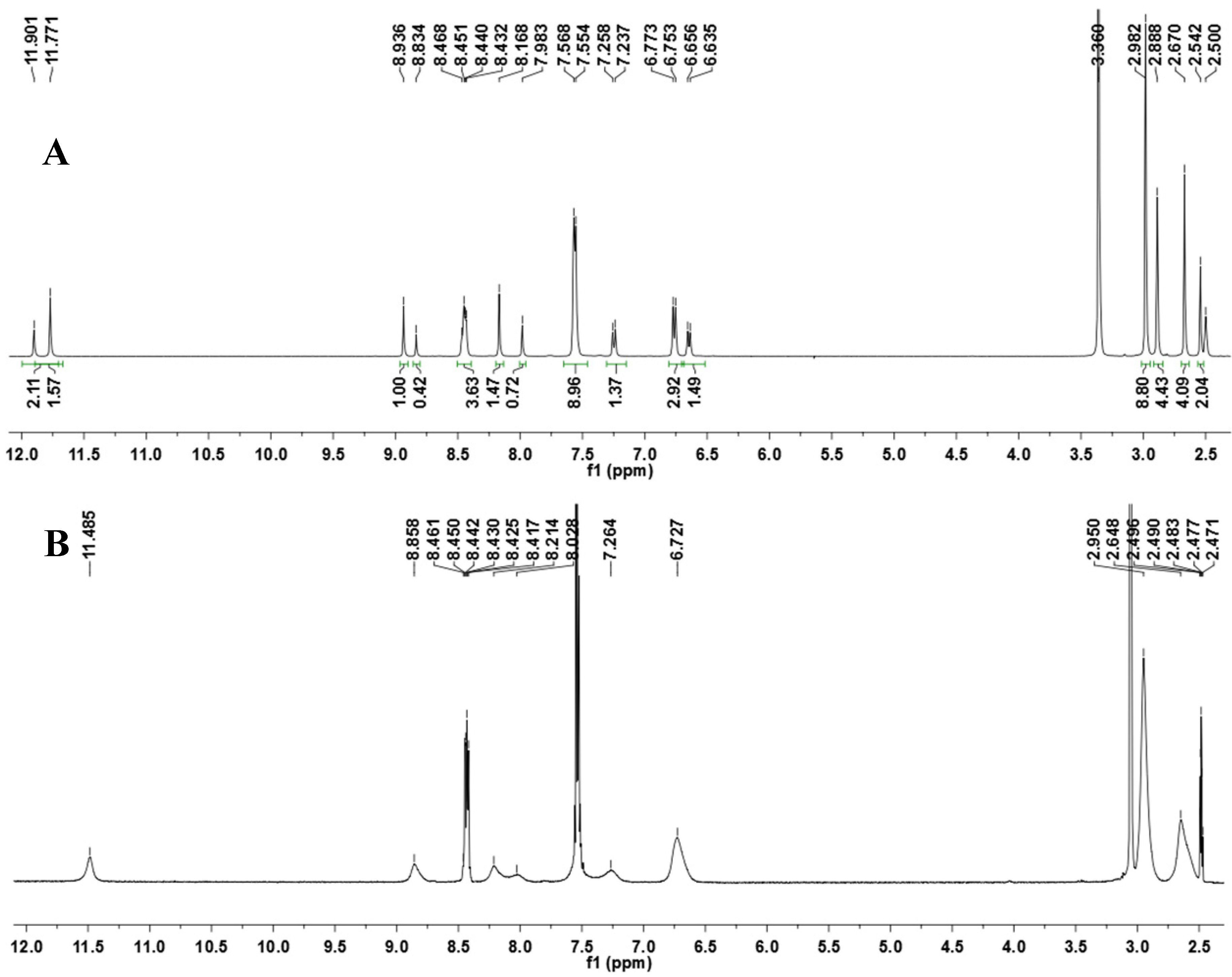
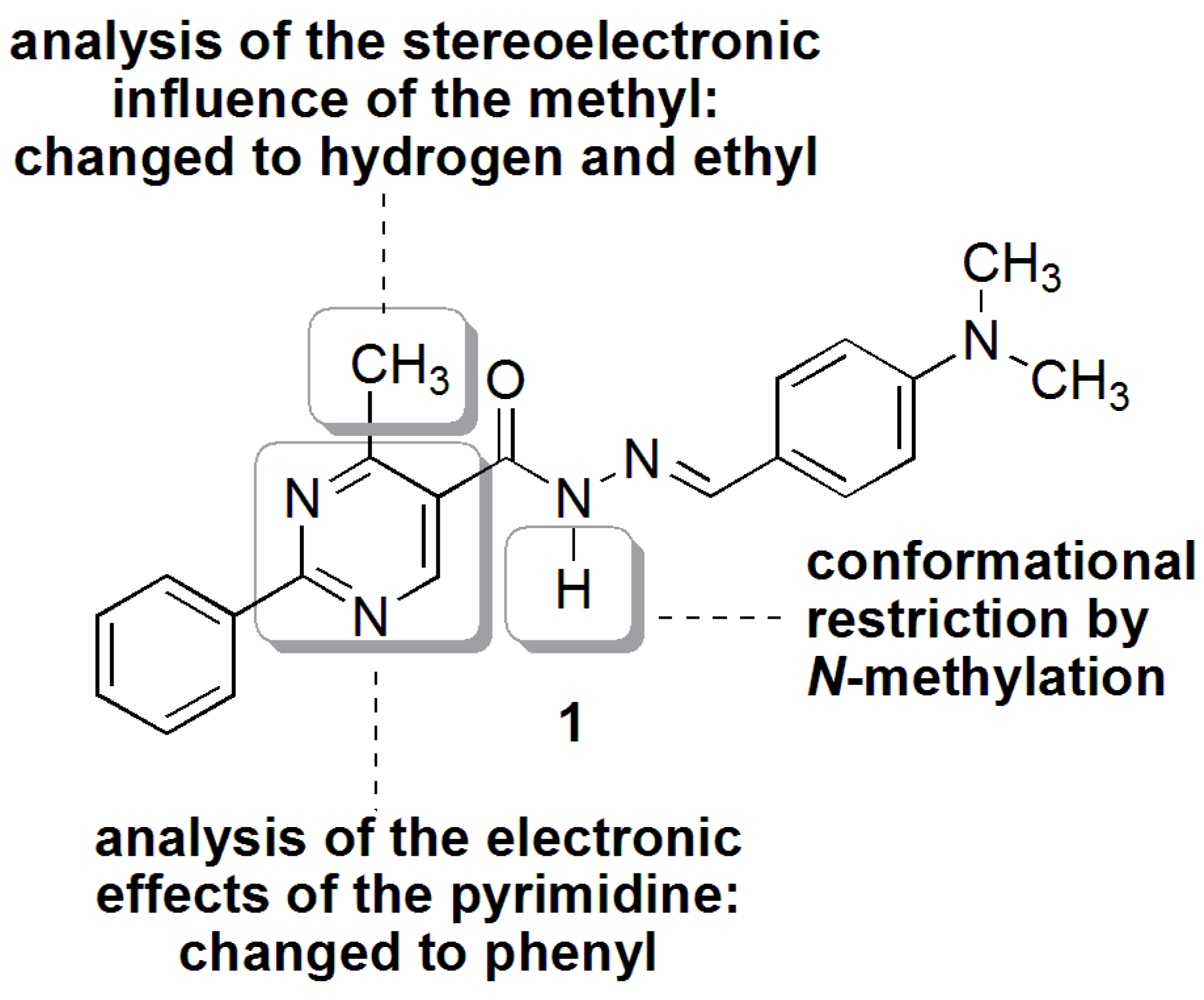
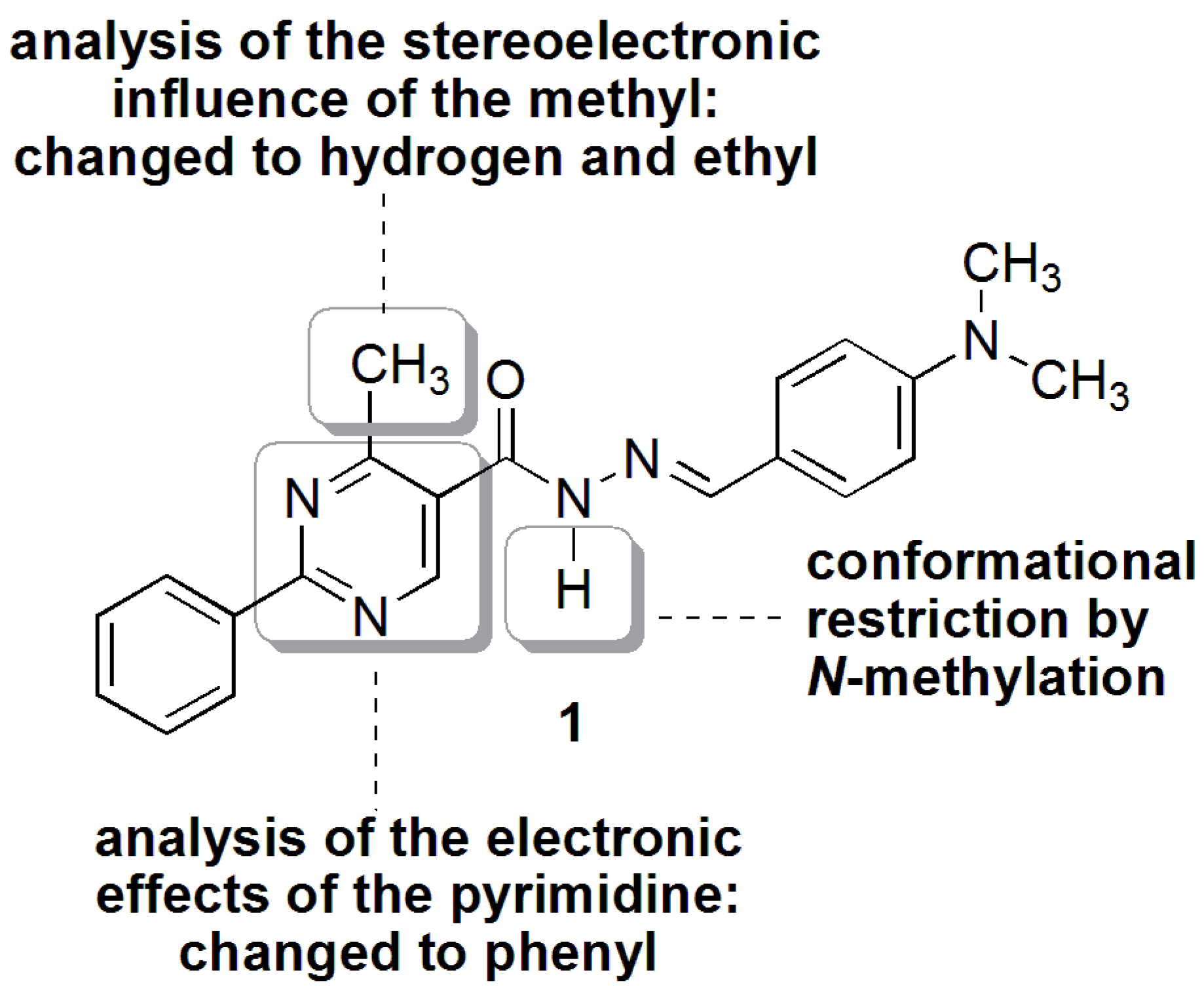
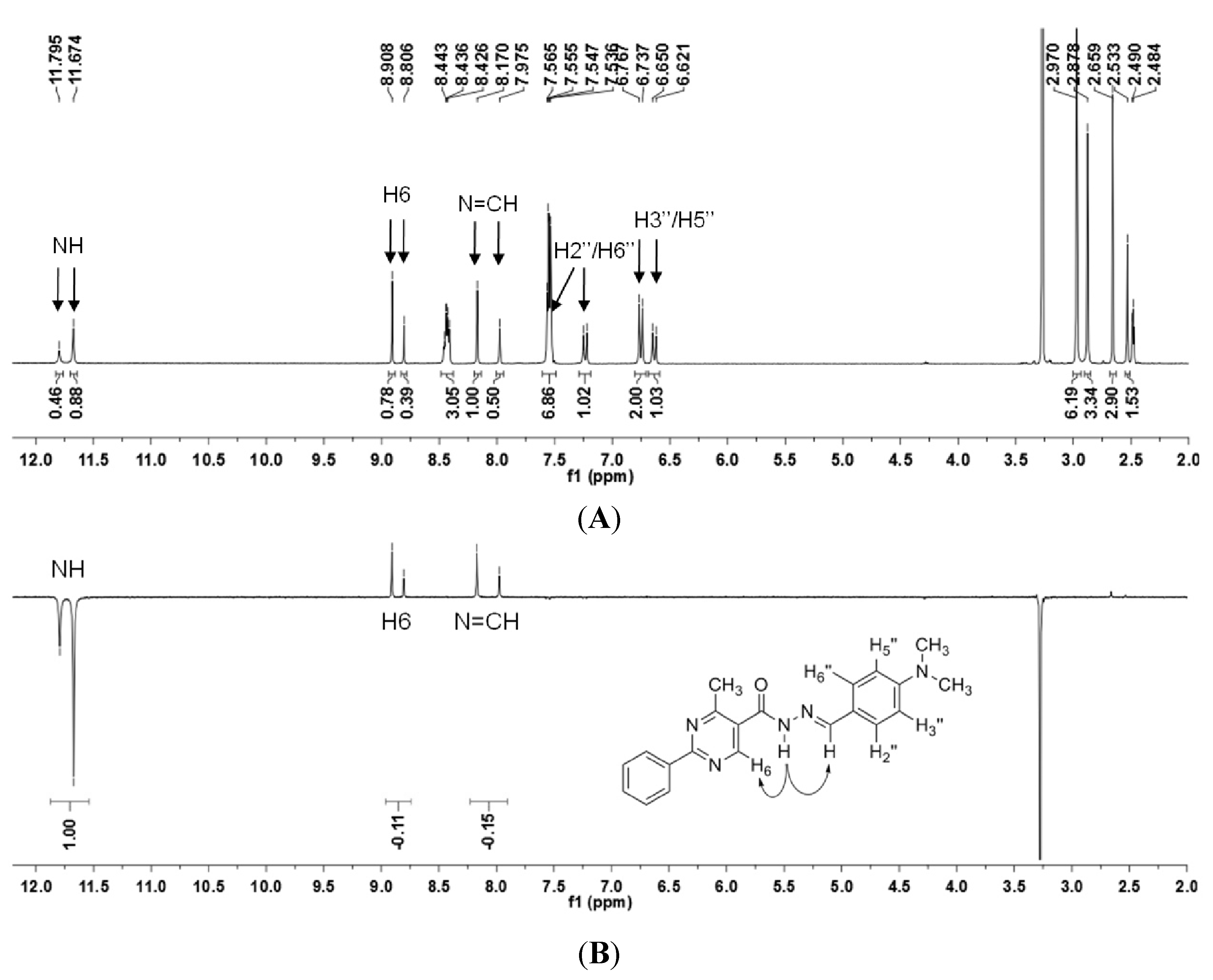
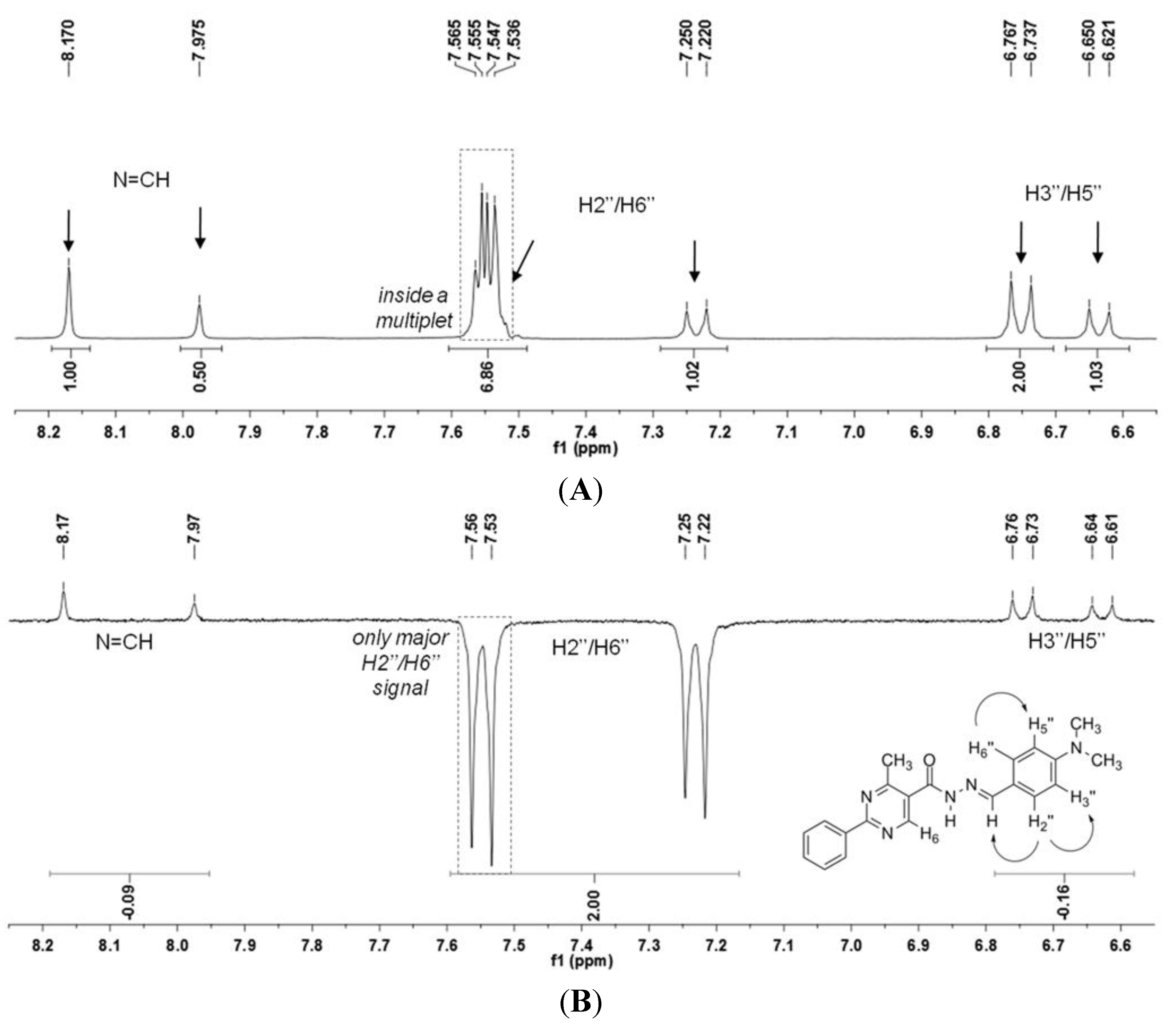
N′-(4-(Dimethylamino)benzylidene)-4-methyl-2-phenylpyrimidine-5-carbohydrazide (1): The condensation of 4-methyl-2-phenylpyrimidine-5-carbohydrazide (11a) and 4-dimethylamino- benzaldehyde, afforded the title compound as a yellow solid (mp. 206 °C) in 99% yield. 1H-NMR (200 MHz, DMSO-d6) δ 11.79, 11.67 (2s, 1H, NH); 8.91, 8.81 (2s, 1H, H6het); 8.45–8.41 (m, 2H, H2′Ph, H6′Ph); 8.17, 7.98 (2s, 1H, N=CH); 7.56–7.54 (m, 5H, H3′Ph–H5′Ph, H2′′ap, H6′′ap); 7.24 (d, 2H, H2′′sp, H6′′sp, J = 8.7 Hz); 6.76, 6.64 (2d, 2H, H3′′, H5′′, J = 8.7 Hz, 9.0 Hz); 2.97, 2.88 (2s, 6H, N(CH3)2); 2.66, 2.53 (2s, 3H, CH3). 13C-NMR (50 MHz, DMSO-d6) δ 167.42, 165.96 (C=O); 164.37, 163.48 (C2het); 162.95, 161.59 (C4het); 156.37, 156.10 (C6het); 151.97, 151.73 (C4′′); 149.60, 145.96 (N=CH); 136.97, 136.84 (C1′Ph); 131.47, 131.34 (C4′Ph); 128.99 (C3′Ph, C5′Ph); 128.26 (C2′Ph, C6′Ph, C2′′, C6′′); 127.64, 126.72 (C5het); 121.09, 121.03 (C1′′); 111.73 (C3′′, C5′′); 39.87 (N(CH3)2); 23.02, 22.89 (CH3). Anal. Calcd for C21H21N5O: C: 70.18; H: 5.89; N: 19.48. Found: C: 70.17; H: 5.89; N: 19.62.
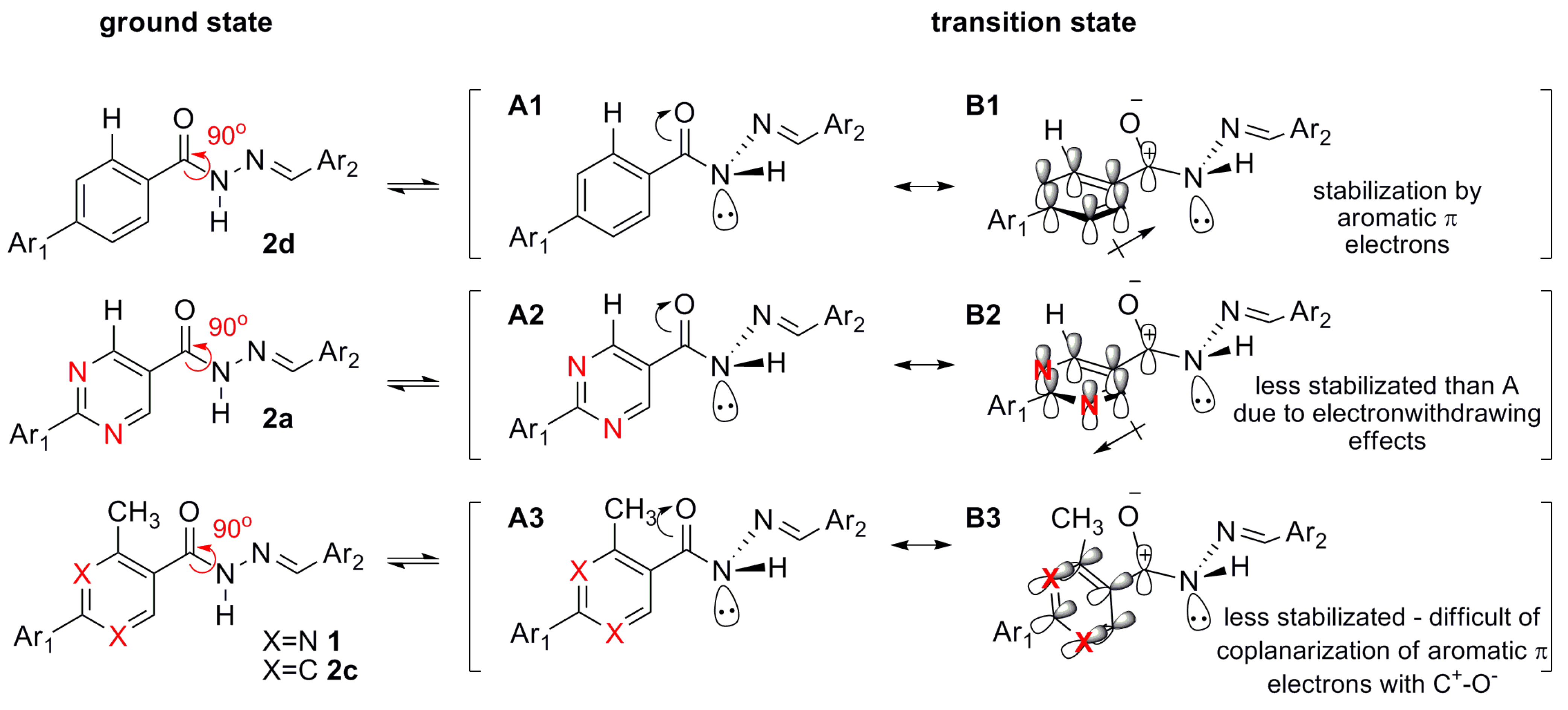
N′-(4-(Dimethylamino)benzylidene)-2-phenylpyrimidine-5-carbohydrazide (2a): The condensation of 2-phenylpyrimidine-5-carbohydrazide (11c) and 4-dimethylaminobenzaldehyde, afforded the title compound as a yellow amorphous solid (mp. 269 °C) in 99% yield. 1H-NMR (300 MHz, DMSO-d6/TMS) δ 11.79 (broad s, 1H, NH); 9.28 (s, 2H, H4het, H6het); 8.47–8.44 (m, 2H, H2′Ph, H6′Ph); 8.30, 8.01 (2s, 1H, N=CH); 7.57–7.55 (m, 5H, H3′Ph, H4′Ph, H5′Ph, H2′′, H6′′); 7.39 (d, 2H, H2′′, H6′′, J = 4.0 Hz); 6.77–6.69 (m, 2H, H3′′, H5′′); 2.97, 2.92 (2s, 6H, N(CH3)2). 13C-NMR (75 MHz, DMSO-d6/TMS) δ 165.47, 165.33 (C=O); 164.66, 160.02 (C2het); 158.90, 157.43 (C4, C6); 152.45, 152.22 (C4′′); 150.42, 146.48 (N=CH); 137.11 (C1′Ph); 132.26 (C4′Ph); 129.54 (C3′Ph, C5′Ph); 129.35 (C2′′, C6′′); 128.90 (C2′Ph, C6′Ph); 126.70, 125.96 (C5het); 121.88 (C1′′); 112.50 (C3′′, C5′′); 40.56 (N(CH3)2). Anal. Calcd for C20H19N5O: C: 69.55; H: 5.54; N: 20.28. Found: C: 69.20; H: 5.54; N: 20.02.
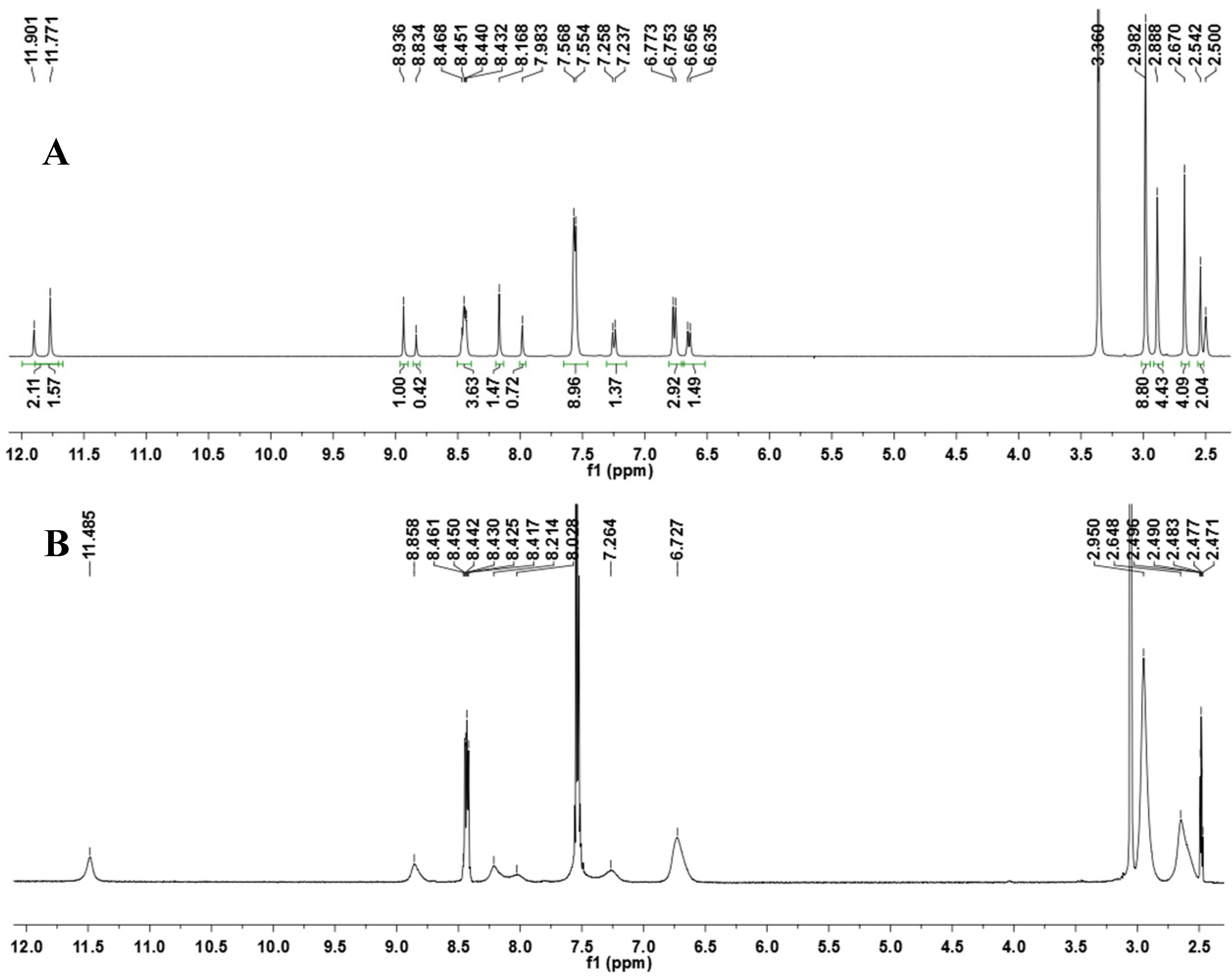
N′-(4-(Dimethylamino)benzylidene)-4-ethyl-2-phenylpyrimidine-5-carbohydrazide (2b): The condensation of 4-ethyl-2-phenylpyrimidine-5-carbohydrazide (11b) and 4-dimethylamino- benzaldehyde, afforded the title compound as a yellow solid (mp. 240 °C) in 85% yield. 1H-NMR (200 MHz, DMSO-d6) δ 11.86, 11.74 (2s, 1H, NH); 8.90, 8.79 (2s, 1H, H6het); 8.47–8.43 (m, 2H, H2′Ph, H6’Ph); 8.15, 7.96 (2s, 1H, N=CH); 7.56–7.53 (m, 5H, H3′Ph–H5′Ph, H2′′, H6′′); 7.21 (d, 2H, H2′′, H6′′, J = 6.6 Hz); 6.75 (d, 2H, H3′′, H5′′, J = 6.6 Hz); 6.62 (d, 2H, H3′′, H5′′, J = 6.6 Hz); 2.99–2.94 (m, 8H, CH2, N(CH3)2); 2.87 (s, 6H, N(CH3)2); 2.81 (q, 2H, CH2, J = 7.5 Hz); 1.32–1.25 (m, 3H, CH3). 13C-NMR (50 MHz, DMSO-d6) δ 169.75, 167.93 (C=O); 167.22, 163.24 (C4het); 162.65, 161.25 (C2het); 155.93, 155.80 (C6het); 151.61, 151.36 (C4′′); 149.18, 145.50 (N=CH); 136.81, 136.67 (C1′Ph); 131.15, 131.00 (C4′Ph); 128.70, 128.56 (C3′Ph, C5′Ph); 127.92, 127.08 (C2′′, C6′′); 126.19 (C2′Ph, C6′Ph); 121.03 (C1′′), 120.95 (C5het); 111.69 (C3′′, C5′′); 39.64, 39.46 (N(CH3)2); 28.30, 28.08 (CH2); 12.49, 12.03 (CH3). Anal. Calcd for C22H23N5O: C: 70.76; H: 6.21; N: 18.75. Found: C: 70.62; H: 6.19; N: 18.80.
(E)-N′-(4-(Dimethylamino)benzylidene)-3-methylbiphenyl-4-carbohydrazide (2c): The condensation of 3-methyl-biphenyl-4-carbohydrazide (11d) and 4-dimethylaminobenzaldehyde, afforded the title compound as a yellow amorphous solid (mp. 238 °C) in 94% yield. 1H-NMR (300 MHz, DMSO-d6/TMS) δ 11.41 (s, 1H, NH); 8.17, 7.94 (2s, 1H, N=CH); 7.72–7.67 (m, 2H, H2Ph, H5Ph); 7.58–7.35 (m, 8H, H6Ph, H2′Ph-H6′Ph, H2′′, H6′′); 7.22 (d, 2H, H2′′, H6′′, J = 8.4 Hz); 6.74, 6.62 (2d, 2H, H3′′, H5′′, J = 8.9 Hz, J = 8.4 Hz); 2.96, 2.87 (2s, 6H, N(CH3)2); 2.45, 2.33 (2s, 3H, CH3). 13C-NMR (75 MHz, DMSO-d6/TMS) δ 164.83 (C=O); 151.74, 151.48 (C4′′); 148.44, 144.70 (N=CH); 141.66, 140.62 (C1Ph); 139.69 (C1′); 136.84, 135.66 (C3Ph); 134.82 (C4Ph); 129.17, 129.06 (C3′Ph, C5′Ph, C2′′, C6′′); 128.63, 128.32 (C2′Ph, C6′Ph); 127.85 (C4′Ph); 126.97 (C2Ph, C5Ph); 124.03, 123.46 (C6Ph); 121.84 (C1′′); 112.03, 111.29 (C3′′, C5′′); 39.95 (N(CH3)2); 19.69 (CH3). Anal. Calcd for C23H23N3O: C: 77.28; H: 6.49; N: 11.76. Found: C: 76.92; H: 6.47; N: 11.59.
(E)-N′-(4-(Dimethylamino)benzylidene)-biphenyl-4-carbohydrazide (2d): The condensation of biphenyl-4-carbohydrazide (11e) and 4-dimethylaminobenzaldehyde, following the general procedure for the synthesis of N-acylhydrazones, afforded the title compound as a yellow amorphous solid (mp. 289 °C) in 99% yield. 1H-NMR (300 MHz, DMSO-d6/TMS) δ 11.58 (s, 1H, NH); 8.32 (s, 1H, N=CH); 7.98 (d, 2H, H3Ph, H5Ph, J = 8.0 Hz); 7.8 (d, 2H, H2Ph, H6Ph, J = 8.0 Hz); 7.74–7.72 (m, 2H, H2′Ph, H6′Ph); 7.55–7.47 (m, 4H, H2′′, H6′′, H3′Ph, H5′Ph); 7.42–7.38 (m, 1H, H4′Ph); 6.75 (d, 2H, H3′′, H5′′, J = 8.0 Hz); 2.96 (s, 6H, N(CH3)2). 13C-NMR (75 MHz, DMSO-d6/TMS) δ 162.27 (C=O); 151.52 (C4′′); 148.64 (N=CH); 142.97 (C1Ph); 139.12 (C1′Ph); 132.53 (C4Ph); 129.02 (C3Ph, C5Ph); 128.42 (C3′Ph, C5′Ph); 128.14 (C2′′, C6′′); 126.85 (C2Ph, C6Ph, C4′Ph); 126.58 (C2′Ph, C6′Ph); 121.62 (C1′′); 111.81 (C3′′, C5′′). Anal. Calcd for C22H21N3O: C: 76.94; H: 6.16; N: 12.24. Found: C: 76.65; H: 6.15; N: 11.96.
3.12. (E)-N′-(4-(Dimethylamino)Benzylidene)-N,4-Dimethyl-2-Phenylpyrimidine-5-Carbohydrazide (3a)
To a solution of N-acylhydrazone 1 (0.14 mmol) and potassium carbonate (0.42 mmol) in acetone (5 mL) in an ice-bath was added methyl iodide (0.42 mmol). After, the reaction mixture was stirred at 43 °C for 24 h. The addition of 0.42 mmol of methyl iodide was needed, and then the reaction mixture was stirred at the same temperature for more 20 h. The reaction mixture was concentrated under vacuum, ethanol (1 mL) was added and the resulting mixture poured into ice. After filtering, the solid was heated into petroleum ether, and then refiltered to give the title compound as a yellow amorphous solid (mp. 188–189 °C) in 77% yield. 1H-NMR (300 MHz, DMSO-d6/TMS) δ 8.73 (s, 1H, H6het); 8.46–8.42 (m, 2H, H2′Ph, H6′Ph); 7.94 (s, 1H, N=CH); 7.54–7.52 (m, 3H, H3′Ph-H5′Ph); 7.22 (d, 2H, J = 9.0 Hz, H2′′, H6′′); 6.61 (d, 2H, J = 9.0 Hz, H3′′, H5′′); 3.50 (s, 3H, N-CH3); 2.85 (s, 6H, N(CH3)2); 2.43 (s, 3H, CH3). 13C-NMR (75 MHz, DMSO-d6/TMS) δ 167.81 (C=O); 164.18 (C2het); 163.12 (C4het); 156.09 (C6het); 152.13 (C4′′); 143.46 (N=CH); 137.53 (C1′Ph); 131.65 (C4′Ph); 129.57 (C3′Ph, C5′Ph); 129.36 (C2′′, C6′′); 128.76 (C2′Ph, C6′Ph); 128.62 (C5het); 122.34 (C1′′); 112.50 (C3′′, C5′′); 40.32 (N(CH3)2; 28.92 (N-CH3); 23.29 (CH3). Anal. Calcd for C22H23N5O: C: 70.76; H: 6.21; N: 18.75. Found: C: 70.41; H: 6.05; N: 18.43.
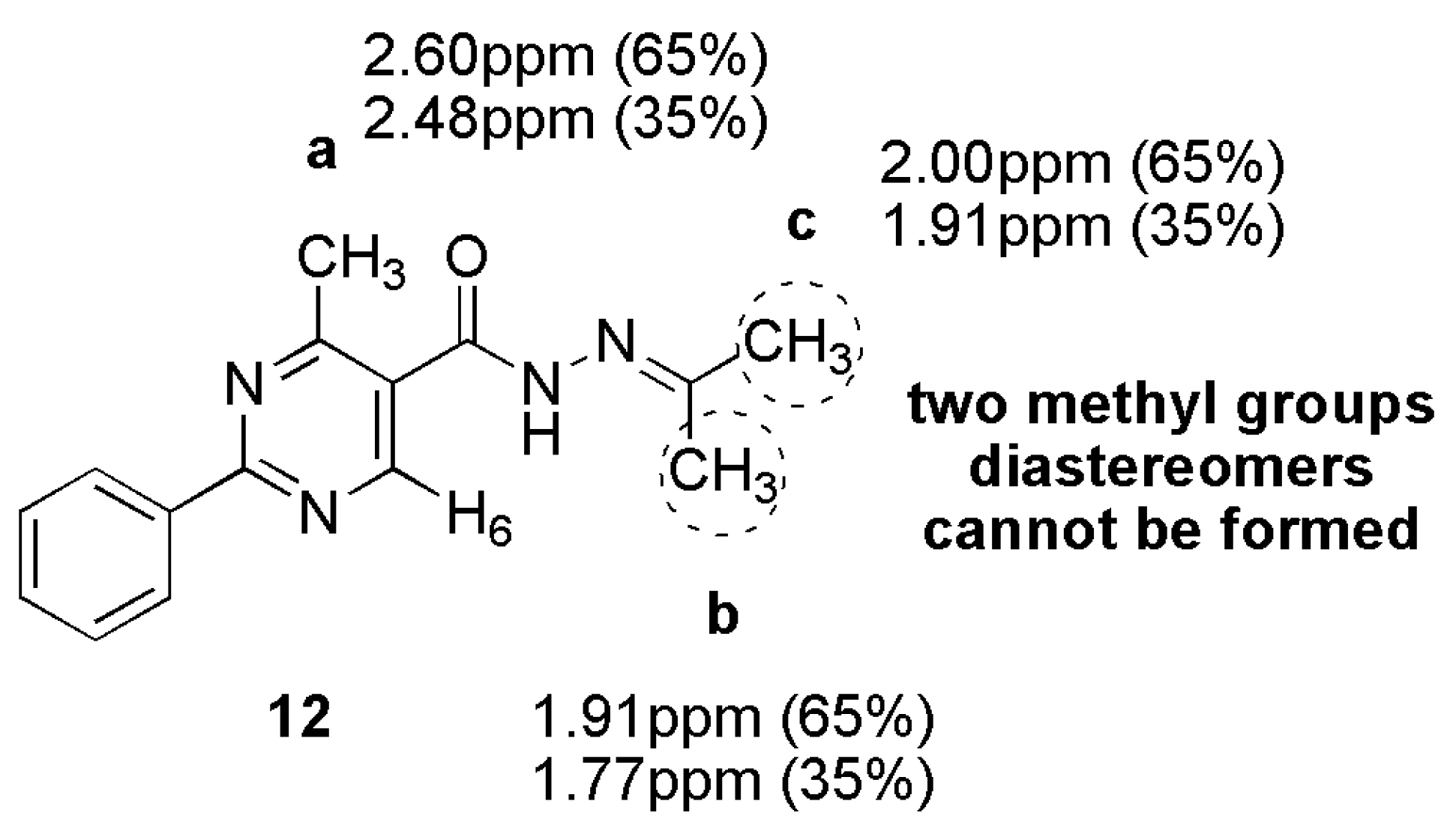
3.13. 4-Methyl-2-Phenyl-N’-(Propan-2-Ylidene)Pyrimidine-5-Carbohydrazide (12)
A mixture of 4-methyl-2-phenylpyrimidine-5-carbohydrazide (11a) 0.07 g, 0.31 mmol) in acetone (7 mL) was stirred for 15 h at 45 °C. After, the solvent was evaporated to give a white solid in 85% yield (mp. 186–188 °C). 1H-NMR (400 MHz, DMSO-d6/TMS) δ 10.9, 10.6 (2s, 1H, NH); 8.83, 8.70 (2s, 1H, H6het); 8.42–8.40 (m, 2H, H2′Ph, H6′Ph); 7.54–7.53 (m, 3H, H3′Ph-H5′Ph); 2.60, 2.48 (2s, 3H, CH3); 1.99, 1.91, 1.77 (3s, 6H, N=C-(CH3)2). 13C-NMR (125 MHz, DMSO-d6/TMS) δ 168.57, 165.73 (C=O); 164.32, 163.47 (N=C); 162.97, 162.39 (C2het); 159.30, 153.61 (C4het); 156.47, 156.30 (C6het); 137.15, 137.12 (C1′Ph); 131.72, 131.60 (C4′Ph); 129.31, 129.25 (C3′Ph, C5′Ph); 128.43, 128.38 (C2′Ph, C6′Ph); 127.43 (C5het); 25.62, 25.56 (N=C-CH3); 23.14, 23.09 (CH3het); 18.55, 17.98 (N=C-CH3). Anal. Calcd for C15H16N4O: C: 67.15; H: 6.01; N: 20.88. Found: C: 66.93; H: 6.05; N: 20.52.
3.14. Molecular Modeling
The sketching, geometry optimization and conformational search of compounds were performed in PC Spartan Pro 1.0.5 software. They were subjected to structural minimization by the use of molecular mechanics, using the base MMFF. Subsequently, the conformational analysis was performed, using the semi-empirical AM1.
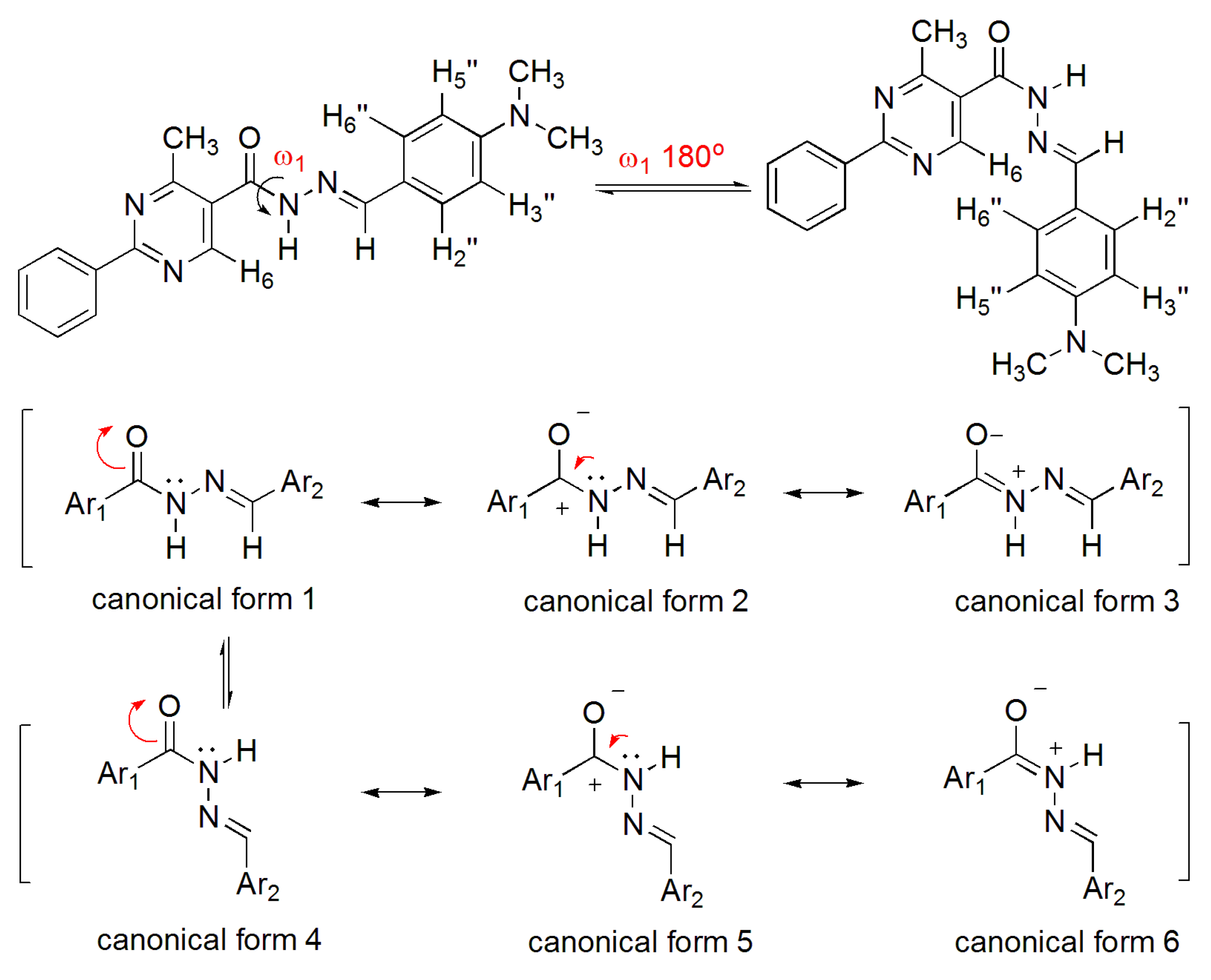
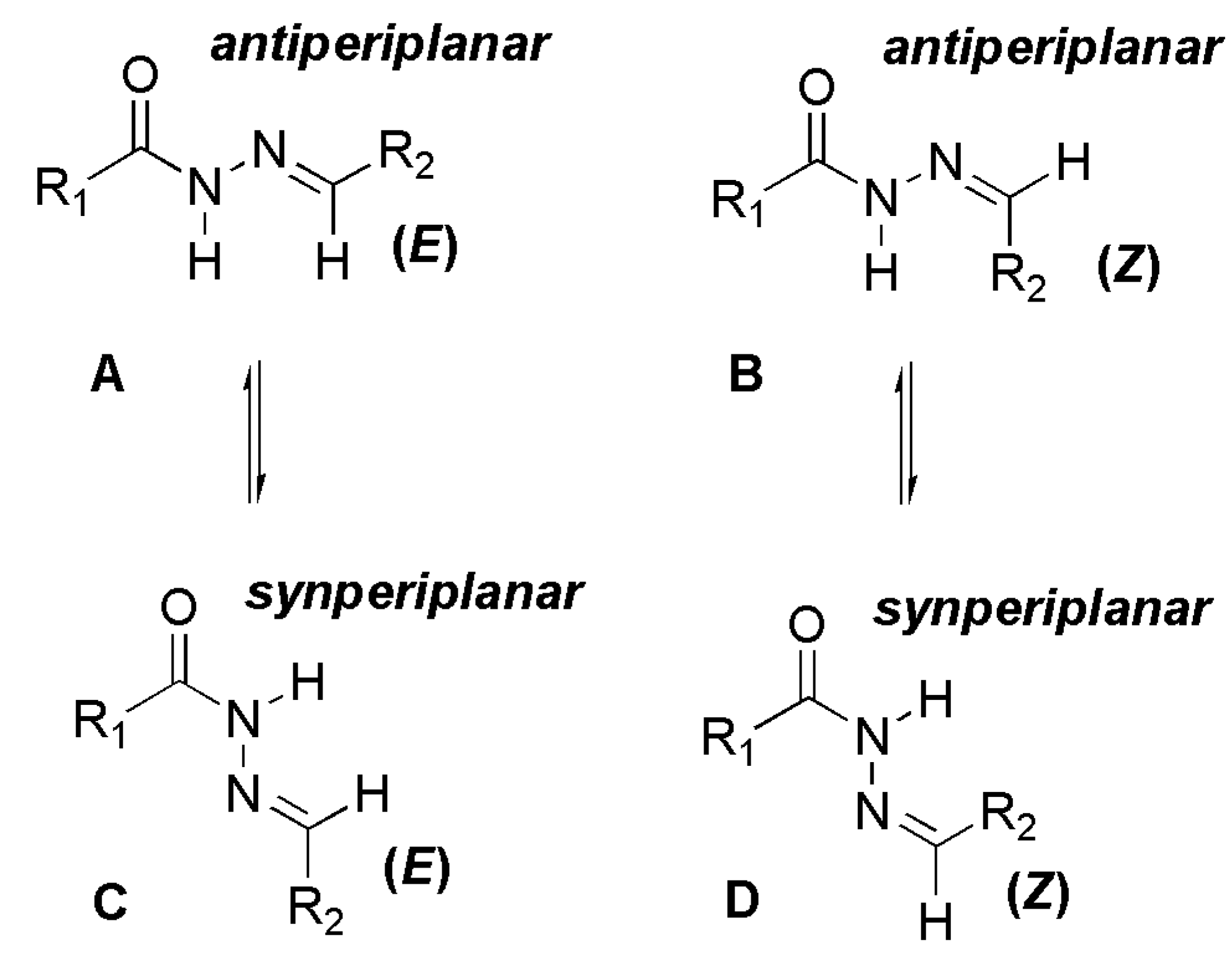
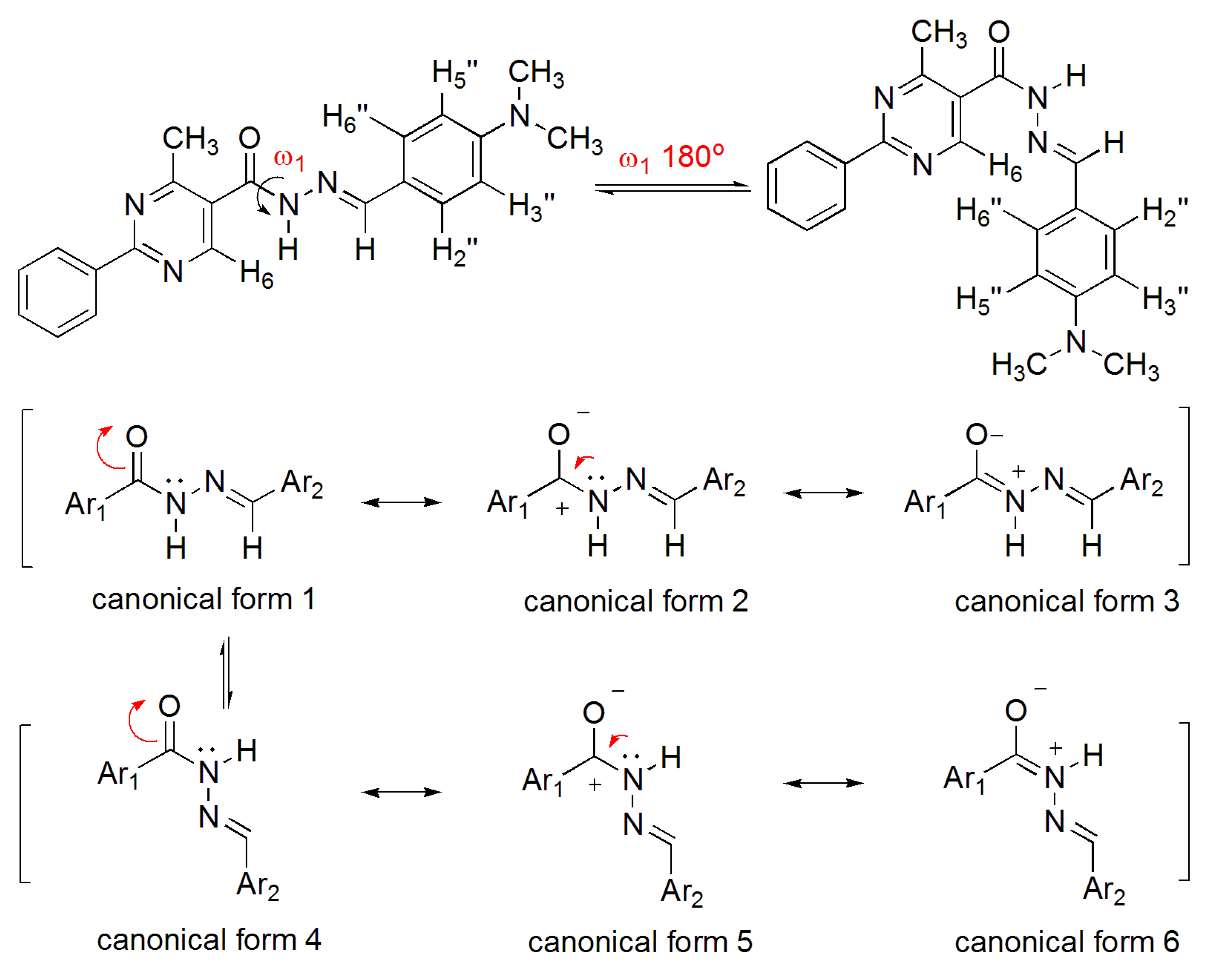
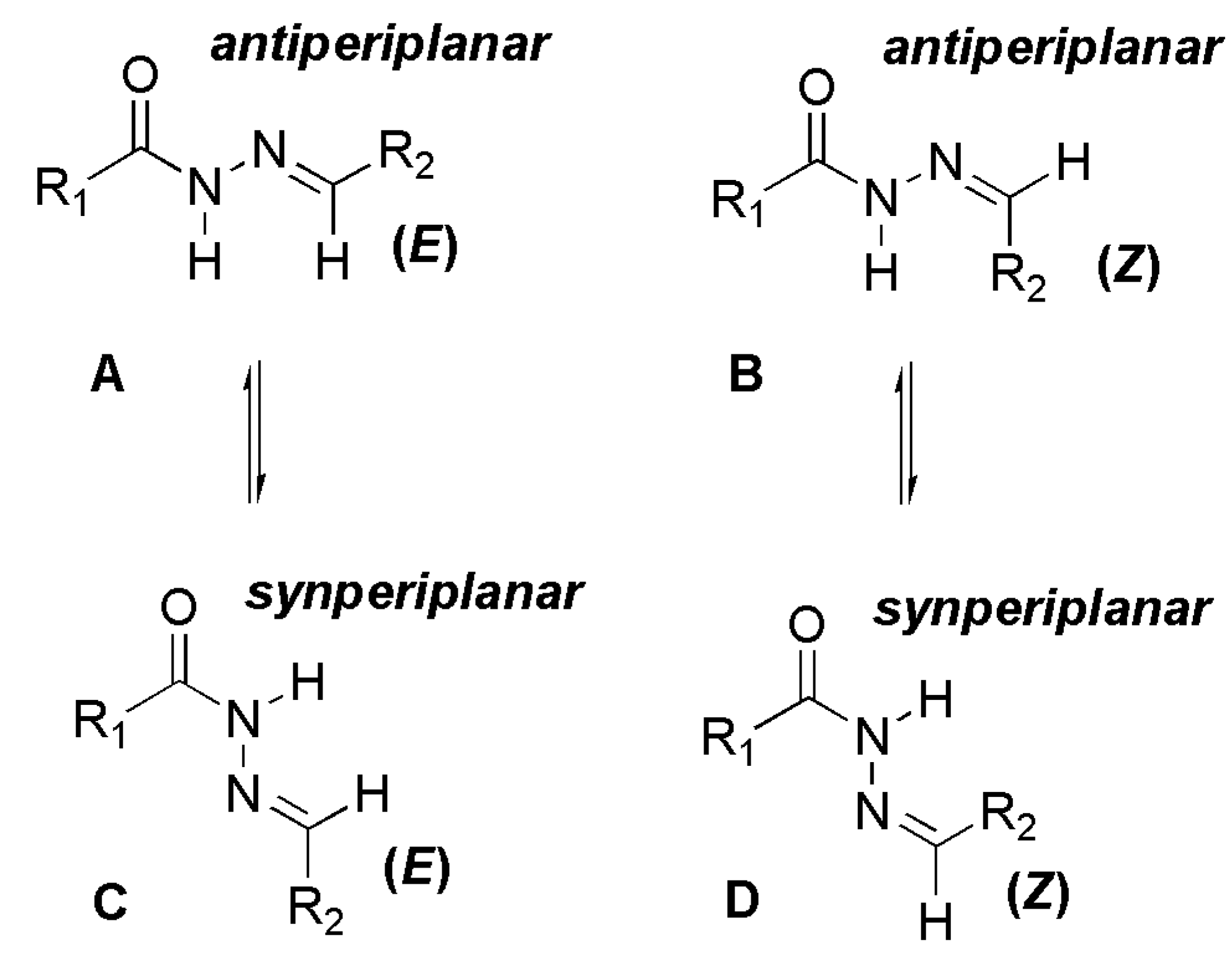
The NOE prediction study was performed by selecting the minimum conformation of each stereoisomer (E/Z), and additionally the election of a second conformer (B) for the E isomer which ΔHf = 114.68 kcal/mol. Conformers that have the carbonyl group synperiplanar related to NH hydrogen were obtained by a dihedral angle search in 12 steps of 30° on the amide bond CO-NH, leaving the other angles free. Then we selected the lowest energy conformer, generated by the conformational analysis with the semi-empirical AM1 method, which presented the carbonyl synperiplanar related to the NH. The NOE calculations were carried out in the MSpin 1.03 software from the selected structures from the conformational analysis.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}