
Immobilization of Neutral Protease from Bacillus subtilis for Regioselective Hydrolysis of Acetylated Nucleosides: Application to Capecitabine Synthesis

 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
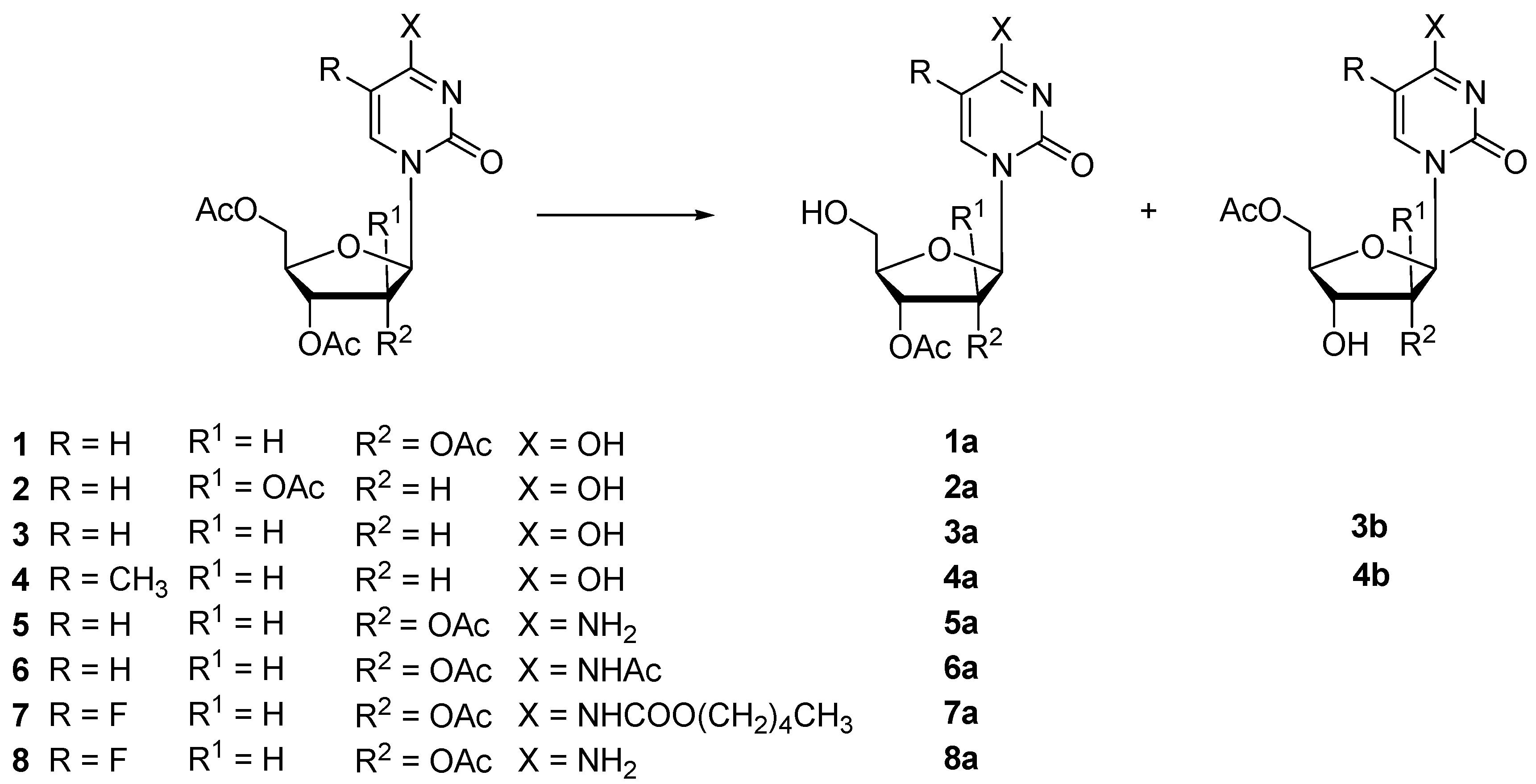
2.1. Screening of Different Hydrolases in the Enzymatic Deacylation of Peracetylated Nucleosides
2.2. Covalent Immobilization of Protease N
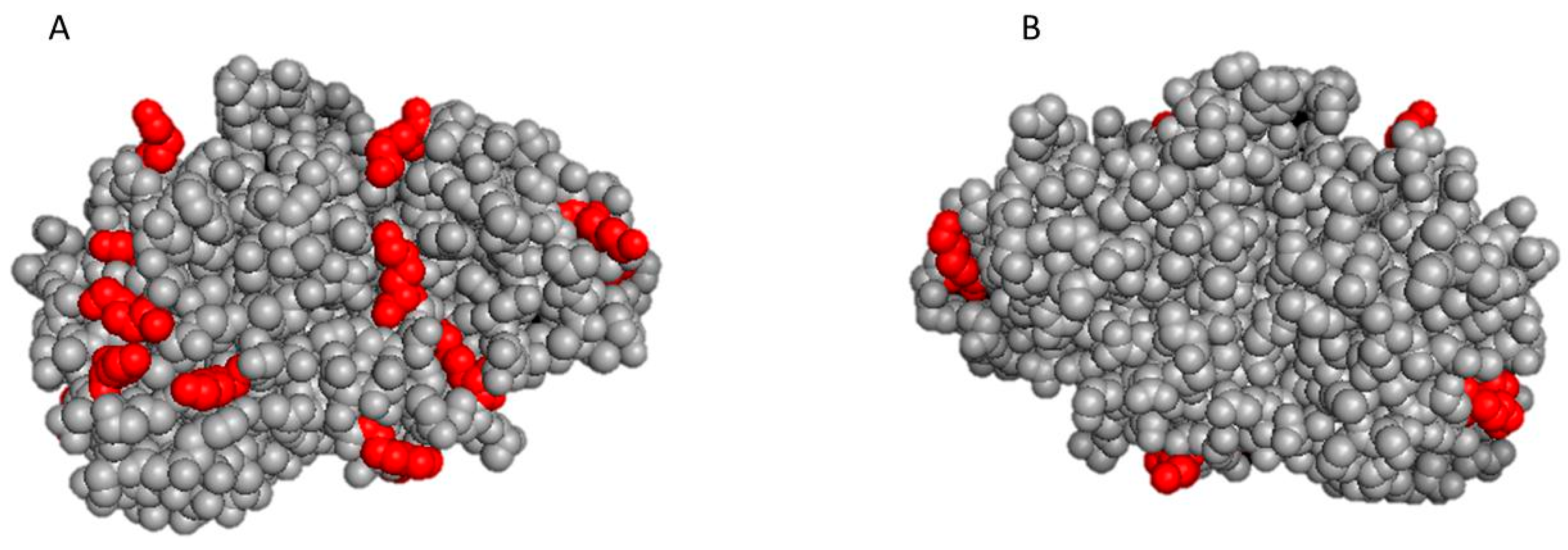
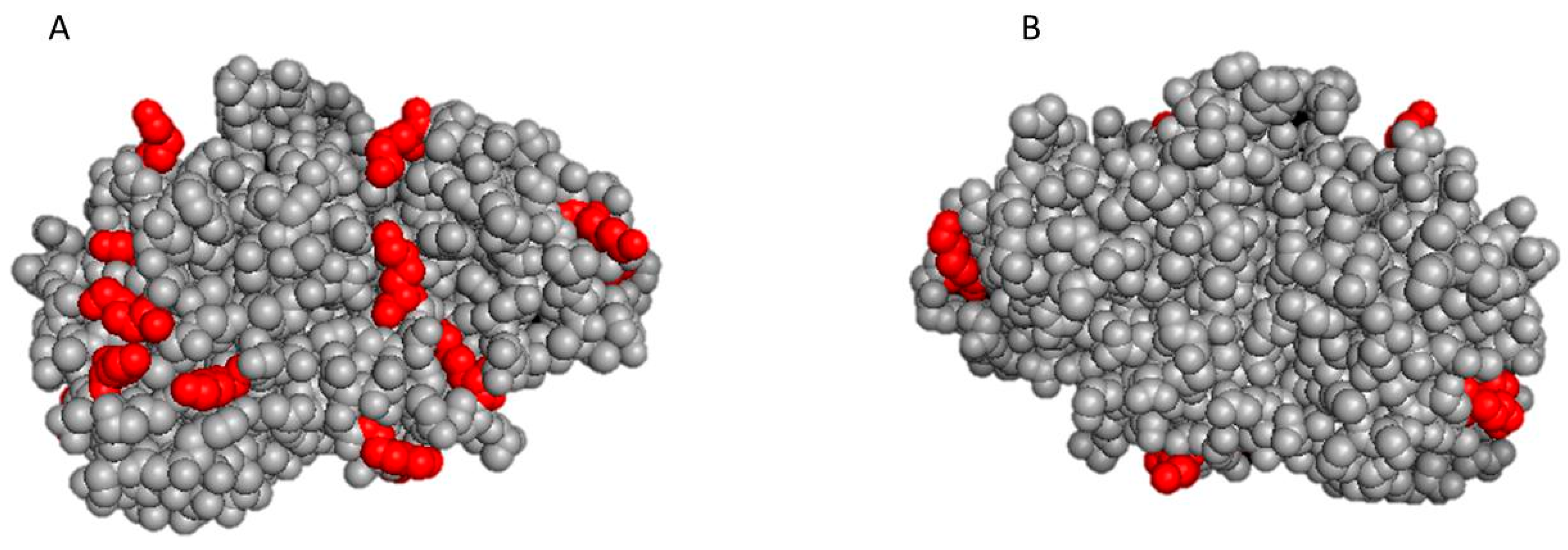
2.3. In Silico Evaluation of the 3D Structure of Protease N
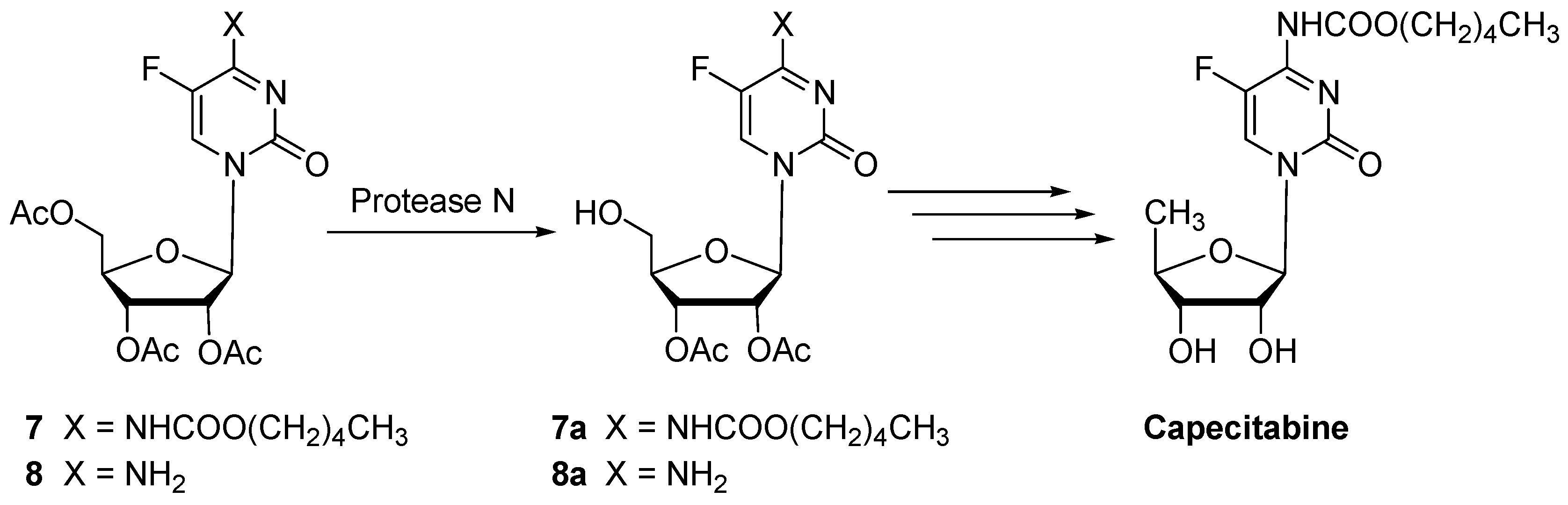
2.4. Optimization of Enzymatic Hydrolysis of 1 and 5
3. Materials and Methods
3.1. General Procedures
3.2. Enzyme Immobilization
3.2.1. Immobilization of Esterase Fraction Contained in the Crude Extract of Aspergillus niger Lipase
3.2.2. Immobilization of Esterase PPL and Acylase on Eupergit® C
3.2.3. Immobilization of Protease N on Eupergit® C and Sepabeads EC-EP
3.2.4. Immobilization of Protease N on Glyoxyl Agarose and Agarose Glutaraldehyde
3.2.5. Immobilization of Protease N on Cyanogen Bromide-Activated Sepharose® 4B
3.3. Activity Assay
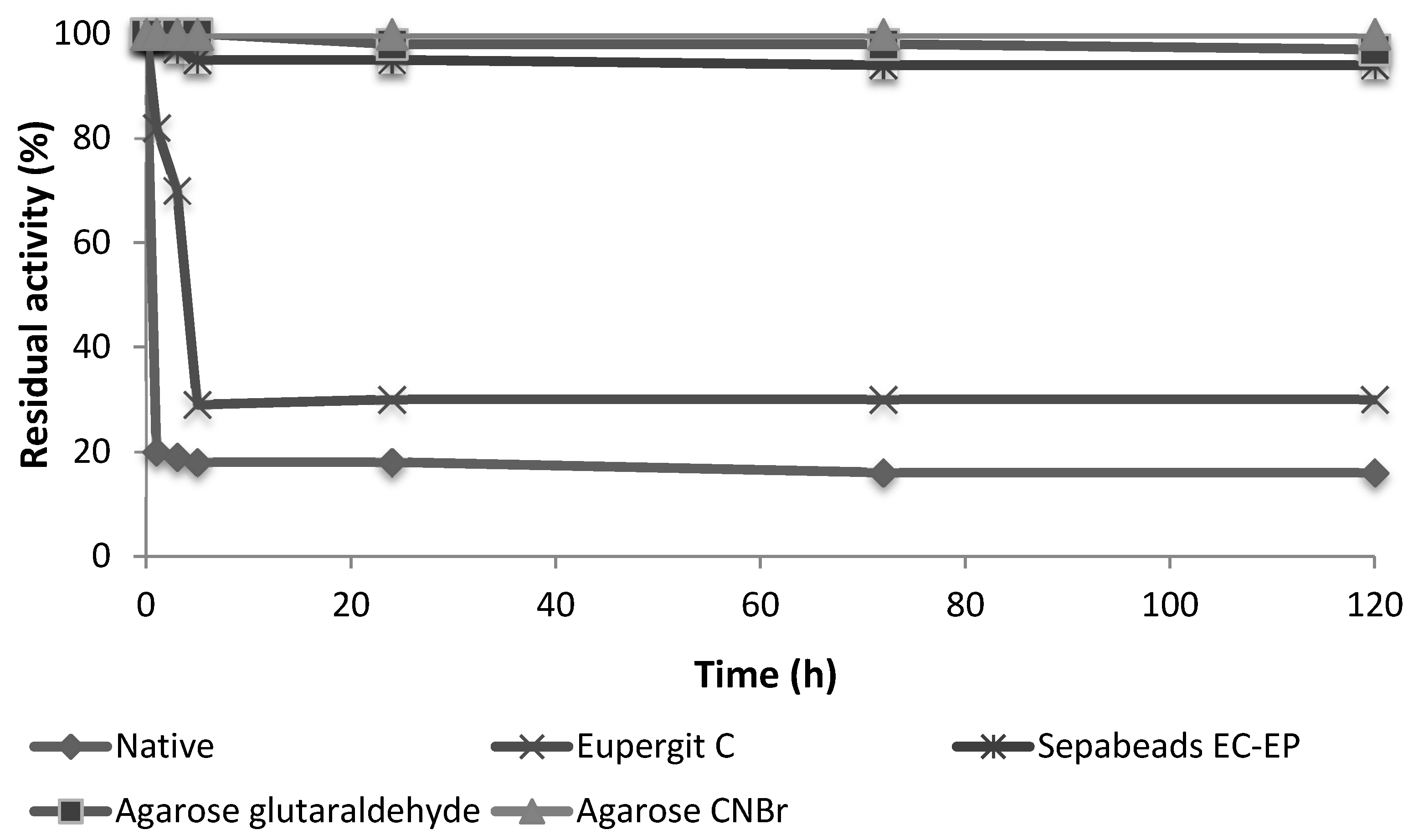
3.4. Enzyme Stability
3.5. Evaluation of the Attachment between the Enzyme and the Support
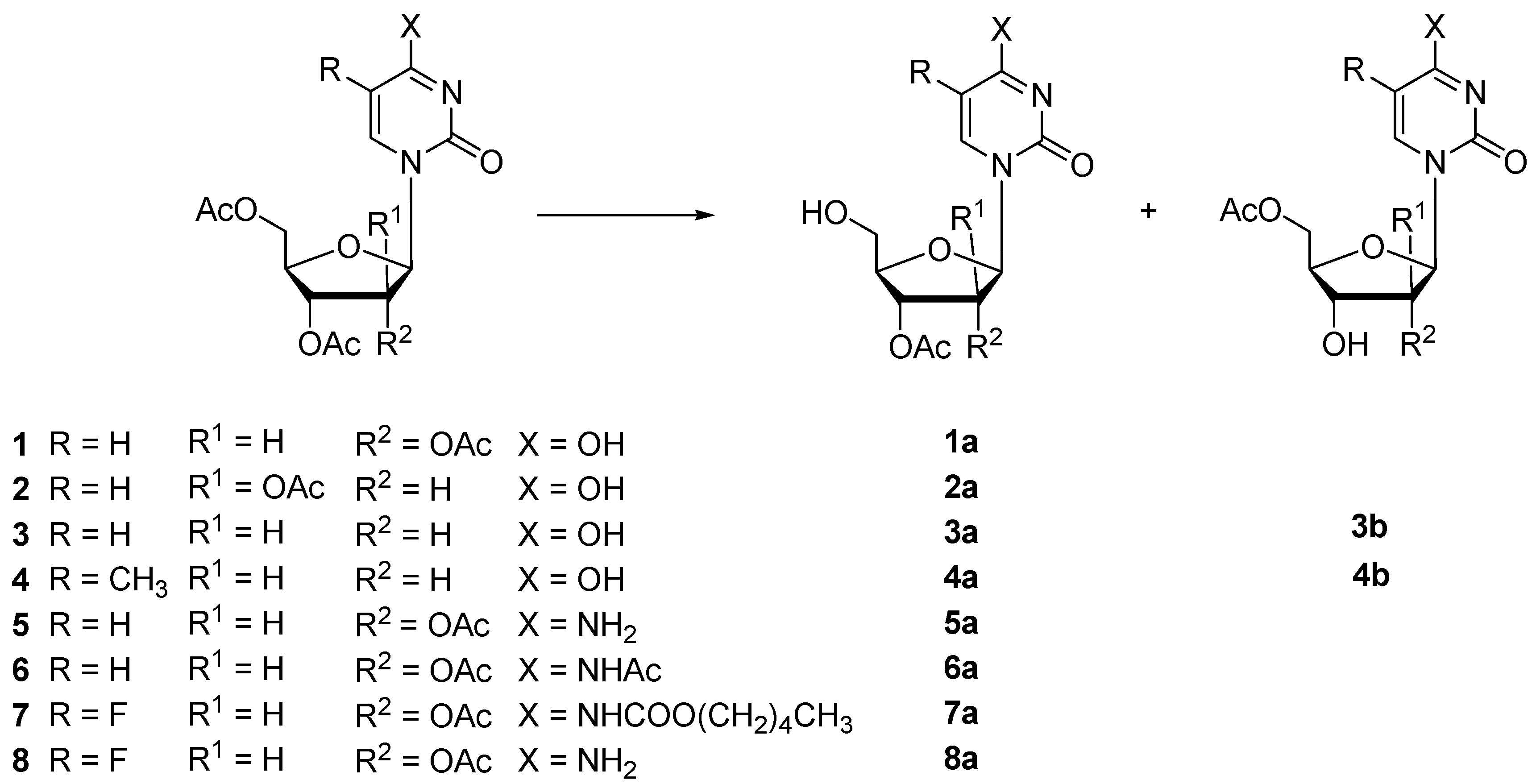
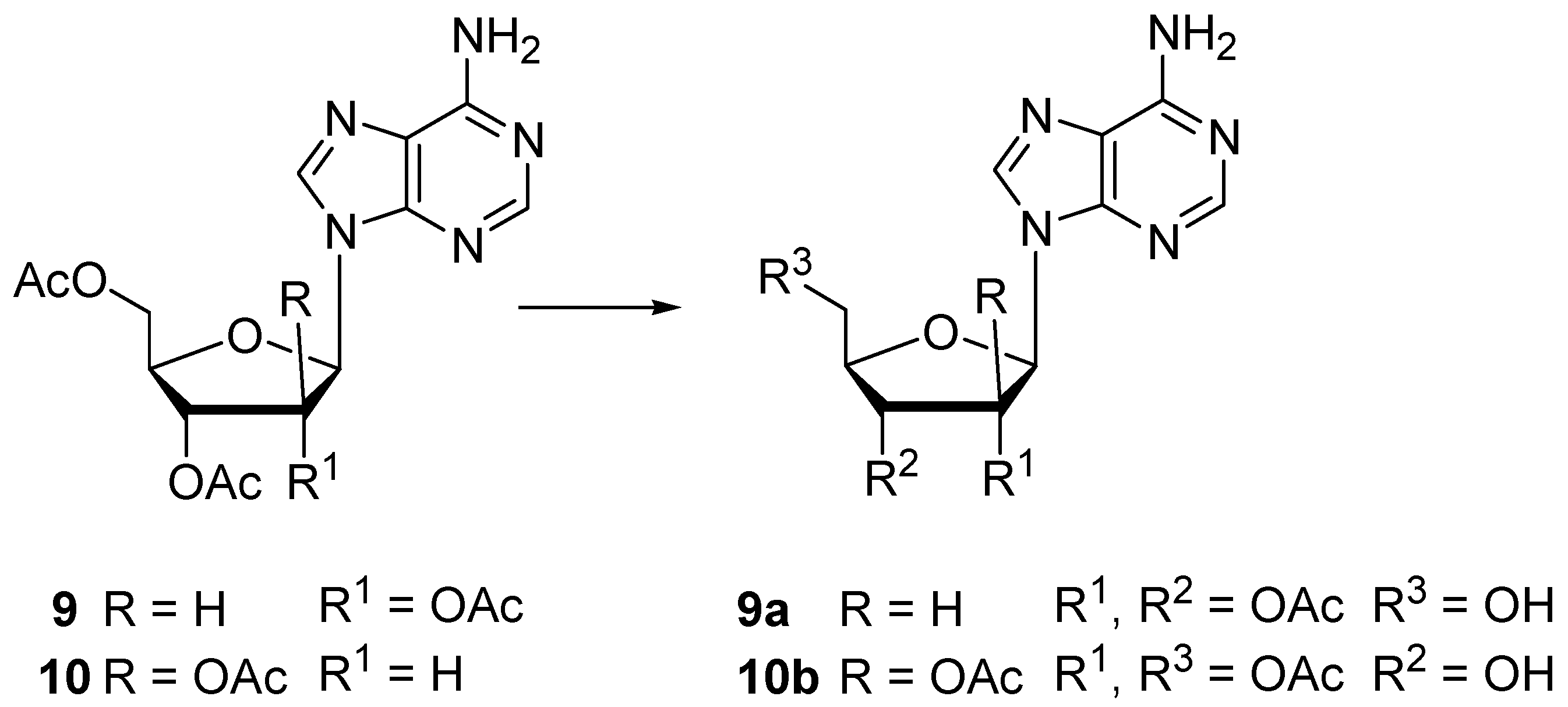
3.6. Enzymatic Hydrolysis of Peracetylated Nucleosides 1–10
3.7. Semi-preparative Method
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bott, R. Development of new proteases for detergents. Surfactant Sci. Ser. 1997, 69, 75–91. [Google Scholar]
- Sumantha, A.; Larroche, C.; Pandey, A. Microbiology and industrial biotechnology of food-grade proteases: A perspective. Food Technol. Biotechnol. 2006, 44, 211–220. [Google Scholar]
- Tufvesson, P.; Lima-Ramos, J.; Nordblad, M.; Woodley, J.M. Guidelines and cost analysis for catalyst production in biocatalytic processes. Org. Process Res. Dev. 2011, 15, 266–274. [Google Scholar]
- Cao, L. Immobilised enzymes: Science or art? Curr. Opin. Chem. Biol. 2005, 9, 217–226. [Google Scholar] [PubMed]
- Rodrigues, R.C.; Ortiz, C.; Berenguer-Murcia, A.; Torres, R.; Fernández-Lafuente, R. Modifying enzyme activity and selectivity by immobilization. Chem. Soc. Rev. 2013, 42, 6290–6307. [Google Scholar] [PubMed]
- Fernandez-Lafuente, R. Stabilization of multimeric enzymes: Strategies to prevent sub-unit dissociation. Enzym. Microb. Technol. 2009, 45, 405–418. [Google Scholar] [CrossRef]
- Guzik, U.; Hupert-Kocurek, K.; Wojcieszynska, D. Immobilization as a strategy for improving enzyme properties-application to oxidoreductases. Molecules 2014, 19, 8995–9018. [Google Scholar] [CrossRef] [PubMed]
- Cantone, S.; Ferrario, V.; Corici, L.; Ebert, C.; Fattor, D.; Spizzo, P.; Gardossi, L. Efficient immobilisation of industrial biocatalysts: Criteria and constraints for the selection of organic polymeric carriers and immobilisation methods. Chem. Soc. Rev. 2013, 42, 6262–6276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, R.C.; Berenguer-Murcia, Á.; Fernandez-Lafuente, R. Coupling chemical modification and immobilization to improve the catalytic performance of enzymes. Adv. Synth. Catal. 2011, 353, 2216–2238. [Google Scholar] [CrossRef]
- Bonomi, P.; Bavaro, T.; Serra, I.; Tagliani, A.; Terreni, M.; Ubiali, D. Modulation of the microenvironment surrounding the active site of Penicillin G Acylase immobilized on acrylic carriers improves the enzymatic synthesis of cephalosporins. Molecules 2013, 18, 14349–14365. [Google Scholar] [CrossRef] [PubMed]
- Bavaro, T.; Rocchietti, S.; Ubiali, D.; Filice, M.; Terreni, M.; Pregnolato, M. A versatile synthesis of 5’-functionalized nucleosides through regioselective enzymatic hydrolysis of their peracetylated precursors. Eur. J. Org. Chem. 2009, 1967–1975. [Google Scholar] [CrossRef]
- Bavaro, T.; Ubiali, D.; Brocca, S.; Rocchietti, S.; Nieto, I.; Pregnolato, M.; Lotti, M.; Terreni, M. Recombinant lipase from Candida rugosa for regioselective hydrolysis of peracetylated nucleosides. A comparison with commercial non recombinant lipases. Biocatal. Biotransf. 2010, 28, 108–116. [Google Scholar] [CrossRef]
- Bastida, A.; Sabuquillo, P.; Armisen, P.; Fernàndez-Lafuente, R.; Huguet, J.; Guisàn, J.M. A single step purification, immobilization, and hyperactivation of lipases via interfacial adsorption on strongly hydrophobic supports. Biotechnol. Bioeng. 1998, 58, 486–493. [Google Scholar] [CrossRef]
- Guisan, J.M.; Betancor, L.; Fernandez-Lorente, G. Encyclopedia of Industrial Biotechnology: Bioprocess, Bioseparation, and Cell Technology, 1st ed.; John Wiley & Sons: Chichester, UK, 2009; pp. 1–16. [Google Scholar]
- Mateo, C.; Palomo, J.M.; Fuentes, M.; Betancor, L.; Grazu, V.; López-Gallego, F.; Pessela, B.C.C.; Hidalgo, A.; Fernández-Lorente, G.; Fernández-Lafuente, R.; et al. Glyoxyl agarose: A fully inert and hydrophilic support for immobilization and high stabilization of proteins. Enzym. Microb. Technol. 2006, 39, 274–280. [Google Scholar] [CrossRef]
- Uemura, A.; Nozaki, K.; Yamashita, J.; Yasumoto, M. Lipase-catalyzed regioselective acylation of sugar moieties of nucleosides. Tetrahedron Lett. 1989, 30, 3817–3818. [Google Scholar] [CrossRef]
- Haribansh, K.; Singh, G.; Cote, L.; Sikors, S.R. Enzymatic regioselective deacylation of 2′,3′,5′-tri-O-acylribonucleosides: Enzymatic synthesis of 2′,3′-di-O-acylribonucleosides. Tetrahedron Lett. 1993, 34, 5201–5204. [Google Scholar]
- Panero, J.; Trelles, J.; Rodano, V.; Montserrat, J.M.; Iglesias, L.E.; Lewkowicz, E.S.; Iribarren, A.M. Microbial hydrolysis of acetylated nucleosides. Biotechnol. Lett. 2006, 8, 1077–1081. [Google Scholar] [CrossRef] [PubMed]
- Tran, L.; Wu, X.; Wong, S. Cloning and expression of a novel protease gene encoding an extracellular neutral protease from Bacillus subtilis. J. Bacteriol. 1991, 173, 6364–6372. [Google Scholar] [CrossRef] [PubMed]
- Mcconn, J.D.; Tsuru, D.; Yasunobu, K.T. Bacillus subtilis neutral proteinase. I. A zinc enzyme of high specific. J. Biol. Chem. 1964, 239, 3706–3715. [Google Scholar] [PubMed]
- Tsuru, D.; Mcconn, J.D.; Yasunobu, K.T. Bacillus subtilis neutral proteinase. II. Some physicochemical. J. Biol. Chem. 1965, 240, 2415–2420. [Google Scholar] [PubMed]
- Yang, M.Y.; Ferrari, E.; Henner, D.J. Cloning of the neutral protease gene of Bacillus subtilis and the use of the cloned gene to create an in vitro-derived deletion mutation. J. Bacteriol. 1984, 160, 15–21. [Google Scholar] [PubMed]
- Marques, D.; Pessela, B.C.; Betancor, L.; Monti, R.; Carrascosa, A.V.; Rocha-Martin, J.; Guisan, J.M.; Fernandez-Lorente, G. Protein hydrolysis by immobilized and stabilized trypsin. Biotechnol. Prog. 2011, 27, 677–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazawa, T.; Masaki, S.; Tanaka, K.; Yamada, T. Peptide syntheses mediated by Bacillus subtilis protease. Lett. Pept. Sci. 2003, 10, 83–87. [Google Scholar] [CrossRef]
- Pregnolato, M.; Terreni, M.; Ubiali, D.; Bavaro, T. 2′,3′-Di-O-Acyl-5-fluoronucleosides. Patent PCT/IB2008/000482, 12 September 2008. [Google Scholar]
- Di Costanzo, F.; Sdrobolini, A.; Gasperoni, S. Capecitabine, a new oral fluoropyrimidine for the treatment of colorectal cancer. Crit. Rev. Oncol. Hematol. 2000, 35, 101–108. [Google Scholar] [CrossRef]
- Ghattas, N.; Filice, M.; Abidi, F.; Guisàn, J.M.; Ben Salah, A. Purification and improvement of the functional properties of Rhizopus oryzae lipase using immobilization techniques. J. Mol. Catal. B Enzym. 2014, 110, 111–116. [Google Scholar] [CrossRef]
- Mateo, C.; Abian, O.; Bernedo, M.; Cuenca, E.; Fuentes, M.; Fernandez-Lorente, G.; Palomo, J.M.; Grazu, V.; Pessela, B.C.C.; Giacomini, C.; et al. Some special features of glyoxyl supports to immobilize proteins. Enzym. Microb. Technol. 2005, 37, 456–462. [Google Scholar] [CrossRef]
- Barbosa, O.; Ortiz, C.; Berenguer-Murcia, A.; Torres, R.; Rodrigues, R.C.; Fernandez-Lafuente, R. Glutaraldehyde in bio-catalysts design: A useful crosslinker and a versatile tool in enzyme immobilization. RSC Adv. 2014, 4, 1583–1600. [Google Scholar] [CrossRef]
- Bavaro, T.; Torres-Salas, P.; Antonioli, N.; Morelli, C.F.; Speranza, G.; Terreni, M. Regioselective deacetylation of disaccharides via immobilized Aspergillus niger esterase(s)-catalyzed hydrolysis in aqueous and non-aqueous media. ChemCatChem 2013, 5, 2925–2931. [Google Scholar] [CrossRef]
- Chung, R.T.; Gale, M., Jr.; Polyak, S.J.; Lemon, S.M.; Liang, T.J.; Hoofnagle, J.H. Mechanisms of action of interferon and ribavirin in chronic hepatitis C: Summary of a workshop. Hepatology 2008, 47, 306–320. [Google Scholar] [CrossRef] [PubMed]
- Mateo, C.; Grazù, V.; Palomo, J.M.; Lopez-Gallego, F.; Fernàndez-Lafuente, R.; Guisàn, J.M. Immobilization of enzymes on heterofunctional epoxy supports. Nat. Protoc. 2007, 2, 1022–1033. [Google Scholar] [CrossRef] [PubMed]
- Fernàndez-Lafuente, R.; Rosell, C.M.; Rodriguez, V.; Santana, C.; Soler, G.; Bastida, A.; Guisàn, J.M. Preparation of activated supports containing low pK amino groups. A new tool for protein immobilization via the carboxyl coupling method. Enzym. Microb. Technol. 1993, 15, 546–550. [Google Scholar] [CrossRef]
- HHpred-Homology Detection & Structure Prediction by HMM-HMM Comparison. Available online: https://toolkit.tuebingen.mpg.de/hhpred (accessed on 24 November 2016).
- Modeller. Available online: https://toolkit.tuebingen.mpg.de/modeller (accessed on 24 November 2016).
- Serra, I.; Serra, C.D.; Rocchietti, S.; Ubiali, D.; Terreni, M. Stabilization of thymidine phosphorylase from Escherichia coli by immobilization and post immobilization techniques. Enzym. Microb. Technol. 2011, 49, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Temporini, C.; Bonomi, P.; Serra, I.; Tagliani, A.; Bavaro, T.; Ubiali, D.; Massolini, G.; Terreni, M. Characterization and study of the orientation of immobilized enzymes by tryptic digestion and HPLC-MS: Design of an efficient catalyst for the synthesis of cephalosporins. Biomacromolecules 2010, 11, 1623–1632. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Sample Availability: Samples of immobilized protease N on agarose CNBr carrier are available from the Authors.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | Enzyme | t (h) | Vh 1 | Conversion (%) | Products (Yield %) | ||
|---|---|---|---|---|---|---|---|
| 5′-OH | 3′-OH | ||||||
| 1 | Protease N | 22 | 0.52 | 100 | 1a (93) | n.i. | |
| Esterase PPL | 96 | 0.02 | 79 | 1a (52) | n.i. | ||
| Esterase ANL | 5 | 3.13 | 98 | 1a (20) | n.i. | ||
| Acylase | 4 | 0.66 | 97 | 1a (56) | n.i. | ||
| 2 | Protease N | 48 | 0.04 | 48 | 2a (34) | n.i. | |
| Esterase PPL | 120 | 0.02 | 47 | 2a (28) | n.i. | ||
| Esterase ANL | 5 | 5.66 | 99 | 2a (35) | n.i. | ||
| Acylase | 24 | 0.10 | 97 | 2a (14) | n.i. | ||
| 3 | Protease N | 48 | 0.10 | 93 | 3a (64) | 3b (13) | |
| Esterase PPL | 48 | 0.01 | 28 | 3a (23) | 3b (5) | ||
| Esterase ANL | 5 | 4.46 | 99 | 3a (44) | 3b (2) | ||
| Acylase | 5 | 0.68 | 98 | 3a (49) | 3b (5) | ||
| 4 | Protease N | 24 | 0.71 | 98 | 4a (74) | 4b (6) | |
| Esterase PPL | 48 | 0.36 | 91 | 4a (41) | 4b (8) | ||
| Esterase ANL | 3 | 2.07 | 92 | 4a (42) | 4b (10) | ||
| Acylase | 5 | 0.07 | 92 | 4a (68) | 4b (9) | ||
| 5 | Protease N | 48 | 0.16 | 94 | 5a (84) | n.i. | |
| Esterase PPL | 48 | 0.90 | 88 | 5a (51) | n.i. | ||
| Esterase ANL | 5 | 5.10 | 98 | 5a (13) | n.i. | ||
| Acylase | 3 | 0.89 | 97 | 5a (49) | n.i. | ||
| 6 | Protease N | 48 | 0.19 | 82 | 6a (77) | n.i. | |
| Esterase PPL | 48 | 0.09 | 99 | 6a (54) | n.i. | ||
| Esterase ANL | 3 | 6.14 | 99 | 6a (22) | n.i. | ||
| Acylase | 7 | 0.35 | 94 | 6a (73) | n.i. | ||
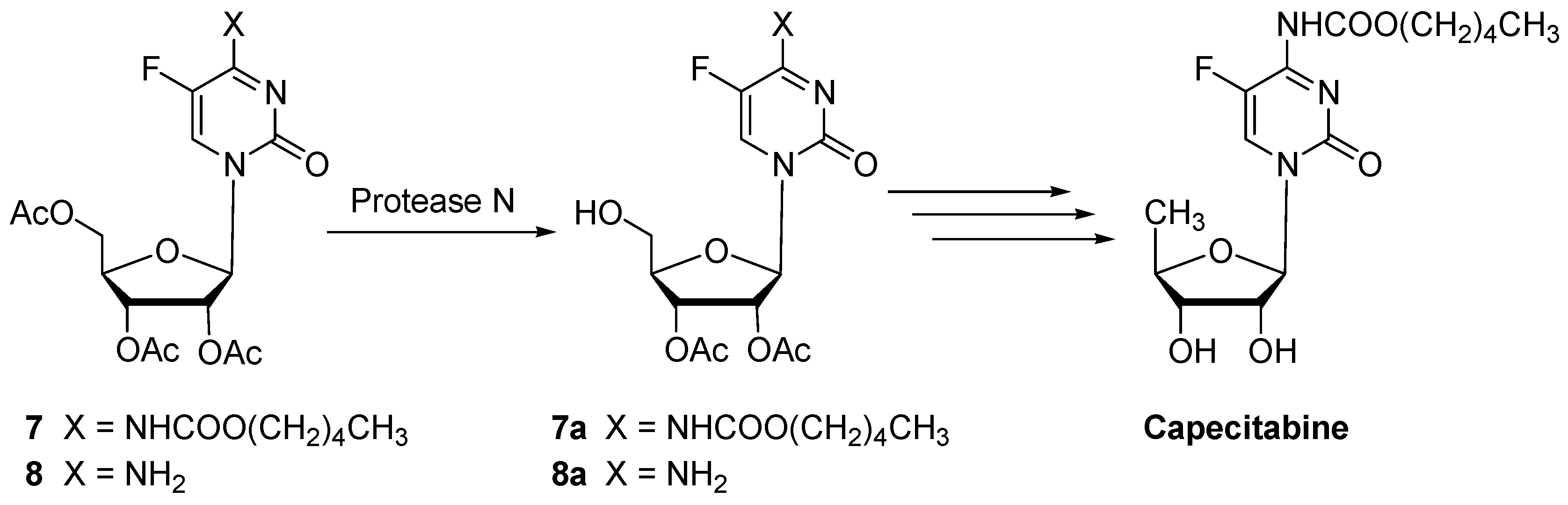
| 7 | Protease N | 24 | 0.10 | 97 | 7a (92) | n.i. | |
| Esterase PPL | 24 | 0.15 | 92 | 7a (48) | n.i. | ||
| Esterase ANL | 5 | 4.30 | 94 | 7a (66) | n.i. | ||
| Acylase | 18 | 0.18 | 90 | 7a (78) | n.i. | ||
| CRL | 3 | 3.3 | 98 | 7a (39) | n.i. | ||
| 8 | Protease N | 30 | 0.18 | 99 | 8a (89) | n.i. | |
| CRL | 30 | 0.12 | 93 | 8a (80) | n.i. | ||
| 9 | Protease N | 48 | 0.12 | 93 | 9a (80) | n.i. | |
| Esterase PPL | 48 | 0.03 | 72 | 9a (72) | n.i. | ||
| Esterase ANL | 3 | 4.42 | 97 | 9a (21) | n.i. | ||
| Acylase | 3 | 1.11 | 99 | 9a (42) | n.i. | ||
| 10 | Protease N | 24 | 0.09 | 55 | n.i. | 10b (42) | |
| Esterase PPL | 24 | 0.04 | 36 | n.i. | 10b (34) | ||
| Esterase ANL | 5 | 3.61 | 99 | n.i. | 10b (57) | ||
| Acylase | 4 | 0.77 | 99 | n.i. | 10b (12) | ||
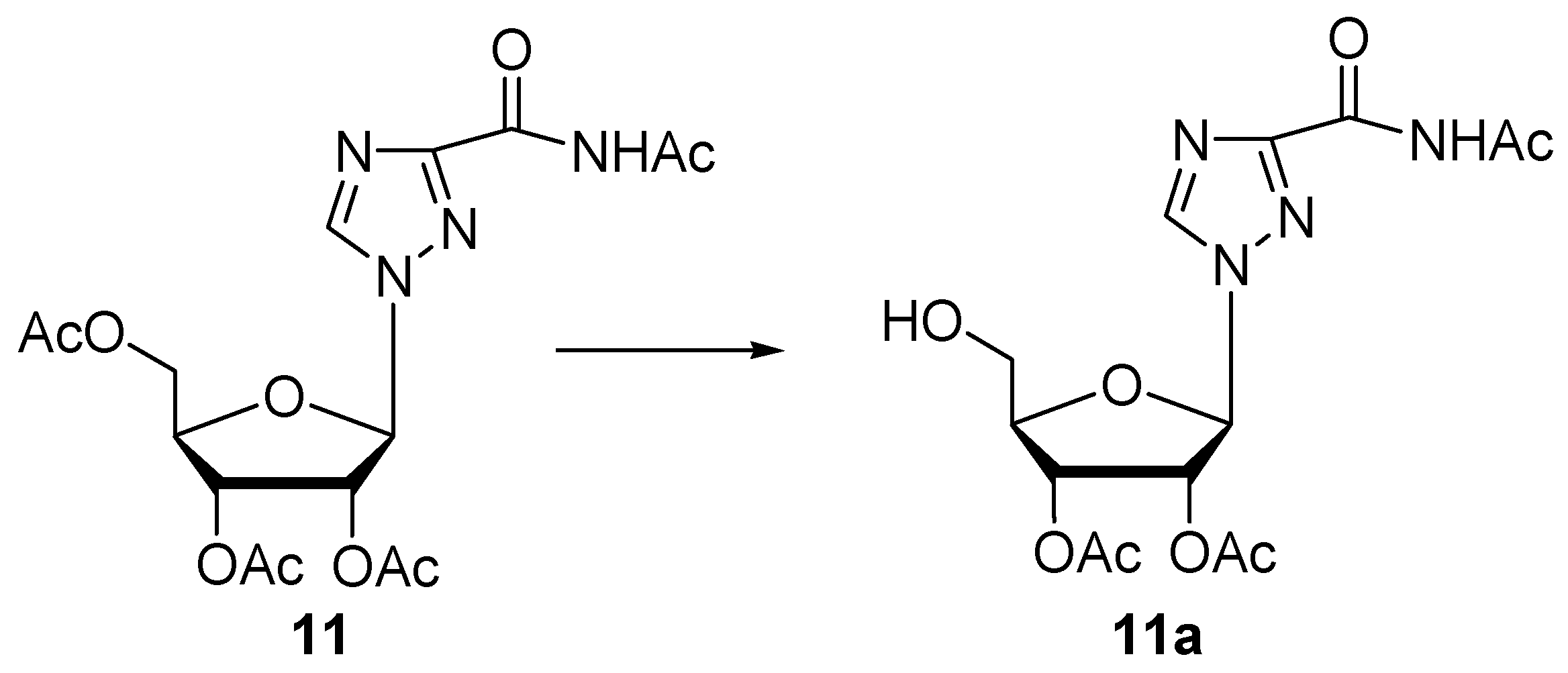
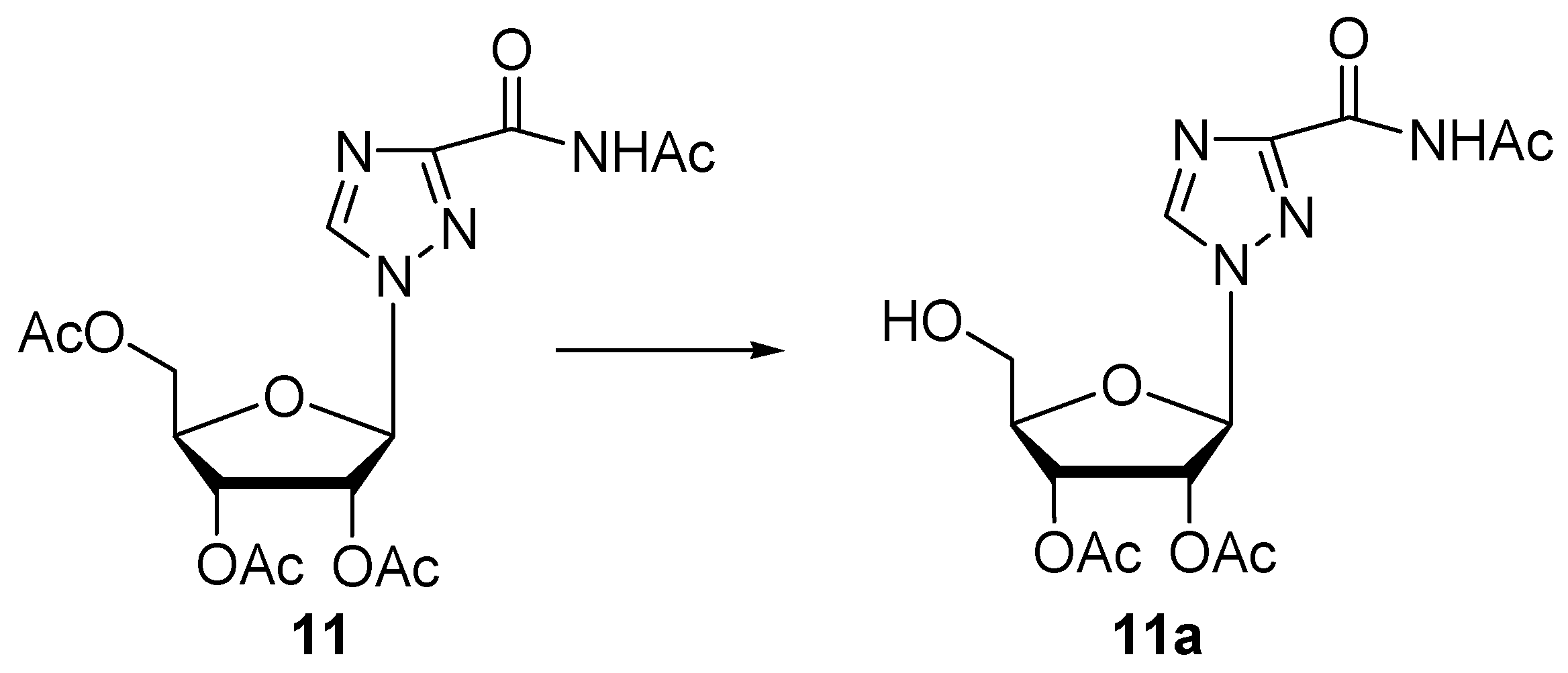
| 11 | Protease N | 48 | 0.05 | 62 | 11a (31) | n.i. | |
| Esterase PPL | 48 | 0.10 | 58 | 11a (31) | n.i. | ||
| Esterase ANL | 4 | 3.16 | 98 | 11a (26) | n.i. | ||
| Acylase | 4 | 0.76 | 100 | 11a (3) | n.i. | ||
| Support | Activation | Loading 1 mg Protein/g Support | Imm. Protein 2 (%) (SD) | Activity 3 (IU·g−1) (SD) | Yield 4 (%) (SD) |
|---|---|---|---|---|---|
| Eupergit® C | Epoxy | 10 | 62 (8) | 1 (0.5) | 20 (10) |
| Sepabeads EC-EP | Epoxy | 10 | 58 (11) | 0.8 (0.4) | 16 (8) |
| Glyoxyl agarose | Aldehyde | 10 | 90 (6) | 1.6 (0.3) | 32 (6) |
| Agarose glutaraldehyde | Aldehyde | 10 | 98 (4) | 1.1 (0.2) | 22 (4) |
| Agarose CNBr | Isocyanate | 10 | 96 (3) | 1.2 (0.3) | 24 (6) |
| Agarose CNBr | Isocyanate | 50 | 65 (5) | 7.8 (0.4) | 31 (2) |
| Substrate | Support | t (h) | Vh | Conversion (%) | Product (Yield %) 5′-OH |
|---|---|---|---|---|---|
| 1 | Native enzyme | 22 | 0.52 | 100 | 1a (93) |
| 1 | Agarose CNBr | 15 | 0.14 | 99 | 1a (94) |
| 1 | Agarose glutaraldehyde | 24 | 0.14 | 81 | 1a (69) |
| 5 | Native enzyme | 48 | 0.16 | 92 | 5a (84) |
| 5 | Agarose CNBr | 48 | 0.05 | 94 | 5a (66) |
| 5 | Agarose glutaraldehyde | 216 | 0.06 | 88 | 5a (54) |
| Substrate | Solvent | pH Temperature | t (h) | Vh 1 | Conversion (%) | Product (Yield %), 5′-OH |
|---|---|---|---|---|---|---|
| 1 | MeOH | pH 7, 25 °C | 7 | 0.12 | 96 | 1a (74) |
| 1 | Acetone | pH 7, 25 °C | 23 | 0.09 | 99 | 1a (82) |
| 1 | CH3CN | pH 7, 25 °C | 15 | 0.14 | 99 | 1a (94) |
| 1 | CH3CN | pH 7, 4 °C | 144 | 0.02 | 99 | 1a (90) |
| 1 | CH3CN | pH 5.5, 25 °C | 216 | 0.01 | 95 | 1a (82) |
| 1 | CH3CN | pH 8.5, 25 °C | 27 | 0.08 | 97 | 1a (72) |
| 5 | MeOH | pH 7, 25 °C | 48 | 0.07 | 88 | 5a (42) |
| 5 | Acetone | pH 7, 25 °C | 120 | 0.02 | 85 | 5a (58) |
| 5 | CH3CN | pH 7, 25 °C | 48 | 0.09 | 94 | 5a (66) |
| 5 | CH3CN | pH 7, 4 °C | 360 | 0.01 | 82 | 5a (63) |
| 5 | CH3CN | pH 5.5, 25 °C | 192 | 0.005 | 92 | 5a (62) |
| 5 | CH3CN | pH 8.5, 25 °C | 120 | 0.02 | 93 | 5a (50) |
| Substrate | Conc. (mM) | t (h) | Vh 1 | Conversion (%) (SD) | Product (Yield %) 5′-OH (SD) |
|---|---|---|---|---|---|
| 1 | 10 | 24 | 0.48 | 94 (5) | 1a (94) (5) |
| 1 | 20 | 48 | 0.30 | 92 (4) | 1a (86) (6) |
| 1 | 40 | 72 | 0.28 | 90 (8) | 1a (79) (10) |
| 5 | 10 | 48 | 0.09 | 94 (3) | 5a (66) (7) |
| 5 | 40 | 72 | - | 88 (9) | 5a (85) (6) |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bavaro, T.; Cattaneo, G.; Serra, I.; Benucci, I.; Pregnolato, M.; Terreni, M. Immobilization of Neutral Protease from Bacillus subtilis for Regioselective Hydrolysis of Acetylated Nucleosides: Application to Capecitabine Synthesis. Molecules 2016, 21, 1621. https://doi.org/10.3390/molecules21121621
Bavaro T, Cattaneo G, Serra I, Benucci I, Pregnolato M, Terreni M. Immobilization of Neutral Protease from Bacillus subtilis for Regioselective Hydrolysis of Acetylated Nucleosides: Application to Capecitabine Synthesis. Molecules. 2016; 21(12):1621. https://doi.org/10.3390/molecules21121621
Chicago/Turabian StyleBavaro, Teodora, Giulia Cattaneo, Immacolata Serra, Ilaria Benucci, Massimo Pregnolato, and Marco Terreni. 2016. "Immobilization of Neutral Protease from Bacillus subtilis for Regioselective Hydrolysis of Acetylated Nucleosides: Application to Capecitabine Synthesis" Molecules 21, no. 12: 1621. https://doi.org/10.3390/molecules21121621