Differential Proteomics Analysis of Bacillus amyloliquefaciens and Its Genome-Shuffled Mutant for Improving Surfactin Production

Abstract
:
1. Introduction
2. Results
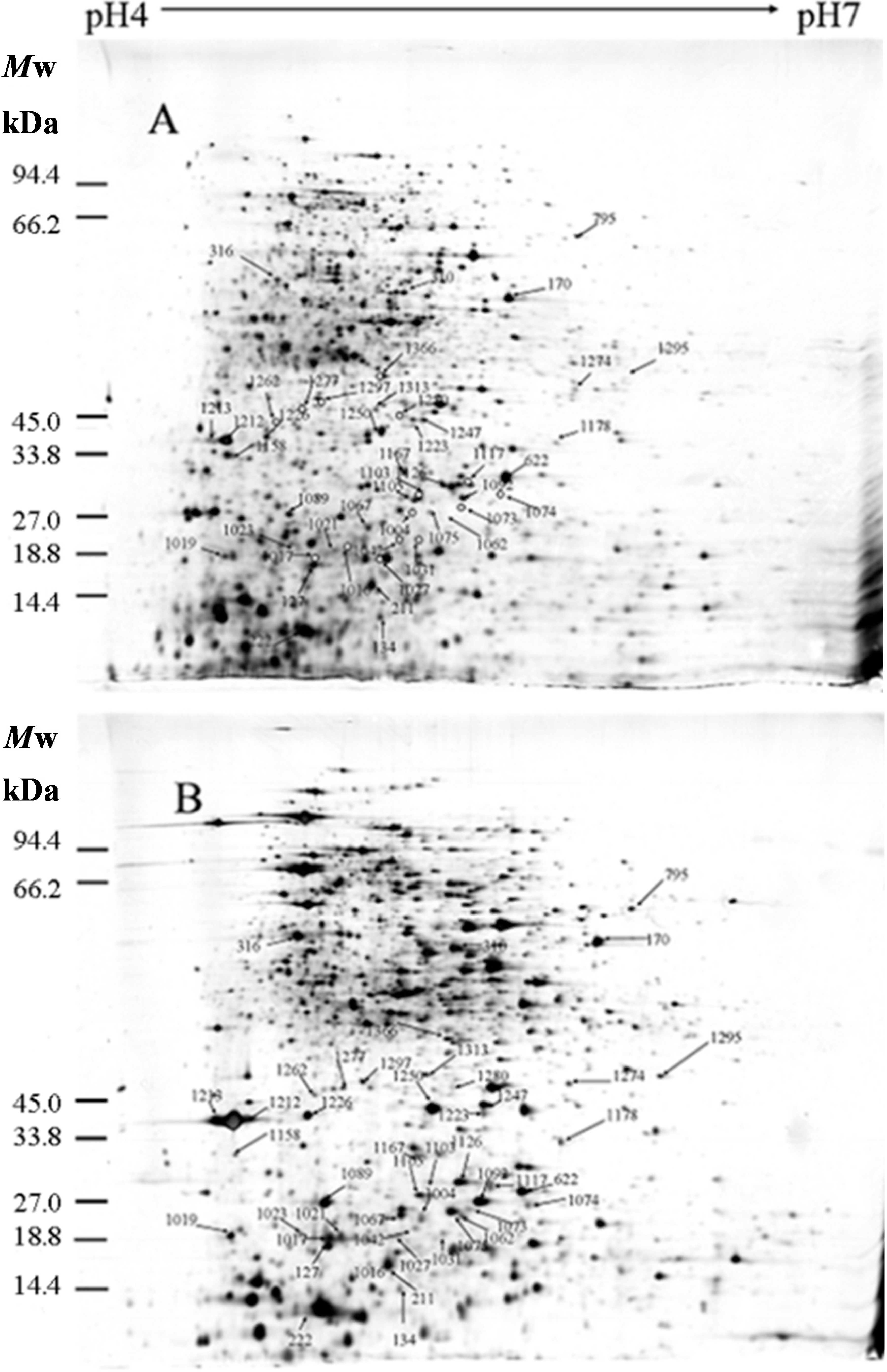
2.1. Identification of Differentially Expressed Proteins
2.2. Isoelectric Point and Molecular Weight Analysis of Theoretically and Experimentally Identified Proteins

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spot No. a | Protein Name b | Accession No. c | Locus d | Gene e | Theor. f Mr/pI | Exper. g Mr/pI | Protein Score h | Sequence Coverage (%) i | Fold Change j (p < 0.05) |
|---|---|---|---|---|---|---|---|---|---|
| 127 | Alkyl hydroperoxide reductase small subunit | gi|308175696 | YP_003922401 | ahpC | 20,669/4.51 | 21,013/4.65 | 104 | 44 | +15.3 |
| 134 | Conserved hypothetical protein | gi|315173048 | EFU17065 | – | 14,048/5.12 | 13,978/5.10 | 158 | 55 | −18.4 |
| 170 | DNA primase | gi|228983124 | ZP_04143383 | – | 55,979/5.93 | 56,328/5.79 | 115 | 48 | −19.8 |
| 211 | Hypothetical protein RBAM_036960 | gi|154688095 | YP_001423256 | ahpC | 20,683/4.51 | 17,985/4.62 | 126 | 50 | +34.7 |
| 222 | Hypothetical protein KSO_14324 | gi|363725374 | EHM05512 | – | 6927/4.56 | 6843/4.49 | 120 | 70 | +2.7 |
| 310 | Hypothetical protein RBAM_029780 | gi|154687379 | YP_001422540 | yurX | 48,265/5.30 | 48,965/5.33 | 109 | 32 | +29.1 |
| 316 | GroEL gene product | gi|311067075 | YP_003971998 | groEL | 57,385/4.75 | 56,789/4.78 | 190 | 43 | +6. 6 |
| 622 | Response regulator DegU | gi|157693950 | YP_003974978 | degU | 25,893/5.65 | 27,124/5.67 | 249 | 92 | −6.1 |
| 795, 816 | Vegetative catalase 1 | gi|89097371 | ZP_01170260 | – | 54,421/6.11 | 55,135/5.89 | 192 | 40 | +7.5, +4.7 |
| 1004, 1056 | Hypothetical protein RBAM_023340 | gi|154686764 | YP_001421925 | sodA | 22,365/5.21 | 23,214/5.43 | 316 | 80 | in FMB38 |
| 1016 | S-ribosylhomocysteinase | gi|154687196 | YP_001422357 | luxS | 17,913/5.27 | 18,324/5.09 | 98 | 66 | in FMB38 |
| 1017 | Thiol peroxidase | gi|154687070 | YP_001422231 | tpx | 18,262/4.99 | 17,321/4.89 | 180 | 89 | in FMB38 |
| 1019 | Hypothetical protein RBAM_026720 | gi|154687100 | YP_001422261 | yraA | 18,672/4.94 | 17,652/4.63 | 110 | 72 | +26.2 |
| 1021 | Hypothetical protein RBAM_008040 | gi|154685258 | YP_001420419 | yfkM | 18,877/4.83 | 18,896/4.85 | 163 | 85 | +17.9 |
| 1023 | Hypothetical protein RBAM_028130 | gi|154687215 | YP_001422376 | yuaE | 19,112/5.46 | 19,431/4.94 | 112 | 56 | +4.7 |
| 1027 | ATP-dependent Clp protease proteolytic subunit | gi|154687585 | YP_001422746 | clpP | 21,874/4.96 | 19,543/5.12 | 154 | 60 | in FMB38 |
| 1031 | DNA-directed DNA polymerase III α subunit | gi|325684283 | EGD26456 | dnaE | 128,931/8.83 | 19,678/5.34 | 81 | 18 | in FMB38 |
| 1042 | Methionine aminopeptidase, type I | gi|229010975 | ZP_04168170 | – | 27,381/4.89 | 190 | 49 | in FMB38 | |
| 1062, 1065 | Transaldolase | gi|154687826 | YP_001422987 | tal | 23,055/5.23 | 23,336/5.31 | 86 | 40 | +17.9, +6.5 |
| 1067 | Hypothetical protein RBAM_036480 | gi|154688047 | YP_001423208 | deoC | 23,111/4.90 | 23,352/5.08 | 94 | 40 | +24.0 |
| 1073 | Hypothetical protein KSO_05864 | gi|363723690 | EHM03828 | – | 23,280/4.62 | 242,110/5.41 | 112 | 56 | in FMB38 |
| 1074 | 2-Aminoethylphosphonate—Pyruvate transaminase | gi|229166259 | ZP_04294018 | – | 41,797/5.29 | 27,312/5.61 | 220 | 50 | in FMB38 |
| 1075 | Recombinase protein | gi|339764913 | AEK01094 | recA | 24,141/6.39 | 24,234/5.31 | 139 | 74 | +8.3 |
| 1089 | ComA | gi|154687277 | YP_001422438 | comA | 24,371/5.19 | 24,351/4.91 | 280 | 51 | +18.7 |
| 1092 | Transcriptional repressor CodY | gi|154686033 | YP_001421194 | codY | 29,038/4.90 | 28,213/5.4 | 110 | 47 | +7.7 |
| 1103 | Pyrroline-5-carboxylate reductase | gi|228921645 | ZP_04084963 | – | 29,356/5.37 | 28,531/5.31 | 77 | 45 | in FMB38 |
| 1105 | NAD synthetase NadE | gi|302671492 | YP_003831452 | nadE | 29,690/5.00 | 28,921/5.12 | 87 | 37 | +6.7 |
| 1117, 1120 | Fructose-bisphosphate aldolase | gi|154687827 | YP_001422988 | fbaA | 30,537/5.26 | 29,314/5.41 | 103 | 34 | in FMB38 |
| 1126 | Hypothetical protein RBAM_036720 | gi|154688071 | YP_001423232 | iolG | 38,449/5.14 | 37,111/5.41 | 225 | 38 | +18.6 |
| 1158 | Putative glycerol-3-phosphate acyltransferase PlsX | gi|326941622 | AEA17518 | plsX | 35,512/6.27 | 35,212/5.81 | 79 | 51 | +5.1 |
| 1167 | Glyceraldehyde-3-phosphate dehydrogenase (phosphorylating) | gi|157693809 | YP_001488271 | gapA | 35,822/5.03 | 35,344/5.13 | 92 | 29 | +4.0 |
| 1178 | Unnamed protein product | gi|311069921 | YP_003974844 | – | 35,900/5.10 | 35,212/5.82 | 93 | 33 | +164.7 |
| 1212 | Hypothetical protein RBAM_006650 | gi|154685120 | YP_001420281 | ydjL | 37,552/5.09 | 35,242/4.57 | 91 | 29 | +3.5, +4.9 |
| 1213, 1220 | YdjL | gi|363726006 | EHM06144 | – | 37,580/5.04 | 36,899/4.59 | 86 | 31 | +14.6 |
| 1223 | Multifunctional SOS repair factor | gi|308173657 | YP_003920362 | recA | 37,938/5.05 | 37,432/5.21 | 148 | 57 | +53.3 |
| 1226 | 30S ribosomal protein S1 | gi|328552793 | AEB23285 | rpsA | 41,960/4.82 | 41,556/4.79 | 81 | 22 | +16.8 |
| 1247 | Elongation factor Tu | gi|154684631 | YP_001419792 | tufA | 43,500/4.84 | 43,211/5.23 | 96 | 37 | +5.5 |
| 1250 | Hypothetical protein Dtox_1245 | gi|258514528 | YP_003190750 | – | 44,823/5.15 | 44,112/5.16 | 81 | 37 | +12.1 |
| 1262 | Site-specific recombinase XerD | gi|295101383 | CBK98928 | – | 46,358/9.08 | 45,212/4.81 | 77 | 43 | in FMB38 |
| 1274 | Plasmid recombination protein | gi|10956056 | NP_042279 | pre | 49,739/5.24 | 48,677/5.84 | 136 | 48 | +11.5 |
| 1277 | Hypothetical protein RBAM_007590 | gi|154685214 | YP_001420375 | yfmT | 53,319/5.26 | 52,695/4.82 | 113 | 29 | in FMB38 |
| 1280 | F0F1 ATP synthase subunit α | gi|154687798 | YP_001422959 | atpA | 54,804/5.34 | 113 | 26 | in FMB38 | |
| 1295 | Galactose-1-phosphate uridylyltransferase | gi|363725414 | EHM05552 | – | 56,804/6.27 | 56,123/6.16 | 301 | 54 | +9.2 |
| 1297 | Phosphopyruvate hydratase | gi|154687527 | YP_001422688 | eno | 46,645/4.68 | 46,131/4.85 | 300 | 61 | in FMB38 |
| 1313 | Dak2 domain fusion protein ylov | gi|312135039 | YP_004002377 | – | 60,930/4.99 | 43,123/5.10 | 77 | 23 | +7.0 |
| 1366 | M6 family metalloprotease | gi|172058939 | YP_001815399 | – | 86,115/5.33 | 48,531/5.21 | 91 | 21 | in FMB38 |

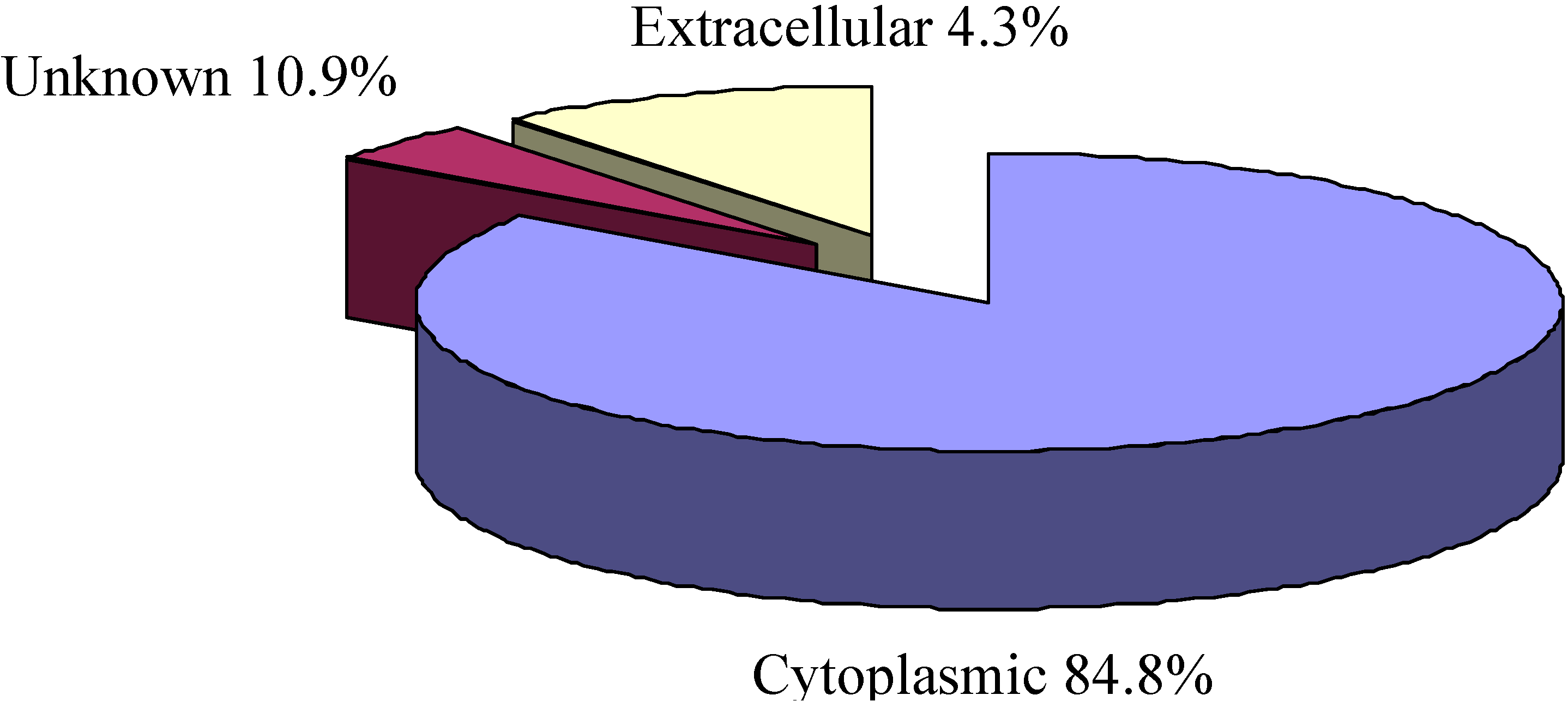
2.3. Cellular Localization Analysis of Experimentally Identified Proteins

2.4. Classification and Functional Analysis of Differential Proteins
| Spot No. a | Protein Name b | COG c | Cellular Localization d | Biological Process e | Molecular Functional Annotation f |
|---|---|---|---|---|---|
| Energy Production and Conversion | |||||
| 1277 | Hypothetical protein RBAM_007590 | C | Cytoplasmic | Unknown | Oxidoreductase activity, acting on the aldehyde or oxo group of donors, NAD or NADP as acceptor |
| 1280 | F0F1 ATP synthase subunit α | C | Cytoplasmic | ATP hydrolysis coupled proton transport, plasma membrane ATP synthesis Coupled proton transport | Hydrogen ion transporting ATP synthase activity, rotational mechanism; hydrolase activity |
| Cell Division and Chromosome Partitioning | |||||
| 1274 | Plasmid recombination protein | D | Cytoplasmic | DNA recombination | DNA binding |
| Amino Acid Transport and Metabolism | |||||
| 1074 | 2-Aminoethylphosphonate—Pyruvate transaminase | E | Cytoplasmic | Organic phosphonate catabolic process | 2-Aminoethylphosphonate-pyruvate transaminase activity, pyridoxal phosphate binding |
| 1103 | Pyrroline-5-carboxylate reductase | E | Cytoplasmic | Proline biosynthetic process | Nucleotide binding, oxidoreductase activity, acting on the CH–OH group of donors, NAD or NADP as acceptor, pyrroline-5-carboxylate reductase activity |
| 1212 | Hypothetical protein RBAM_006650 | ER | Cytoplasmic | Unknown | Nucleotide binding, oxidoreductase activity, zinc ion binding |
| 1213, 1220 | YdjL | ER | Cytoplasmic | Unknown | Nucleotide binding, oxidoreductase activity, zinc ion binding |
| Nucleotide Transport and Metabolism | |||||
| 1067 | Hypothetical protein RBAM_036480 | F | Cytoplasmic | Deoxyribonucleotide catabolic process | Deoxyribose-phosphate aldolase activity |
| Carbohydrate Transport and Metabolism | |||||
| 1062, 1065 | Transaldolase | G | Cytoplasmic | Pentose-phosphate shunt | Sedoheptulose-7-phosphate: d-glyceraldehyde-3-phosphate glyceronetransferase activity |
| 1117, 1120 | Fructose-bisphosphate aldolase | G | Cytoplasmic | Fructose 1,6-bisphosphate metabolic process, glycolysis, sporulation resulting in formation of a cellular spore | Fructose-bisphosphate aldolase activity, zinc ion binding |
| 1167 | Glyceraldehyde-3-phosphate dehydrogenase (phosphorylating) | G | Cytoplasmic | Glycolysis | NAD binding; NADP binding, glyceraldehyde-3-phosphate dehydrogenase (NAD+) (phosphorylating) activity |
| 1178 | Unnamed protein product | G | Cytoplasmic | Unknown | Glyceraldehyde-3-phosphate dehydrogenase/erythrose-4-phosphate dehydrogenase |
| 1297 | Phosphopyruvate hydratase | G | Cytoplasmic | Glycolysis | Magnesium ion binding, phosphopyruvate hydratase activity |
| Coenzyme Metabolism | |||||
| 1105 | NAD synthetase NadE | H | Unknown | NAD biosynthetic process, response to stress, sporulation resulting in formation of a cellular spore | ATP binding, NAD+ synthase (glutamine-hydrolyzing) activity, NAD+ synthase activity |
| Lipid Metabolism | |||||
| 1158 | Putative glycerol-3-phosphate acyltransferase PlsX | I | Unknown | Phospholipid biosynthetic process | Transferase activity, transferring acyl groups other than amino-acyl groups |
| Translation, Ribosomal Structure, and Biogenesis | |||||
| 1042 | Methionine aminopeptidase, type I | J | Cytoplasmic | Protein initiator methionine removal, proteolysis | Ferrous iron binding, metalloaminopeptidase activity |
| 1226 | 30S ribosomal protein S1 | J | Cytoplasmic | Translation | RNA binding, structural constituent of ribosome |
| 1247 | Elongation factor Tu | JE | Cytoplasmic | Response to antibiotic | GTP binding, GTPase activity, protein binding, translation elongation factor activity |
| DNA Replication | |||||
| 170 | DNA primase | L | Cytoplasmic | DNA replication, synthesis of RNA primer | ATP binding; DNA binding, DNA helicase activity, DNA primase activity, zinc ion binding |
| 1031 | DNA-directed DNA polymerase III α subunit | L | Cytoplasmic | Transcription | Nucleotidyltransferase, transferase |
| 1075 | Recombinase protein | L | Cytoplasmic | DNA recombination, DNA repair, SOS response | ATP binding, DNA-dependent ATPase activity, single-stranded DNA binding |
| 1223 | Multifunctional SOS repair factor | L | Cytoplasmic | DNA recombination, DNA repair, SOS response | ATP binding, DNA-dependent ATPase activity, damaged DNA binding, single-stranded DNA binding |
| 1262 | Site-specific recombinase XerD | L | Cytoplasmic | DNA integration, DNA recombination | DNA binding |
| Cell Motility and Secretion | |||||
| 1027 | ATP-dependent Clp protease proteolytic subunit | NO | Cytoplasmic | Protein metabolic process | ATP binding, ATP-dependent peptidase activity, protein binding |
| Posttranslational Modification | |||||
| 127 | Alkyl hydroperoxide reductase small subunit | O | Cytoplasmic | Unknown | Cytochrome-c peroxidase activity, peroxiredoxin activity |
| 211 | Hypothetical protein RBAM_036960 | O | Cytoplasmic | Unknown | Peroxidase activity; peroxiredoxin activity |
| 316 | GroEL gene product | O | Cytoplasmic | Cellular protein metabolic process | ATP binding |
| Inorganic Ion Transport and Metabolism | |||||
| 795, 816 | Vegetative catalase 1 | P | Cytoplasmic | Response to oxidative stress | Catalase activity, heme binding |
| 1004, 1056 | Hypothetical protein RBAM_023340 | P | Extracellular | Superoxide metabolic process | Metal ion binding, superoxide dismutase activity |
| Secondary Metabolite Biosynthesis, Transport, and Catabolism | |||||
| 1073 | Hypothetical protein KSO_05864 | Q | Cytoplasmic | Peptidyl-pyrromethane cofactor linkage, porphyrin-containing compound biosynthetic process | Hydroxymethylbilane synthase activity |
| General Function Prediction | |||||
| 310 | Hypothetical protein RBAM_029780 | R | Unknown | Iron-sulfur cluster assembly | Unknown |
| 1019 | Hypothetical protein RBAM_026720 | R | Cytoplasmic | Unknown | Hydrolase activity, acting on glycosyl bonds |
| 1021 | Hypothetical protein RBAM_008040 | R | Cytoplasmic | Unknown | Hydrolase activity, acting on glycosyl bonds |
| 1126 | Hypothetical protein RBAM_036720 | R | Cytoplasmic | Inositol Catabolic Process | Inositol 2-dehydrogenase activity, nucleotide binding |
| 1313 | Dak2 domain fusion protein ylov | R | Cytoplasmic | Glycerol metabolic process | Glycerone kinase activity |
| Signal Transduction Mechanisms | |||||
| 622 | Response regulator DegU | TK | Cytoplasmic | Transcription, DNA-dependent | Sequence-specific DNA binding, sequence-specific DNA binding transcription factor activity, two-component response regulator activity |
| 1016 | S-ribosylhomocysteinase | T | Cytoplasmic | Quorum Sensing | iron ion binding, lyase activity |
| 1089 | ComA | TK | Cytoplasmic | Transcription, DNA-dependent | Sequence-specific DNA binding, sequence-specific DNA binding transcription factor activity, two-component response regulator activity |
| Others | |||||
| 134 | Conserved hypothetical protein | – | Unknown | Growth of symbiont in host, protein omooligomerization | ATP binding, ATPase activity, protein binding |
| 222 | Hypothetical protein KSO_14324 | – | Unknown | Unknown | Electron carrier activity, heme binding |
| 1017 | Thiol peroxidase | – | Cytoplasmic | Cellular response to oxidative stress | Thioredoxin peroxidase activity |
| 1023 | Hypothetical protein RBAM_028130 | – | Cytoplasmic | Unknown | Unknown |
| 1092 | Transcriptional repressor CodY | – | Cytoplasmic | Transcription, DNA-dependent | DNA binding, GTP binding, sequence-specific DNA binding, transcription factor activity |
| 1250 | Hypothetical protein Dtox_1245 | – | Cytoplasmic | Unknown | Unknown |
| 1295 | Galactose-1-phosphate uridylyltransferase | – | Cytoplasmic | Galactose Metabolic Process | UDP-glucose: hexose-1-phosphate uridylyltransferase activity |
| 1366 | M6 family metalloprotease | – | Extracellular | Proteolysis | Metallopeptidase activity |
2.5. Gene Expression Verification by qRT-PCR

3. Discussion
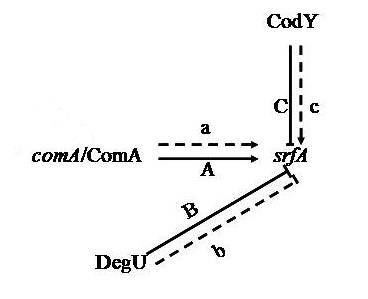
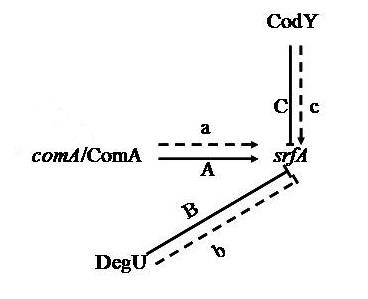
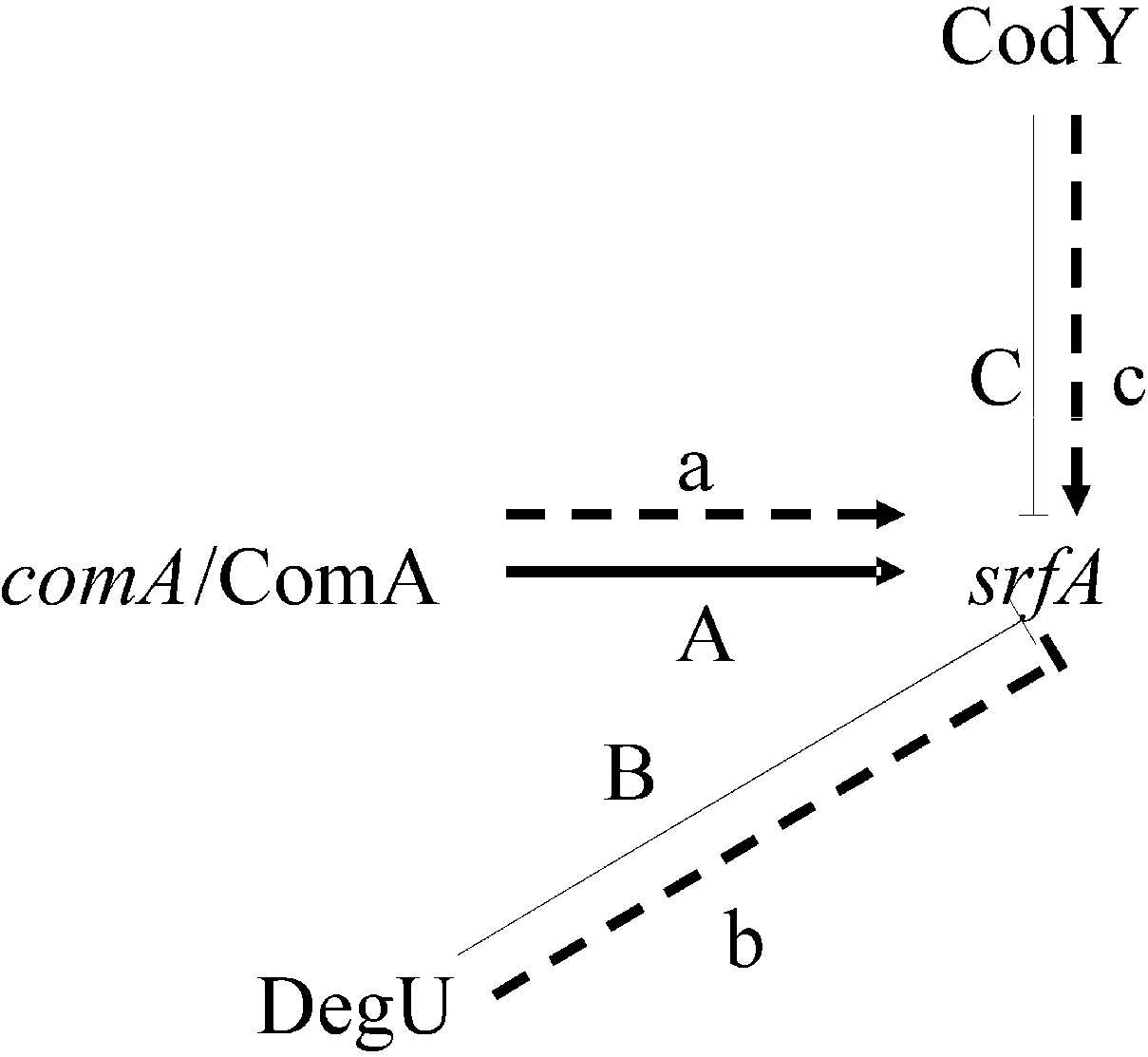
3.1. Proteins Related to Surfactin Synthesis


3.2. Metabolism-Related Proteins
3.3. Proteins Related to Energy Generation and Transformation
3.4. Proteins Related to DNA Replication, Recombination and Repair
3.5. Proteins Related to Translation and Post-Translational Modifications
3.6. Proteins Related to Cell Secretion and Signal Transduction Mechanisms
3.7. Hypothetical and Unknown Proteins
4. Experimental Section
4.1. Strains and Culture Conditions
4.2. Protein Sample Preparation
4.3. 2-DE and Staining
4.4. Image Acquisition and Data Analysis
4.5. Protein In-Gel Digestion
4.6. Protein Identification by MALDI-TOF and Database Searches
4.7. RT-PCR Analysis
| Gene | Forward Primer (5'→3') | Reverse Primer (5'→3') |
|---|---|---|
| 16S rDNA | CCTACGGGAGGCAGCAG | ATTACCGCGGCTGCTGG |
| comA | TCAAAGTGAGCAGGATCGGTTAA | CTTCTGTACGGGAGCCGACAT |
| codY | GGCAGGCAAACCCGTAAACT | ACTGGCGGTCTTCCAGCATT |
| degU | CACCCGAAAGTAACCCACAAT | AGCACTTCACATTCCCGTCTC |
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Carrillo, C.; Teruel, J.A.; Aranda, F.J.; Ortiz, A. Molecular mechanism of membrane permeabilization by the peptide antibiotic surfactin. Biochim. Biophys. Acta 2003, 1611, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Ongena, M.; Jacques, P. Bacillus lipopeptides: Versatile weapons for plant disease bio-control. Trends Microbiol. 2008, 16, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Kowall, M.; Vater, J.; Kluge, B.; Stein, T.; Franke, P. Separation and characterization of surfactin isoforms produced by Bacillus subtilis OKB 105. J. Colloid Interface Sci. 1998, 204, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bie, X.M.; Lu, Z.X.; Lu, F.X.; Zeng, X.X. Screening the main factors affecting extraction of the antimicrobial substance from Bacillus sp. fmbJ using Plackett–Burman method. World J. Microbiol. Biotechnol. 2005, 21, 925–928. [Google Scholar] [CrossRef]
- Kanlayavattanakul, M.; Lourith, N. Lipopeptides in cosmetics. Int. J. Cosmet. Sci. 2010, 32, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Schaller, K.D.; Fox, S.L.; Bruhn, D.F.; Noah, K.S.; Bala, G.A. Characterization of surfactin from Bacillus subtilis for application as an agent for enhanced oil recovery. Appl. Biochem. Biotechnol. 2004, 115, 827–836. [Google Scholar] [CrossRef]
- Mulligan, C.N.; Yong, R.N.; Gibbs, B.F. Heavy metal removal from sediments by biosurfactants. J. Hazard. Mater. 2001, 85, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Jacques, P.; Hbid, C.; Destain, J.; Razafindralambo, H.; Paquot, M. Optimization of biosurfactant lipopeptide production from Bacillus subtilis S499 by Plackett-Burman design. Appl. Biochem. Biotechnol. 1999, 77, 223–233. [Google Scholar] [CrossRef]
- Dimitrov, K.; Gancel, F.; Montastruc, L.; Nikov, I. Liquid membrane extraction of bioactive amphiphilic substances:Recovery of surfactin. Biochem. Eng. J. 2008, 42, 248–253. [Google Scholar] [CrossRef] [Green Version]
- Cao, G.Q.; Zhang, X.H.; Zhong, L.; Lu, Z.X. A modified electro-transformation method for Bacillus subtilis and its application in the production of antimicrobial lipopeptides. Biotechnol. Lett. 2010, 33, 1047–1051. [Google Scholar] [CrossRef]
- Sun, H.G.; Bie, X.M.; Lu, F.X.; Lu, Y.P.; Wu, Y. Enhancement of surfactin production of Bacillus subtilis fmbR by replacement of the native promoter with the Pspac promoter. Can. J. Microbiol. 2009, 55, 1003–1009. [Google Scholar] [CrossRef] [PubMed]
- Stein, T. Bacillus subtilis antibiotics: Structures, syntheses and specific functions. Mol. Microbiol. 2005, 56, 845–857. [Google Scholar]
- Hamoen, L.W.; Venema, G.; Kuipers, O.P. Controlling competence in Bacillus subtilis: Shared use of regulators. Microbiology 2003, 149, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Tsuge, K.; Ohata, Y.; Shoda, M. Gene yerP, involved in surfactin self-resistance in Bacillus subtilis. Anti-Microb. Agents Chemother. 2001, 45, 3566–3573. [Google Scholar] [CrossRef]
- Zhao, J.F.; Li, Y.H.; Zhang, C.; Yao, Z.Y.; Zhang, L. Genome shuffling of Bacillus amyloliquefaciens for improving antimicrobial lipopeptide production and an analysis of relative gene expression using FQ RT-PCR. J. Ind. Microbiol. Biotechnol. 2012, 39, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.W.; Ma, H.Q.; Chen, S.; Ji, M.; Perl, A. Stress response proteins’ differential expression in embryogenic and non-embryogenic callus of Vitis. vinifera L. cv. Cabernet Sauvignon-A proteomic approach. Plant. Sci. 2009, 177, 103–113. [Google Scholar] [CrossRef]
- Marahiel, M.A.; Nakano, M.M.; Zuber, P. Regulation of peptide antibiotic production in Bacillus. Mol. Microbiol. 1997, 7, 631–636. [Google Scholar] [CrossRef]
- Hahn, J.; Dubnau, D. Growth stage signal transduction and the requirements for srfA induction in development of competence. J. Bacteriol. 1991, 173, 7275–7282. [Google Scholar] [PubMed]
- Serror, P.; Sonenshein, A.L. CodY is required for nutritional repression of Bacillus subtilis genetic competence. J. Bacteriol. 1996, 178, 5910–5915. [Google Scholar] [PubMed]
- Duitman, E.H.; Wyczawski, D.; Boven, L.G.; Venema, G.; Kuipers, O.P. Novel methods for genetic transformation of natural Bacillus subtilis isolates used to study the regulation of the mycosubtilin and surfactin synthetases. Appl. Environ. Microbiol. 2007, 73, 3490–3496. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, W.L. The Bacillus subtilis ydjL (bdhA) gene encodes acetoin reductase/2,3-butanedioldehydrogenase. Appl. Environ. Microbiol. 2008, 74, 6832–6838. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, H.S.; Broomall, S.M.; McNew, L.A.; Daligault, H.; Chapman, C. Genomic signatures of strain selection and enhancement in Bacillus atrophaeus var. globigii, a historical biowarfare simulant. PLoS One 2011, 6, e17836. [Google Scholar] [CrossRef]
- Mukhopadhyay, A.; Redding, A.M.; Joachimiak, M.P.; Arkin, A.P.; Borglin, S.E. Cell-wide responses to low-oxygen exposure in Desulfovibrio vulgaris Hildenborough. J. Bacteriol. 2007, 189, 5996–6010. [Google Scholar] [CrossRef] [PubMed]
- Borriss, R.; Chen, X.; Rueckert, C.; Blom, J.; Becker, A. Relationship of Bacillus amyloliquefaciens clades associated with strains DSM7T and FZB42T: A proposal for Bacillus amyloliquefaciens sub sp. amyloliquefaciens subsp. nov. and Bacillus amyloliquefaciens subsp. plantarum subsp. nov. based on their discriminating complete genome sequences. Int. J. Syst. Evol. Microbiol. 2011, 61, 1786–1801. [Google Scholar]
- Chen, X.H.; Koumoutsi, A.; Scholz, R.; Eisenreich, A.; Schneider, K. Comparative analysis of the complete genome sequence of the plant growth-promoting bacterium Bacillus amyloliquefaciens FZB42. Nat. Biotechnol. 2007, 25, 1007–1014. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.J.; Lu, Z.X.; Bie, X.M.; Lu, F.X.; Yang, S.Y. Isolation and characterization of a co-producer of fengycins and surfactins, endophytic Bacillus amyloliquefaciens ES-2, from Scutellaria. Baicalensis Georgi. World J. Microbiol. Biotechnol. 2006, 22, 1259–1266. [Google Scholar] [CrossRef]
- Fang, C.J.; Lu, Z.X.; Sun, L.J.; Lu, F.X.; Bie, X.M. Study on mutation breeding and fermentation of antimicrobial lipopeptides yielding bacterium with 20 keV N+ ion beam implantation. J. Radiat. Res. Radiat. Process. 2006, 24, 333–336. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, J.; Cao, L.; Zhang, C.; Zhong, L.; Lu, J.; Lu, Z. Differential Proteomics Analysis of Bacillus amyloliquefaciens and Its Genome-Shuffled Mutant for Improving Surfactin Production. Int. J. Mol. Sci. 2014, 15, 19847-19869. https://doi.org/10.3390/ijms151119847
Zhao J, Cao L, Zhang C, Zhong L, Lu J, Lu Z. Differential Proteomics Analysis of Bacillus amyloliquefaciens and Its Genome-Shuffled Mutant for Improving Surfactin Production. International Journal of Molecular Sciences. 2014; 15(11):19847-19869. https://doi.org/10.3390/ijms151119847
Chicago/Turabian StyleZhao, Junfeng, Lin Cao, Chong Zhang, Lei Zhong, Jing Lu, and Zhaoxin Lu. 2014. "Differential Proteomics Analysis of Bacillus amyloliquefaciens and Its Genome-Shuffled Mutant for Improving Surfactin Production" International Journal of Molecular Sciences 15, no. 11: 19847-19869. https://doi.org/10.3390/ijms151119847