Hedgehog Signaling in Prostate Cancer and Its Therapeutic Implication

Abstract
:1. Introduction
2. Hedgehog Signaling
2.1. Hedgehog Signaling Pathway
2.2. Hedgehog Signaling in Cancer
2.3. Hedgehog Signaling in Prostate Cancer
3. Therapeutic Application of Hedgehog Inhibition
3.1. Hh Inhibitors
3.2. Hh Signaling Inhibition as Monotherapy
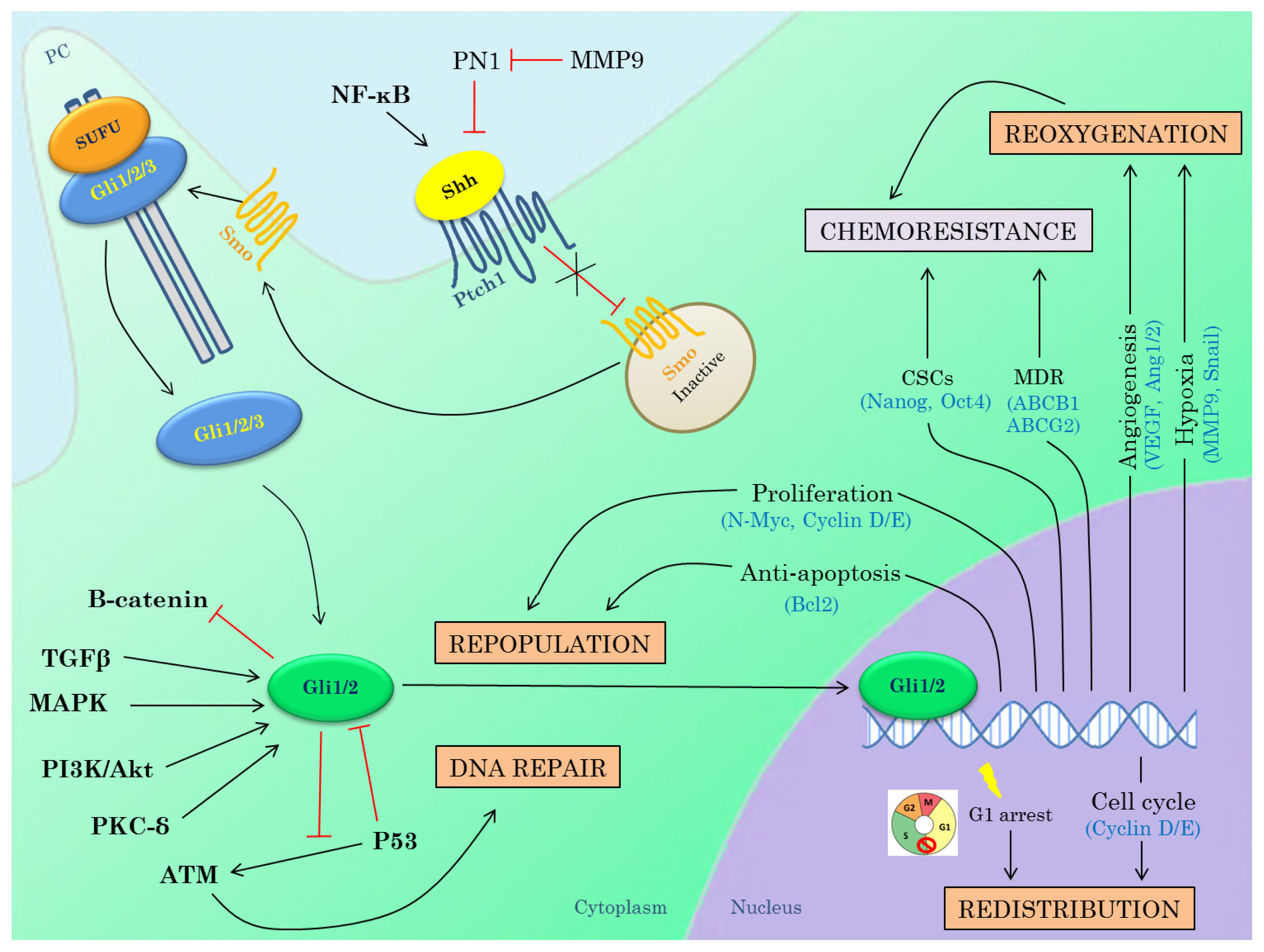
3.3. Hedgehog Inhibitors in Combination with Radiotherapy
3.3.1. DNA Repair
3.3.2. Repopulation
3.3.3. Redistribution
3.3.4. Reoxygenation
3.3.5. Interactions between Hh Pathway and Genes Known to Induce Radioresistance
3.4. Hedgehog Inhibitors in Combination with Chemotherapy
3.5. Hedgehog Inhibitors in Combination with Other Molecular Targeted Agents (MTAs)
3.5.1. PI3K Inhibitors
3.5.2. EGFR Inhibitors
3.5.3. Androgen Deprivation Therapy
3.5.4. Others
4. Conclusions and Future Perspectives
Acknowledgments
Conflict of Interest
References
- Karhadkar, S.S.; Bova, G.S.; Abdallah, N.; Dhara, S.; Gardner, D.; Maitra, A.; Isaacs, J.T.; Berman, D.M.; Beachy, P.A. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature 2004, 431, 707–712. [Google Scholar]
- Teglund, S.; Toftgard, R. Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim. Biophys. Acta 2010, 1805, 181–208. [Google Scholar]
- Beachy, P.A.; Karhadkar, S.S.; Berman, D.M. Tissue repair and stem cell renewal in carcinogenesis. Nature 2004, 432, 324–331. [Google Scholar]
- Lai, K.; Kaspar, B.K.; Gage, F.H.; Schaffer, D.V. Sonic hedgehog regulates adult neural progenitor proliferation in vitro and in vivo. Nat. Neurosci 2003, 6, 21–27. [Google Scholar]
- Pasca di, M.M.; Hebrok, M. Hedgehog signalling in cancer formation and maintenance. Nat. Rev. Cancer 2003, 3, 903–911. [Google Scholar]
- Jiang, J.; Hui, C.C. Hedgehog signaling in development and cancer. Dev. Cell 2008, 15, 801–812. [Google Scholar]
- Yang, L.; Xie, G.; Fan, Q.; Xie, J. Activation of the hedgehog-signaling pathway in human cancer and the clinical implications. Oncogene 2010, 29, 469–481. [Google Scholar]
- Chuang, P.T.; McMahon, A.P. Vertebrate Hedgehog signalling modulated by induction of a Hedgehog-binding protein. Nature 1999, 397, 617–621. [Google Scholar]
- Zhang, W.; Kang, J.S.; Cole, F.; Yi, M.J.; Krauss, R.S. Cdo functions at multiple points in the Sonic Hedgehog pathway, and Cdo-deficient mice accurately model human holoprosencephaly. Dev. Cell 2006, 10, 657–665. [Google Scholar]
- Tenzen, T.; Allen, B.L.; Cole, F.; Kang, J.S.; Krauss, R.S.; McMahon, A.P. The cell surface membrane proteins Cdo and Boc are components and targets of the Hedgehog signaling pathway and feedback network in mice. Dev. Cell 2006, 10, 647–656. [Google Scholar]
- Carpenter, D.; Stone, D.M.; Brush, J.; Ryan, A.; Armanini, M.; Frantz, G.; Rosenthal, A.; de Sauvage, F.J. Characterization of two patched receptors for the vertebrate hedgehog protein family. Proc. Natl. Acad. Sci. USA 1998, 95, 13630–13634. [Google Scholar]
- Stone, D.M.; Hynes, M.; Armanini, M.; Swanson, T.A.; Gu, Q.; Johnson, R.L.; Scott, M.P.; Pennica, D.; Goddard, A.; Phillips, H.; et al. The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature 1996, 384, 129–134. [Google Scholar]
- Corbit, K.C.; Aanstad, P.; Singla, V.; Norman, A.R.; Stainier, D.Y.; Reiter, J.F. Vertebrate Smoothened functions at the primary cilium. Nature 2005, 437, 1018–1021. [Google Scholar]
- Singla, V.; Reiter, J.F. The primary cilium as the cell’s antenna: Signaling at a sensory organelle. Science 2006, 313, 629–633. [Google Scholar]
- Haycraft, C.J.; Banizs, B.; Aydin-Son, Y.; Zhang, Q.; Michaud, E.J.; Yoder, B.K. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet 2005, 1, e53. [Google Scholar]
- Rohatgi, R.; Milenkovic, L.; Scott, M.P. Patched1 regulates hedgehog signaling at the primary cilium. Science 2007, 317, 372–376. [Google Scholar]
- Riobo, N.A.; Manning, D.R. Pathways of signal transduction employed by vertebrate Hedgehogs. Biochem. J 2007, 403, 369–379. [Google Scholar]
- Rohatgi, R.; Milenkovic, L.; Corcoran, R.B.; Scott, M.P. Hedgehog signal transduction by Smoothened: Pharmacologic evidence for a 2-step activation process. Proc. Natl. Acad. Sci. USA 2009, 106, 3196–3201. [Google Scholar]
- Sasaki, H.; Nishizaki, Y.; Hui, C.; Nakafuku, M.; Kondoh, H. Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: Implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development 1999, 126, 3915–3924. [Google Scholar]
- Dai, P.; Akimaru, H.; Tanaka, Y.; Maekawa, T.; Nakafuku, M.; Ishii, S. Sonic Hedgehog-induced activation of the Gli1 promoter is mediated by GLI3. J. Biol. Chem 1999, 274, 8143–8152. [Google Scholar]
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev 2001, 15, 3059–3087. [Google Scholar]
- Kogerman, P.; Grimm, T.; Kogerman, L.; Krause, D.; Unden, A.B.; Sandstedt, B.; Toftgard, R.; Zaphiropoulos, P.G. Mammalian suppressor-of-fused modulates nuclear-cytoplasmic shuttling of Gli-1. Nat. Cell Biol 1999, 1, 312–319. [Google Scholar]
- Yue, S.; Chen, Y.; Cheng, S.Y. Hedgehog signaling promotes the degradation of tumor suppressor Sufu through the ubiquitin-proteasome pathway. Oncogene 2009, 28, 492–499. [Google Scholar]
- Stecca, B.; Ruiz, I.A. Context-dependent regulation of the GLI code in cancer by HEDGEHOG and non-HEDGEHOG signals. J. Mol. Cell Biol 2010, 2, 84–95. [Google Scholar]
- Lauth, M.; Toftgard, R. Non-canonical activation of GLI transcription factors: Implications for targeted anti-cancer therapy. Cell Cycle 2007, 6, 2458–2463. [Google Scholar]
- Onishi, H.; Katano, M. Hedgehog signaling pathway as a therapeutic target in various types of cancer. Cancer Sci 2011, 102, 1756–1760. [Google Scholar]
- Hahn, H.; Wicking, C.; Zaphiropoulous, P.G.; Gailani, M.R.; Shanley, S.; Chidambaram, A.; Vorechovsky, I.; Holmberg, E.; Unden, A.B.; Gillies, S.; et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996, 85, 841–851. [Google Scholar]
- Johnson, R.L.; Rothman, A.L.; Xie, J.; Goodrich, L.V.; Bare, J.W.; Bonifas, J.M.; et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 1996, 272, 1668–1671. [Google Scholar]
- Szkandera, J.; Kiesslich, T.; Haybaeck, J.; Gerger, A.; Pichler, M. Hedgehog signaling pathway in ovarian cancer. Int. J. Mol. Sci 2013, 14, 1179–1196. [Google Scholar]
- Rubin, L.L.; de Sauvage, F.J. Targeting the Hedgehog pathway in cancer. Nat. Rev. Drug Discov 2006, 5, 1026–1033. [Google Scholar]
- Scales, S.J.; de Sauvage, F.J. Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol. Sci 2009, 30, 303–312. [Google Scholar]
- Yauch, R.L.; Gould, S.E.; Scales, S.J.; Tang, T.; Tian, H.; Ahn, C.P.; Marshall, D.; Fu, L.; Januario, T.; Kallop, D.; et al. A paracrine requirement for hedgehog signalling in cancer. Nature 2008, 455, 406–410. [Google Scholar]
- Tian, H.; Callahan, C.A.; DuPree, K.J.; Darbonne, W.C.; Ahn, C.P.; Scales, S.J.; de Sauvage, F.J. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 4254–4259. [Google Scholar]
- Fan, L.; Pepicelli, C.V.; Dibble, C.C.; Catbagan, W.; Zarycki, J.L.; Laciak, R.; Laciak, R.; Gipp, J.; Shaw, A.; Lamm, M.L.G.; et al. Hedgehog signaling promotes prostate xenograft tumor growth. Endocrinology 2004, 145, 3961–3970. [Google Scholar]
- Dierks, C.; Grbic, J.; Zirlik, K.; Beigi, R.; Englund, N.P.; Guo, G.R.; Veelken, H.; Engelhardt, M.; Mertelsmann, R.; Kelleher, J.F.; et al. Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nat. Med 2007, 13, 944–951. [Google Scholar]
- Hegde, G.V.; Peterson, K.J.; Emanuel, K.; Mittal, A.K.; Joshi, A.D.; Dickinson, J.D.; Kollessery, G.J.; Bociek, R.G.; Bierman, P.; Vose, J.M.; et al. Hedgehog-induced survival of B-cell chronic lymphocytic leukemia cells in a stromal cell microenvironment: a potential new therapeutic target. Mol. Cancer Res 2008, 6, 1928–1936. [Google Scholar]
- Huangfu, D.; Anderson, K.V. Cilia and Hedgehog responsiveness in the mouse. Proc. Natl. Acad. Sci. USA 2005, 102, 11325–11330. [Google Scholar]
- Han, Y.G.; Kim, H.J.; Dlugosz, A.A.; Ellison, D.W.; Gilbertson, R.J.; Alvarez-Buylla, A. Dual and opposing roles of primary cilia in medulloblastoma development. Nat. Med 2009, 15, 1062–1065. [Google Scholar]
- Wong, S.Y.; Seol, A.D.; So, P.L.; Ermilov, A.N.; Bichakjian, C.K.; Epstein, E.H., Jr; Dlugosz, A.A.; Reiter, J.F. Primary cilia can both mediate and suppress Hedgehog pathway-dependent tumorigenesis. Nat. Med. 2009, 15, 1055–1061. [Google Scholar]
- Hassounah, N.B.; Bunch, T.A.; McDermott, K.M. Molecular pathways: The role of primary cilia in cancer progression and therapeutics with a focus on Hedgehog signaling. Clin. Cancer Res 2012, 18, 2429–2435. [Google Scholar]
- Podlasek, C.A.; Barnett, D.H.; Clemens, J.Q.; Bak, P.M.; Bushman, W. Prostate development requires Sonic hedgehog expressed by the urogenital sinus epithelium. Dev. Biol 1999, 209, 28–39. [Google Scholar]
- Lamm, M.L.; Catbagan, W.S.; Laciak, R.J.; Barnett, D.H.; Hebner, C.M.; Gaffield, W.; Walterhouse, D.; Iannaccone, P.; Bushman, W. Sonic hedgehog activates mesenchymal Gli1 expression during prostate ductal bud formation. Dev. Biol 2002, 249, 349–366. [Google Scholar]
- Freestone, S.H.; Marker, P.; Grace, O.C.; Tomlinson, D.C.; Cunha, G.R.; Harnden, P.; Thomson, A.A. Sonic hedgehog regulates prostatic growth and epithelial differentiation. Dev. Biol 2003, 264, 352–362. [Google Scholar]
- Berman, D.M.; Desai, N.; Wang, X.; Karhadkar, S.S.; Reynon, M.; Abate-Shen, C.; Beachy, P.A.; Shen, M.M. Roles for Hedgehog signaling in androgen production and prostate ductal morphogenesis. Dev. Biol 2004, 267, 387–398. [Google Scholar]
- Sheng, T.; Li, C.; Zhang, X.; Chi, S.; He, N.; Chen, K.; McCormick, F.; Gatalica, Z.; Xie, J. Activation of the hedgehog pathway in advanced prostate cancer. Mol. Cancer 2004, 3, 29. [Google Scholar]
- Datta, S.; Datta, M.W. Sonic Hedgehog signaling in advanced prostate cancer. Cell Mol. Life Sci 2006, 63, 435–448. [Google Scholar]
- Sanchez, P.; Hernandez, A.M.; Stecca, B.; Kahler, A.J.; DeGueme, A.M.; Barrett, A.; Beyna, M.; Datta, M.W.; Datta, S.; Altaba, A. Inhibition of prostate cancer proliferation by interference with SONIC HEDGEHOG-GLI1 signaling. Proc. Natl. Acad. Sci. USA 2004, 101, 12561–12566. [Google Scholar]
- Tzelepi, V.; Karlou, M.; Wen, S.; Hoang, A.; Logothetis, C.; Troncoso, P.; Efstathiou, E. Expression of hedgehog pathway components in prostate carcinoma microenvironment: shifting the balance towards autocrine signalling. Histopathology 2011, 58, 1037–1047. [Google Scholar]
- Azoulay, S.; Terry, S.; Chimingqi, M.; Sirab, N.; Faucon, H.; Gil Diez de, M.S.; Moutereau, S.; Maille, P.; Soyeux, P.; Abbou, C.; et al. Comparative expression of Hedgehog ligands at different stages of prostate carcinoma progression. J. Pathol 2008, 216, 460–470. [Google Scholar]
- Kim, T.J.; Lee, J.Y.; Hwang, T.K.; Kang, C.S.; Choi, Y.J. Hedgehog signaling protein expression and its association with prognostic parameters in prostate cancer: A retrospective study from the view point of new 2010 anatomic stage/prognostic groups. J. Surg. Oncol 2011, 104, 472–479. [Google Scholar]
- Chen, M.; Carkner, R.; Buttyan, R. The hedgehog/Gli signaling paradigm in prostate cancer. Expert. Rev. Endocrinol. Metab 2011, 6, 453–467. [Google Scholar]
- Gipp, J.; Gu, G.; Crylen, C.; Kasper, S.; Bushman, W. Hedgehog pathway activity in the LADY prostate tumor model. Mol. Cancer 2007, 6, 19. [Google Scholar]
- Bragina, O.; Njunkova, N.; Sergejeva, S.; Jarvekulg, L.; Kogerman, P. Sonic Hedgehog pathway activity in prostate cancer. Oncol. Lett 2010, 1, 319–325. [Google Scholar]
- Efstathiou, E.; Karlou, M.; Wen, S.; Hoang, A.; Pettaway, C.A.; Pisters, L.L.; Maity, S.; Troncoso, P.; Logothetis, C.J. Integrated Hedgehog signaling is induced following castration in human and murine prostate cancers. Prostate 2013, 73, 153–161. [Google Scholar]
- Ibuki, N.; Ghaffari, M.; Pandey, M.; Iu, I.; Fazli, L.; Kashiwagi, M.; Tojo, H.; Nakanishi, O.; Gleave, M.E.; Cox, M.E. TAK-441, a novel investigational smoothened antagonist, delays castration-resistant progression in prostate cancer by disrupting paracrine hedgehog signaling. Int. J. Cancer 2013. [Google Scholar] [CrossRef]
- Shaw, G.; Price, A.M.; Ktori, E.; Bisson, I.; Purkis, P.E.; McFaul, S.; Oliver, R.T.; Prowse, D.M. Hedgehog signalling in androgen independent prostate cancer. Eur. Urol 2008, 54, 1333–1343. [Google Scholar]
- Chen, M.; Tanner, M.; Levine, A.C.; Levina, E.; Ohouo, P.; Buttyan, R. Androgenic regulation of hedgehog signaling pathway components in prostate cancer cells. Cell Cycle 2009, 8, 149–157. [Google Scholar]
- Chen, M.; Feuerstein, M.A.; Levina, E.; Baghel, P.S.; Carkner, R.D.; Tanner, M.J.; Shtutman, M.; Vacherot, F.; Terry, S.; de la Taille, A.; et al. Hedgehog/Gli supports androgen signaling in androgen deprived and androgen independent prostate cancer cells. Mol. Cancer 2010, 9, 89. [Google Scholar]
- Chen, G.; Goto, Y.; Sakamoto, R.; Tanaka, K.; Matsubara, E.; Nakamura, M.; Zheng, H.; Lu, J.; Takayanagi, R.; Nomura, M. GLI1, a crucial mediator of sonic hedgehog signaling in prostate cancer, functions as a negative modulator for androgen receptor. Biochem. Biophys. Res. Commun 2011, 404, 809–815. [Google Scholar]
- Shaw, G.; Prowse, D.M. Inhibition of androgen-independent prostate cancer cell growth is enhanced by combination therapy targeting Hedgehog and ErbB signalling. Cancer Cell Int 2008, 8, 3. [Google Scholar]
- Thiyagarajan, S.; Bhatia, N.; Reagan-Shaw, S.; Cozma, D.; Thomas-Tikhonenko, A.; Ahmad, N.; Spiegelman, V.S. Role of GLI2 transcription factor in growth and tumorigenicity of prostate cells. Cancer Res 2007, 67, 10642–10646. [Google Scholar]
- Chung, M.K.; Kim, H.J.; Lee, Y.S.; Han, M.E.; Yoon, S.; Baek, S.Y.; Kim, B.S.; Kim, J.B.; Oh, S.O. Hedgehog signaling regulates proliferation of prostate cancer cells via stathmin1. Clin. Exp. Med 2010, 10, 51–57. [Google Scholar]
- Bar, E.E.; Chaudhry, A.; Farah, M.H.; Eberhart, C.G. Hedgehog signaling promotes medulloblastoma survival via Bc/II. Am. J. Pathol 2007, 170, 347–355. [Google Scholar]
- Narita, S.; So, A.; Ettinger, S.; Hayashi, N.; Muramaki, M.; Fazli, L.; Kim, Y.; Gleave, M.E. GLI2 knockdown using an antisense oligonucleotide induces apoptosis and chemosensitizes cells to paclitaxel in androgen-independent prostate cancer. Clin. Cancer Res 2008, 14, 5769–5777. [Google Scholar]
- Nanta, R.; Kumar, D.; Meeker, D.; Rodova, M.; Van Veldhuizen, P.J.; Shankar, S.; Srivastava, R.K. NVP-LDE-225 (Erismodegib) inhibits epithelial-mesenchymal transition and human prostate cancer stem cell growth in NOD/SCID IL2Rgamma null mice by regulating Bmi-1 and microRNA-128. Oncogenesis 2013, 2, e42. [Google Scholar]
- McKee, C.M.; Xu, D.; Cao, Y.; Kabraji, S.; Allen, D.; Kersemans, V.; Beech, J.; Smart, S.; Hamdy, F.; Ishkanian, A.; et al. Protease nexin 1 inhibits hedgehog signaling in prostate adenocarcinoma. J. Clin. Invest 2012, 122, 4025–4036. [Google Scholar]
- Ferlay, J.; Parkin, D.M.; Steliarova-Foucher, E. Estimates of cancer incidence and mortality in Europe in 2008. Eur. J. Cancer 2010, 46, 765–781. [Google Scholar]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin 2011, 61, 69–90. [Google Scholar]
- Spahn, M.; Joniau, S.; Gontero, P.; Fieuws, S.; Marchioro, G.; Tombal, B.; Kneitz, B.; Hsu, C.Y.; van der Eeckt, K.; Bader, P.; et al. Outcome predictors of radical prostatectomy in patients with prostate-specific antigen greater than 20 ng/mL: A European multi-institutional study of 712 patients. Eur. Urol 2010, 58, 1–7. [Google Scholar]
- Cooperberg, M.R.; Lubeck, D.P.; Mehta, S.S.; Carroll, P.R. Time trends in clinical risk stratification for prostate cancer: Implications for outcomes (data from CaPSURE). J. Urol 2003, 170, S21–S25. [Google Scholar]
- Mas, C.; Altaba, A. Small molecule modulation of HH-GLI signaling: Current leads, trials and tribulations. Biochem. Pharmacol 2010, 80, 712–723. [Google Scholar]
- Stanton, B.Z.; Peng, L.F. Small-molecule modulators of the Sonic Hedgehog signaling pathway. Mol. Biosyst 2010, 6, 44–54. [Google Scholar]
- Karlou, M.; Lu, J.F.; Wu, G.; Maity, S.; Tzelepi, V.; Navone, N.M.; Hoang, A.; Logothetis, C.J.; Efstathiou, E. Hedgehog signaling inhibition by the small molecule smoothened inhibitor GDC-0449 in the bone forming prostate cancer xenograft MDA PCa 118b. Prostate 2012, 72, 1638–1647. [Google Scholar]
- LoRusso, P.M.; Rudin, C.M.; Reddy, J.C.; Tibes, R.; Weiss, G.J.; Borad, M.J.; Hann, C.L.; Brahmer, J.R.; Chang, I.; Darbonne, W.C.; et al. Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clin. Cancer Res 2011, 17, 2502–2511. [Google Scholar]
- Tang, J.Y.; Mackay-Wiggan, J.M.; Aszterbaum, M.; Yauch, R.L.; Lindgren, J.; Chang, K.; Coppola, C.; Chanana, A.M.; Marji, J.; Bickers, D.R.; et al. Inhibiting the hedgehog pathway in patients with the basal-cell nevus syndrome. N. Engl. J. Med 2012, 366, 2180–2188. [Google Scholar]
- Sekulic, A.; Migden, M.R.; Oro, A.E.; Dirix, L.; Lewis, K.D.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N. Engl. J. Med 2012, 366, 2171–2179. [Google Scholar]
- NCI Clinical Trail Database. Available online: http://www.clinicaltrials.gov (on accessed 13 March 2013).
- Rudin, C.M. Vismodegib. Clin. Cancer Res 2012, 18, 3218–3222. [Google Scholar]
- Kimura, H.; Ng, J.M.; Curran, T. Transient inhibition of the Hedgehog pathway in young mice causes permanent defects in bone structure. Cancer Cell 2008, 13, 249–260. [Google Scholar]
- Rudin, C.M.; Hann, C.L.; Laterra, J.; Yauch, R.L.; Callahan, C.A.; Fu, L.; Holcomb, T.; Stinson, J.; Gould, S.E.; Coleman, B.; et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N. Engl. J. Med 2009, 361, 1173–1178. [Google Scholar]
- Atwood, S.X.; Chang, A.L.; Oro, A.E. Hedgehog pathway inhibition and the race against tumor evolution. J. Cell Biol 2012, 199, 193–197. [Google Scholar]
- Buonamici, S.; Williams, J.; Morrissey, M.; Wang, A.; Guo, R.; Vattay, A.; Hsiao, K.; Yuan, J.; Green, J.; Ospina, B.; et al. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci. Transl. Med 2010, 2, 51r, a70.. [Google Scholar]
- Yauch, R.L.; Dijkgraaf, G.J.; Alicke, B.; Januario, T.; Ahn, C.P.; Holcomb, T.; Pujara, K.; Stinson, J.; Callahan, C.A.; Tang, T.; et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 2009, 326, 572–574. [Google Scholar]
- Coni, S.; Infante, P.; Gulino, A. Control of stem cells and cancer stem cells by Hedgehog signaling: Pharmacologic clues from pathway dissection. Biochem. Pharmacol 2013, 85, 623–628. [Google Scholar]
- Kim, J.; Aftab, B.T.; Tang, J.Y.; Kim, D.; Lee, A.H.; Rezaee, M.; Kim, J.; Chen, B.; King, E.M.; Borodovsky, A.; et al. Itraconazole and arsenic trioxide inhibit Hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell 2013, 23, 23–34. [Google Scholar]
- Chen, Y.J.; Lin, C.P.; Hsu, M.L.; Shieh, H.R.; Chao, N.K.; Chao, K.S. Sonic hedgehog signaling protects human hepatocellular carcinoma cells against ionizing radiation in an autocrine manner. Int. J. Radiat. Oncol. Biol. Phys 2011, 80, 851–859. [Google Scholar]
- Sims-Mourtada, J.; Izzo, J.G.; Apisarnthanarax, S.; Wu, T.T.; Malhotra, U.; Luthra, R.; Liao, Z.; Komaki, R.; van der Kogel, A.; Ajani, J.; et al. Hedgehog: An attribute to tumor regrowth after chemoradiotherapy and a target to improve radiation response. Clin. Cancer Res 2006, 12, 6565–6572. [Google Scholar]
- Shafaee, Z.; Schmidt, H.; Du, W.; Posner, M.; Weichselbaum, R. Cyclopamine increases the cytotoxic effects of paclitaxel and radiation but not cisplatin and gemcitabine in Hedgehog expressing pancreatic cancer cells. Cancer Chemother. Pharmacol 2006, 58, 765–770. [Google Scholar]
- Zeng, J.; Aziz, K.; Chettiar, S.T.; Aftab, B.T.; Armour, M.; Gajula, R.; Gandhi, N.; Salih, T.; Herman, J.M.; Wong, J.; et al. Hedgehog pathway inhibition radiosensitizes non-small cell lung cancers. Int. J. Radiat. Oncol. Biol. Phys 2012, 86, 143–149. [Google Scholar]
- Yoshikawa, R.; Nakano, Y.; Tao, L.; Koishi, K.; Matsumoto, T.; Sasako, M.; Tsujimura, T.; Hashimoto-Tamaoki, T.; Fujiwara, Y. Hedgehog signal activation in oesophageal cancer patients undergoing neoadjuvant chemoradiotherapy. Br. J. Cancer 2008, 98, 1670–1674. [Google Scholar]
- Zhu, W.; You, Z.; Li, T.; Yu, C.; Tao, G.; Hu, M.; Chen, X. Correlation of hedgehog signal activation with chemoradiotherapy sensitivity and survival in esophageal squamous cell carcinomas. Jpn. J. Clin. Oncol 2011, 41, 386–393. [Google Scholar]
- Chaudary, N.; Pintilie, M.; Hedley, D.; Fyles, A.W.; Milosevic, M.; Clarke, B.; Hill, R.P.; Mackay, H. Hedgehog pathway signaling in cervical carcinoma and outcome after chemoradiation. Cancer 2012, 118, 3105–3115. [Google Scholar]
- Leonard, J.M.; Ye, H.; Wetmore, C.; Karnitz, L.M. Sonic Hedgehog signaling impairs ionizing radiation-induced checkpoint activation and induces genomic instability. J. Cell Biol 2008, 183, 385–391. [Google Scholar]
- Frappart, P.O.; Lee, Y.; Russell, H.R.; Chalhoub, N.; Wang, Y.D.; Orii, K.E.; Zhao, J.; Kondo, N.; Baker, S.J.; McKinnon, P.J. Recurrent genomic alterations characterize medulloblastoma arising from DNA double-strand break repair deficiency. Proc. Natl. Acad. Sci. USA 2009, 106, 1880–1885. [Google Scholar]
- Mazumdar, T.; Devecchio, J.; Agyeman, A.; Shi, T.; Houghton, J.A. Blocking Hedgehog survival signaling at the level of the GLI genes induces DNA damage and extensive cell death in human colon carcinoma cells. Cancer Res 2011, 71, 5904–5914. [Google Scholar]
- Pawlik, T.M.; Keyomarsi, K. Role of cell cycle in mediating sensitivity to radiotherapy. Int. J. Radiat. Oncol. Biol. Phys 2004, 59, 928–942. [Google Scholar]
- Onishi, H.; Kai, M.; Odate, S.; Iwasaki, H.; Morifuji, Y.; Ogino, T.; Morisaki, T.; Nakashima, Y.; Katano, M. Hypoxia activates the hedgehog signaling pathway in a ligand-independent manner by upregulation of Smo transcription in pancreatic cancer. Cancer Sci 2011, 102, 1144–1150. [Google Scholar]
- Wang, G.; Zhang, Z.; Xu, Z.; Yin, H.; Bai, L.; Ma, Z.; Decoster, M.A.; Qian, G.; Wu, G. Activation of the sonic hedgehog signaling controls human pulmonary arterial smooth muscle cell proliferation in response to hypoxia. Biochim. Biophys. Acta 2010, 1803, 1359–1367. [Google Scholar]
- Bijlsma, M.F.; Groot, A.P.; Oduro, J.P.; Franken, R.J.; Schoenmakers, S.H.; Peppelenbosch, M.P.; Spek, C.A. Hypoxia induces a hedgehog response mediated by HIF-1alpha. J. Cell Mol. Med 2009, 13, 2053–2060. [Google Scholar]
- Nakamura, K.; Sasajima, J.; Mizukami, Y.; Sugiyama, Y.; Yamazaki, M.; Fujii, R.; Kawamoto, T.; Koizumi, K.; Sato, K.; Fujiya, M.; et al. Hedgehog promotes neovascularization in pancreatic cancers by regulating Ang-1 and IGF-1 expression in bone-marrow derived pro-angiogenic cells. PLoS One 2010, 5, e8824. [Google Scholar]
- Pola, R.; Ling, L.E.; Silver, M.; Corbley, M.J.; Kearney, M.; Blake, P.R.; Shapiro, R.; Taylor, F.R.; Baker, D.P.; Asahara, T.; et al. The morphogen Sonic hedgehog is an indirect angiogenic agent upregulating two families of angiogenic growth factors. Nat. Med 2001, 7, 706–711. [Google Scholar]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar]
- Bahra, M.; Kamphues, C.; Boas-Knoop, S.; Lippert, S.; Esendik, U.; Schuller, U.; Hartmann, W.; Waha, A.; Neuhaus, P.; Heppner, F.; et al. Combination of hedgehog signaling blockage and chemotherapy leads to tumor reduction in pancreatic adenocarcinomas. Pancreas 2012, 41, 222–229. [Google Scholar]
- McKee, C.M.; Xu, D.; Muschel, R.J. Protease nexin 1: A novel regulator of prostate cancer cell growth and neo-angiogenesis. Oncotarget 2013, 4, 1–2. [Google Scholar]
- McKenna, W.G.; Muschel, R.J.; Gupta, A.K.; Hahn, S.M.; Bernhard, E.J. The RAS signal transduction pathway and its role in radiation sensitivity. Oncogene 2003, 22, 5866–5875. [Google Scholar]
- Bussink, J.; van der Kogel, A.J.; Kaanders, J.H. Activation of the PI3-K/AKT pathway and implications for radioresistance mechanisms in head and neck cancer. Lancet Oncol 2008, 9, 288–296. [Google Scholar]
- Meyn, R.E.; Munshi, A.; Haymach, J.V.; Milas, L.; Ang, K.K. Receptor signaling as a regulatory mechanism of DNA repair. Radiother. Oncol 2009, 92, 316–322. [Google Scholar]
- Debucquoy, A.; Machiels, J.P.; McBride, W.H.; Haustermans, K. Integration of epidermal growth factor receptor inhibitors with preoperative chemoradiation. Clin. Cancer Res 2010, 16, 2709–2714. [Google Scholar]
- Stecca, B.; Mas, C.; Clement, V.; Zbinden, M.; Correa, R.; Piguet, V.; Beermann, F.; Ruizi Altaba, A. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 5895–5900. [Google Scholar]
- Riobo, N.A.; Lu, K.; Ai, X.; Haines, G.M.; Emerson, C.P., Jr. Phosphoinositide 3-kinase and Akt are essential for Sonic Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 4505–4510. [Google Scholar]
- Riobo, N.A.; Haines, G.M.; Emerson, C.P., Jr. Protein kinase C-delta and mitogen-activated protein/extracellular signal-regulated kinase-1 control GLI activation in hedgehog signaling. Cancer Res. 2006, 66, 839–845. [Google Scholar]
- Ji, Z.; Mei, F.C.; Xie, J.; Cheng, X. Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. J. Biol. Chem 2007, 282, 14048–14055. [Google Scholar]
- Sigal, A.; Rotter, V. Oncogenic mutations of the p53 tumor suppressor: The demons of the guardian of the genome. Cancer Res 2000, 60, 6788–6793. [Google Scholar]
- Navone, N.M.; Troncoso, P.; Pisters, L.L.; Goodrow, T.L.; Palmer, J.L.; Nichols, W.W.; von Eschenbach, A.C.; Conti, C.J. p53 protein accumulation and gene mutation in the progression of human prostate carcinoma. J. Natl. Cancer Inst 1993, 85, 1657–1669. [Google Scholar]
- Osman, I.; Drobnjak, M.; Fazzari, M.; Ferrara, J.; Scher, H.I.; Cordon-Cardo, C. Inactivation of the p53 pathway in prostate cancer: Impact on tumor progression. Clin. Cancer Res 1999, 5, 2082–2088. [Google Scholar]
- Lu, C.; El-Deiry, W.S. Targeting p53 for enhanced radio- and chemo-sensitivity. Apoptosis 2009, 14, 597–606. [Google Scholar]
- Lehmann, B.D.; McCubrey, J.A.; Terrian, D.M. Radiosensitization of prostate cancer by priming the wild-type p53-dependent cellular senescence pathway. Cancer Biol. Ther 2007, 6, 1165–1170. [Google Scholar]
- Abe, Y.; Oda-Sato, E.; Tobiume, K.; Kawauchi, K.; Taya, Y.; Okamoto, K.; Oren, M.; Tanaka, N. Hedgehog signaling overrides p53-mediated tumor suppression by activating Mdm2. Proc. Natl. Acad. Sci. USA 2008, 105, 4838–4843. [Google Scholar]
- Mahon, K.L.; Henshall, S.M.; Sutherland, R.L.; Horvath, L.G. Pathways of chemotherapy resistance in castration-resistant prostate cancer. Endocr. Relat Cancer 2011, 18, R103–R123. [Google Scholar]
- Balk, S.P. Androgen receptor as a target in androgen-independent prostate cancer. Urology 2002, 60, 132–138. [Google Scholar]
- Holzbeierlein, J.; Lal, P.; LaTulippe, E.; Smith, A.; Satagopan, J.; Zhang, L.; Ryan, C.; Smith, S.; Scher, H.; Scardino, P.; et al. Gene expression analysis of human prostate carcinoma during hormonal therapy identifies androgen-responsive genes and mechanisms of therapy resistance. Am. J. Pathol 2004, 164, 217–227. [Google Scholar]
- Mohler, J.L. Castration-recurrent prostate cancer is not androgen-independent. Adv. Exp. Med. Biol 2008, 617, 223–234. [Google Scholar]
- Schweizer, M.T.; Antonarakis, E.S. Abiraterone and other novel androgen-directed strategies for the treatment of prostate cancer: A new era of hormonal therapies is born. Ther. Adv. Urol 2012, 4, 167–178. [Google Scholar]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med 2012, 367, 1187–1197. [Google Scholar]
- Tredan, O.; Galmarini, C.M.; Patel, K.; Tannock, I.F. Drug resistance and the solid tumor microenvironment. J. Natl. Cancer Inst 2007, 99, 1441–1454. [Google Scholar]
- Minchinton, A.I.; Tannock, I.F. Drug penetration in solid tumours. Nat. Rev. Cancer 2006, 6, 583–592. [Google Scholar]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar]
- Croker, A.K.; Allan, A.L. Cancer stem cells: Implications for the progression and treatment of metastatic disease. J. Cell Mol. Med 2008, 12, 374–390. [Google Scholar]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar]
- Gilbertson, R.J.; Graham, T.A. Cancer: Resolving the stem-cell debate. Nature 2012, 488, 462–463. [Google Scholar]
- Janikova, M.; Skarda, J. Differentiation pathways in carcinogenesis and in chemo- and radioresistance. Neoplasma 2012, 59, 6–17. [Google Scholar]
- Fu, J.; Rodova, M.; Roy, S.K.; Sharma, J.; Singh, K.P.; Srivastava, R.K.; Shankar, S. GANT-61 inhibits pancreatic cancer stem cell growth in vitro and in NOD/SCID/IL2R gamma null mice xenograft. Cancer Lett 2013, 330, 22–32. [Google Scholar]
- Sims-Mourtada, J.; Izzo, J.G.; Ajani, J.; Chao, K.S. Sonic Hedgehog promotes multiple drug resistance by regulation of drug transport. Oncogene 2007, 26, 5674–5679. [Google Scholar]
- Chen, Y.; Bieber, M.M.; Teng, N.N. Hedgehog signaling regulates drug sensitivity by targeting ABC transporters ABCB1 and ABCG2 in epithelial ovarian cancer. Mol. Carcinog. 2013. [Google Scholar] [CrossRef]
- Zhang, Y.; Laterra, J.; Pomper, M.G. Hedgehog pathway inhibitor HhAntag691 is a potent inhibitor of ABCG2/BCRP and ABCB1/Pgp. Neoplasia 2009, 11, 96–101. [Google Scholar]
- Singh, S.; Chitkara, D.; Mehrazin, R.; Behrman, S.W.; Wake, R.W.; Mahato, R.I. Chemoresistance in prostate cancer cells is regulated by miRNAs and Hedgehog pathway. PLoS One 2012, 7, e40021. [Google Scholar]
- Mimeault, M.; Johansson, S.L.; Henichart, J.P.; Depreux, P.; Batra, S.K. Cytotoxic effects induced by docetaxel, gefitinib, and cyclopamine on side population and nonside population cell fractions from human invasive prostate cancer cells. Mol. Cancer Ther 2010, 9, 617–630. [Google Scholar]
- Domingo-Domenech, J.; Vidal, S.J.; Rodriguez-Bravo, V.; Castillo-Martin, M.; Quinn, S.A.; Rodriguez-Barrueco, R.; Bonal, D.M.; Charytonowicz, E.; Gladoun, N.; de la lglesia-Vicente, J.; et al. Suppression of acquired docetaxel resistance in prostate cancer through depletion of notch-and hedgehog-dependent tumor-initiating cells. Cancer Cell 2012, 22, 373–388. [Google Scholar]
- Infinity Pharmaceuticals, Infinity Reports Update from Phase 2 Study of Saridegib Plus Gemcitabine in Patients with Metastatic Pancreatic Cancer. Available online: http://phx.corporate-ir.net/phoenix.zhtml?c=121941&p=irol-newsArticle&ID=1653550&highlight= (on accessed 13 June 2013).
- Kwak, E.L.; Clark, J.W.; Chabner, B. Targeted agents: The rules of combination. Clin. Cancer Res 2007, 13, 5232–5237. [Google Scholar]
- Dancey, J.E.; Chen, H.X. Strategies for optimizing combinations of molecularly targeted anticancer agents. Nat. Rev. Drug Discov 2006, 5, 649–659. [Google Scholar]
- Bitting, R.L.; Armstrong, A.J. Targeting the PI3K/Akt/mTOR pathway in castration-resistant prostate cancer. Endocr. Relat Cancer 2013, 20, R83–R89. [Google Scholar]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar]
- Mulholland, D.J.; Kobayashi, N.; Ruscetti, M.; Zhi, A.; Tran, L.M.; Huang, J.; Gleave, M.; Wu, H. Pten loss and RAS/MAPK activation cooperate to promote EMT and metastasis initiated from prostate cancer stem/progenitor cells. Cancer Res 2012, 72, 1878–1889. [Google Scholar]
- Gioeli, D.; Mandell, J.W.; Petroni, G.R.; Frierson, H.F., Jr; Weber, M.J. Activation of mitogen-activated protein kinase associated with prostate cancer progression. Cancer Res. 1999, 59, 279–284. [Google Scholar]
- Wang, Y.; Ding, Q.; Yen, C.J.; Xia, W.; Izzo, J.G.; Lang, J.Y.; Li, C.W.; Hsu, J.L.; Miller, S.A.; Wang, X.; et al. The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell 2012, 21, 374–387. [Google Scholar]
- Mimeault, M.; Batra, S.K. Recent advances on multiple tumorigenic cascades involved in prostatic cancer progression and targeting therapies. Carcinogenesis 2006, 27, 1–22. [Google Scholar]
- Shi, Y.; Brands, F.H.; Chatterjee, S.; Feng, A.C.; Groshen, S.; Schewe, J.; Lieskovsky, G.; Cote, R.J. Her-2/neu expression in prostate cancer: High level of expression associated with exposure to hormone therapy and androgen independent disease. J. Urol 2001, 166, 1514–1519. [Google Scholar]
- Shah, R.B.; Ghosh, D.; Elder, J.T. Epidermal growth factor receptor (ErbB1) expression in prostate cancer progression: Correlation with androgen independence. Prostate 2006, 66, 1437–1444. [Google Scholar]
- Canil, C.M.; Moore, M.J.; Winquist, E.; Baetz, T.; Pollak, M.; Chi, K.N.; Berry, C.S.; Ernst, D.S.; Douglas, L.; Brundage, M.; et al. Randomized phase II study of two doses of gefitinib in hormone-refractory prostate cancer: A trial of the National Cancer Institute of Canada-Clinical Trials Group. J. Clin. Oncol 2005, 23, 455–460. [Google Scholar]
- Mimeault, M.; Johansson, S.L.; Vankatraman, G.; Moore, E.; Henichart, J.P.; Depreux, P.; Lin, M.F.; Batra, S.K. Combined targeting of epidermal growth factor receptor and hedgehog signaling by gefitinib and cyclopamine cooperatively improves the cytotoxic effects of docetaxel on metastatic prostate cancer cells. Mol. Cancer Ther 2007, 6, 967–978. [Google Scholar]
- Leuprolide Acetate or Goserelin With or Without GDC-0449 Followed by Surgery in Treating Patients With Locally Advanced Prostate Cancer. NCI Clinical Trial Database NCT01163084. Available online: http://www.clinicaltrials.gov/ct2/show/NCT01163084?term=Hedgehog&rank=13 (on accessed 13 March 2013).
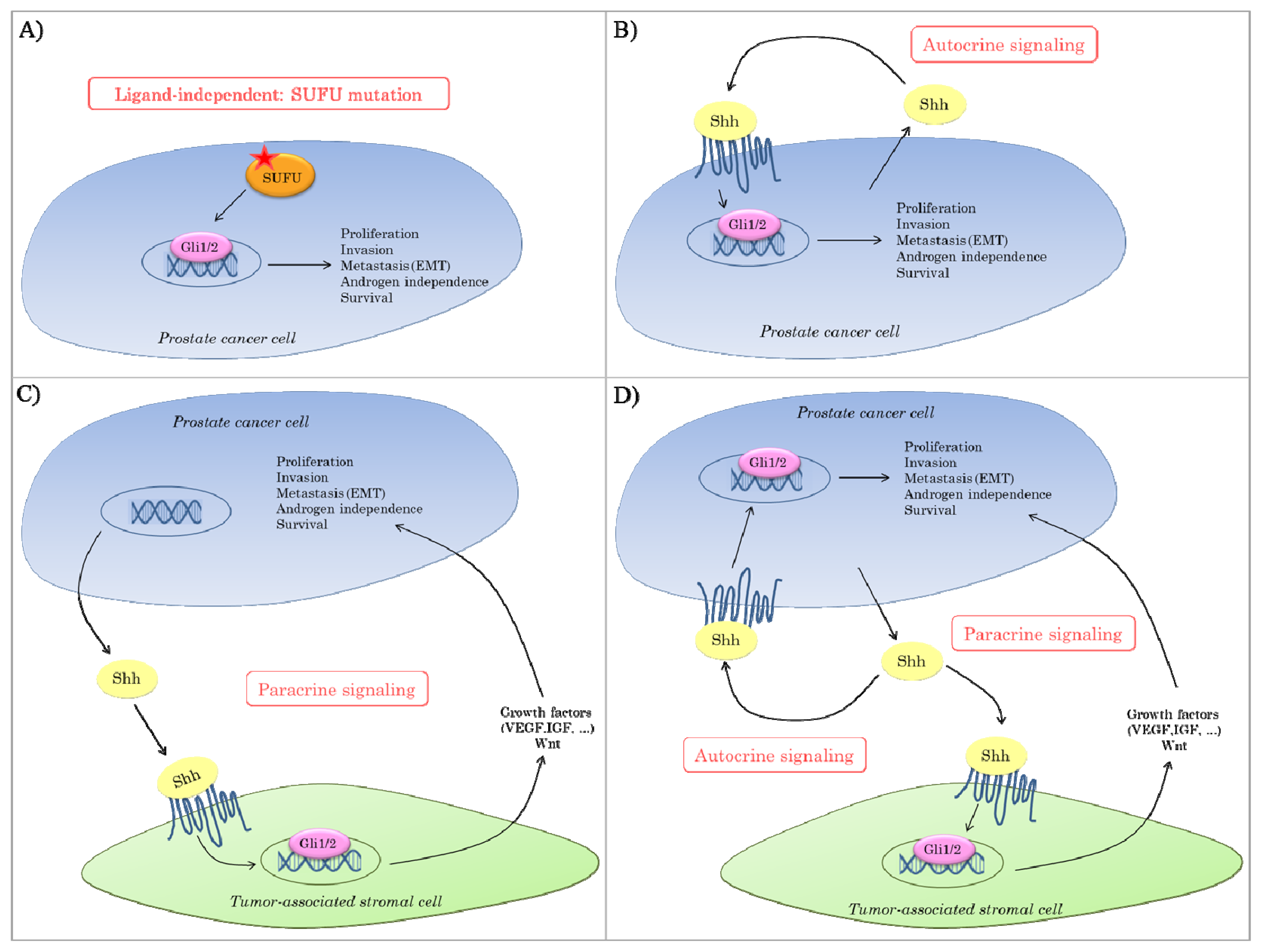

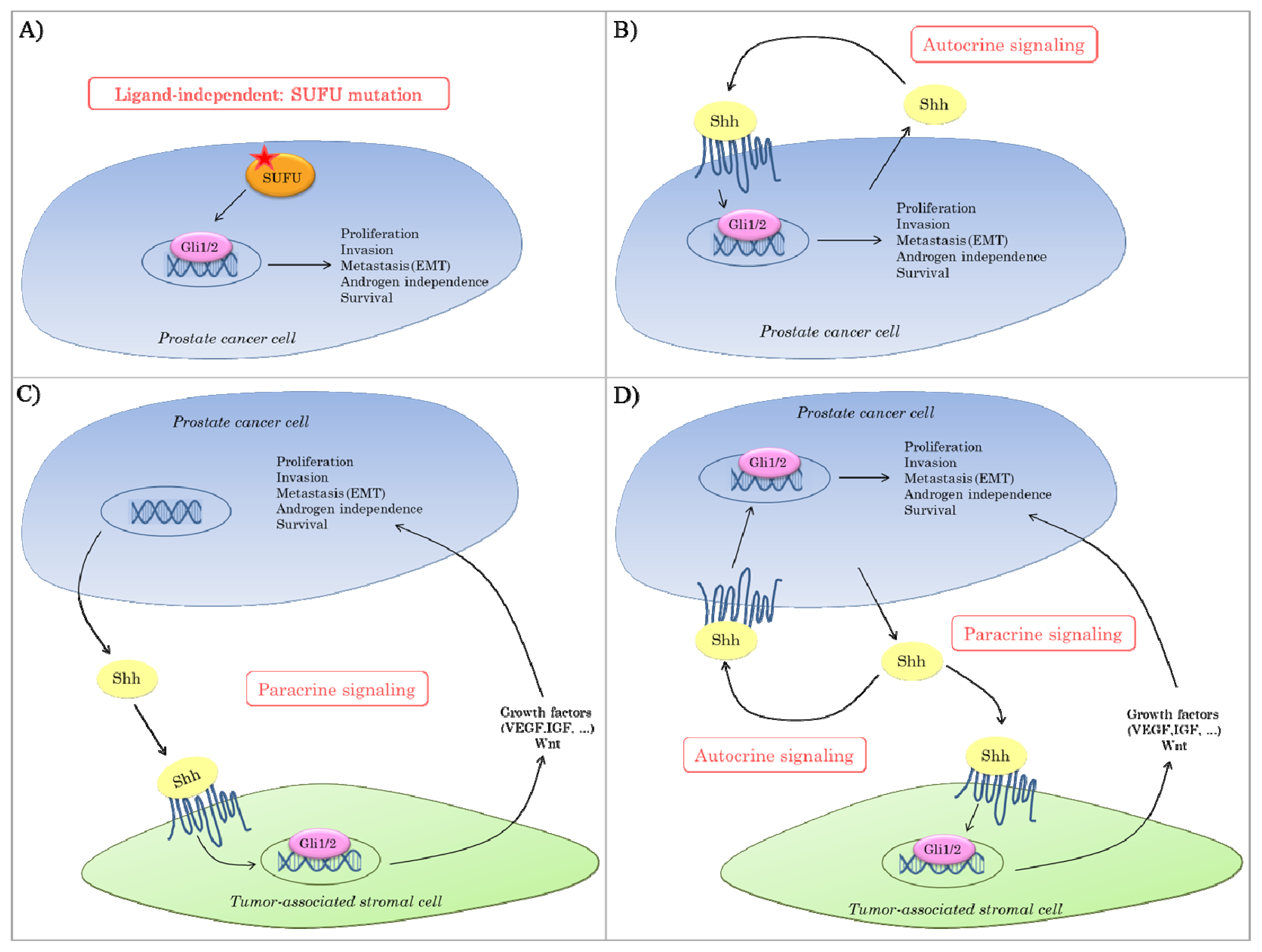{kind=link}
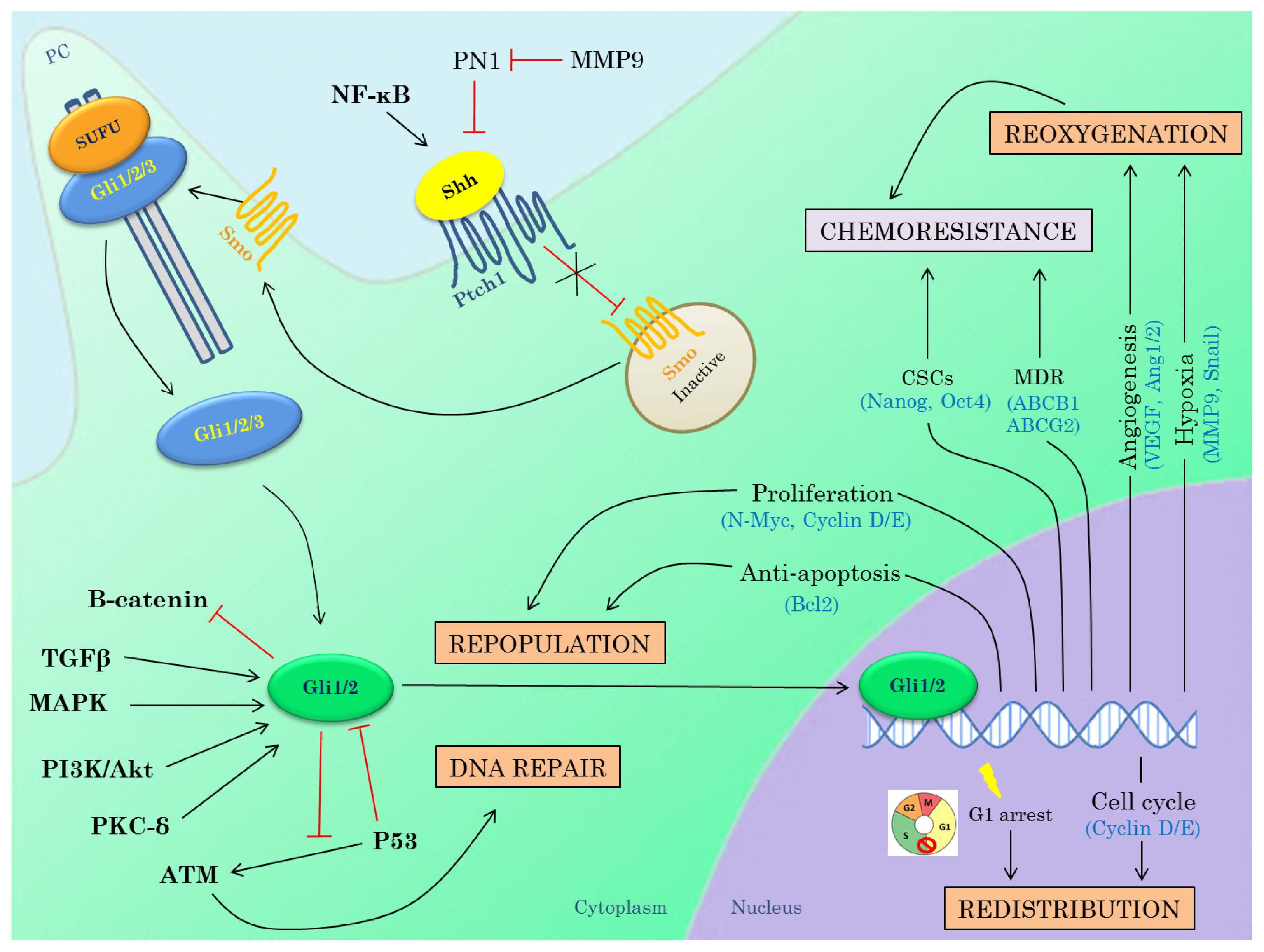{kind=link}
| Study | # Tissue samples | Technique | Key findings | p-value |
|---|---|---|---|---|
| Tzelepi et al. [48] | 141 PCa | IHC | Epithelial Shh, Smo and Ptch up-regulated in T vs. N | <0.001 |
| 53 mPCa | Stromal Ptch, Smo and Gli1 down-regulated in T vs. N | <0.001 | ||
| 119 N | Correlation Ptch1 and tumor grade/stage | <0.001 | ||
| Higher epithelial Ptch expression in metastasis vs. tumor | <0.001 | |||
| Correlation Hh signaling and proliferation (Ki67) and vasculogenesis (VEGF) | <0.001 | |||
| Sanchez et al. [47] | 239 PCa | IHC | Higher Shh expression in T (33%) vs. N (<1%) | <0.001 |
| 15 HGPIN | Correlation Shh and proliferation (Ki67) | 0.0141 | ||
| 135 N | No correlation between Shh and other clinical parameters | |||
| Fan et al. [34] | 6 PCa | qPCR | No significant difference between Hh signaling in T vs. N | |
| 6 BPH | ||||
| 7 N | ||||
| Sheng et al. [45] | 55 PCa | IHC | Hh signaling pathway frequently activated in advanced PCa | |
| 4 mPCa | qPCR | Correlation Ptch1 and Hhip with Gleason score and metastasis | ||
| 55 N | Loss-of-SUFU frequently present in PCa | |||
| Azoulay et al. [49] | 275 PCa | IHC | In HNPC, correlation between epithelial Shh and Gleason, metastatic lymph nodes | <0.05 |
| (231 HNPC) | qPCR | Concomitant absence of stromal Shh and Dhh prognostic factor for PSA recurrence | 0.01 | |
| (20 HTPC) | Dhh expression up-regulated in epithelial HTPC and HRPC vs. HNPC | <0.0001 | ||
| (24 HRPC) | ||||
| Kim et al. [50] | 155 PCa | IHC | Correlation between Shh, Ptch, Smo, Gli and Gleason score | <0.01 |
| 155 N | qPCR | Shh independent prognostic factor for PSA recurrence | <0.001 | |
| Karhadkar et al. [1] | 12 PCa | qPCR | Shh and Ihh present in all prostate samples | |
| 15 mPCa | PTCH1 and GLI1 mRNA expression tenfold higher in metastatic vs. tumor tissues | |||
| 12 N | ||||
| Efstathiou et al. [54] | 79 PCa | IHC | Up-regulated Hh signaling (Gli1, Gli2, Smo, Shh) after ADT or ADT with chemotherapy | <0.05 |
| 26 (ADT) | Nuclear pAKT increased | <0.001 | ||
| 27 (ADT + CT) | Epithelial Bcl2 increased after combination treatment | <0.01 | ||
| 27 (Untreated) | ||||
| Ibuki et al. [55] | 210 PCa | IHC | Dhh expression up-regulated after long-term ADT | |
| (44 ST-ADT) | Shh expression elevated in HRPC specimens | |||
| (76 LT-ADT) | ||||
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gonnissen, A.; Isebaert, S.; Haustermans, K. Hedgehog Signaling in Prostate Cancer and Its Therapeutic Implication. Int. J. Mol. Sci. 2013, 14, 13979-14007. https://doi.org/10.3390/ijms140713979
Gonnissen A, Isebaert S, Haustermans K. Hedgehog Signaling in Prostate Cancer and Its Therapeutic Implication. International Journal of Molecular Sciences. 2013; 14(7):13979-14007. https://doi.org/10.3390/ijms140713979
Chicago/Turabian StyleGonnissen, Annelies, Sofie Isebaert, and Karin Haustermans. 2013. "Hedgehog Signaling in Prostate Cancer and Its Therapeutic Implication" International Journal of Molecular Sciences 14, no. 7: 13979-14007. https://doi.org/10.3390/ijms140713979