Effects of Antidepressants on IP-10 Production in LPS-Activated THP-1 Human Monocytes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
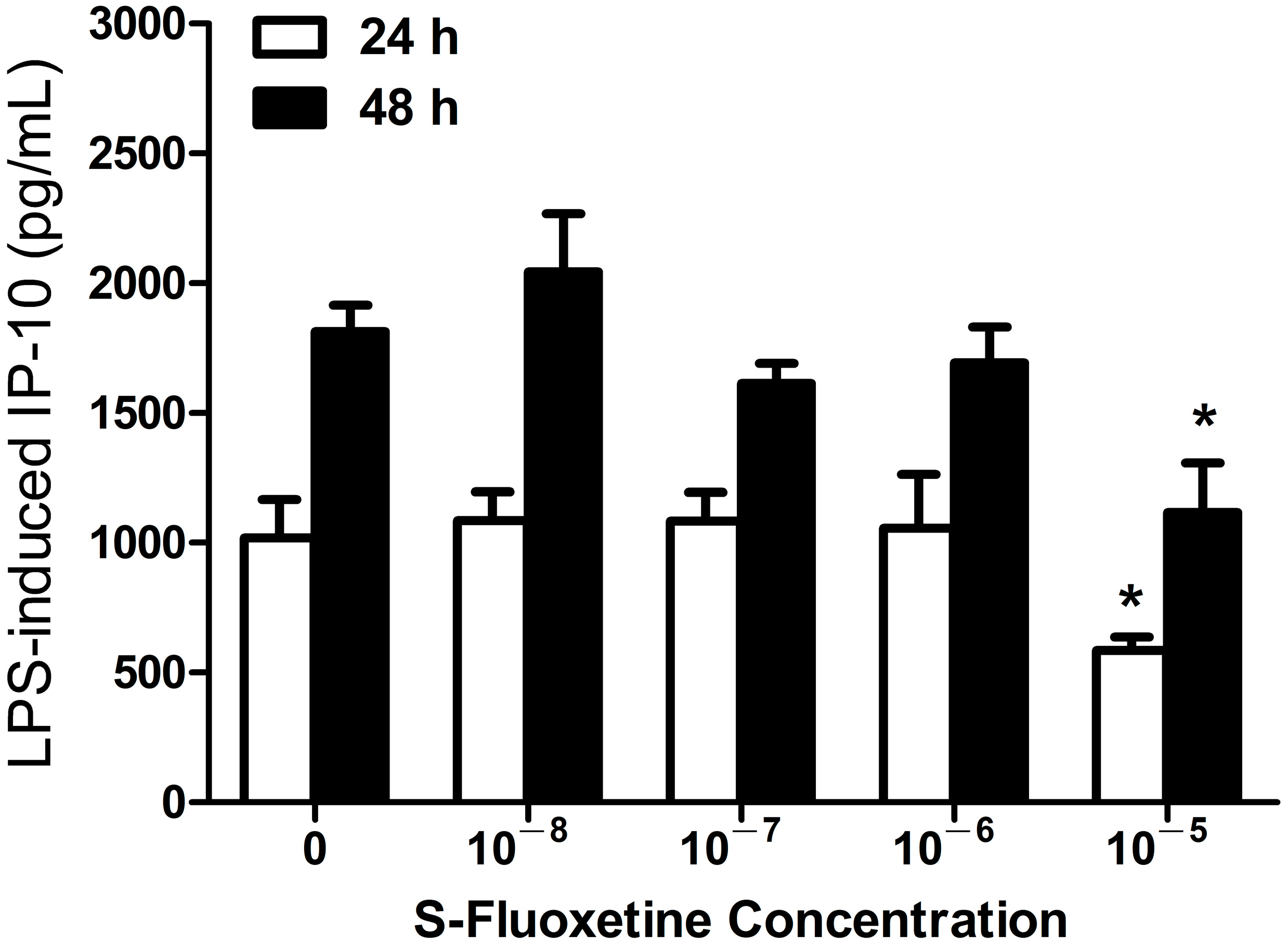
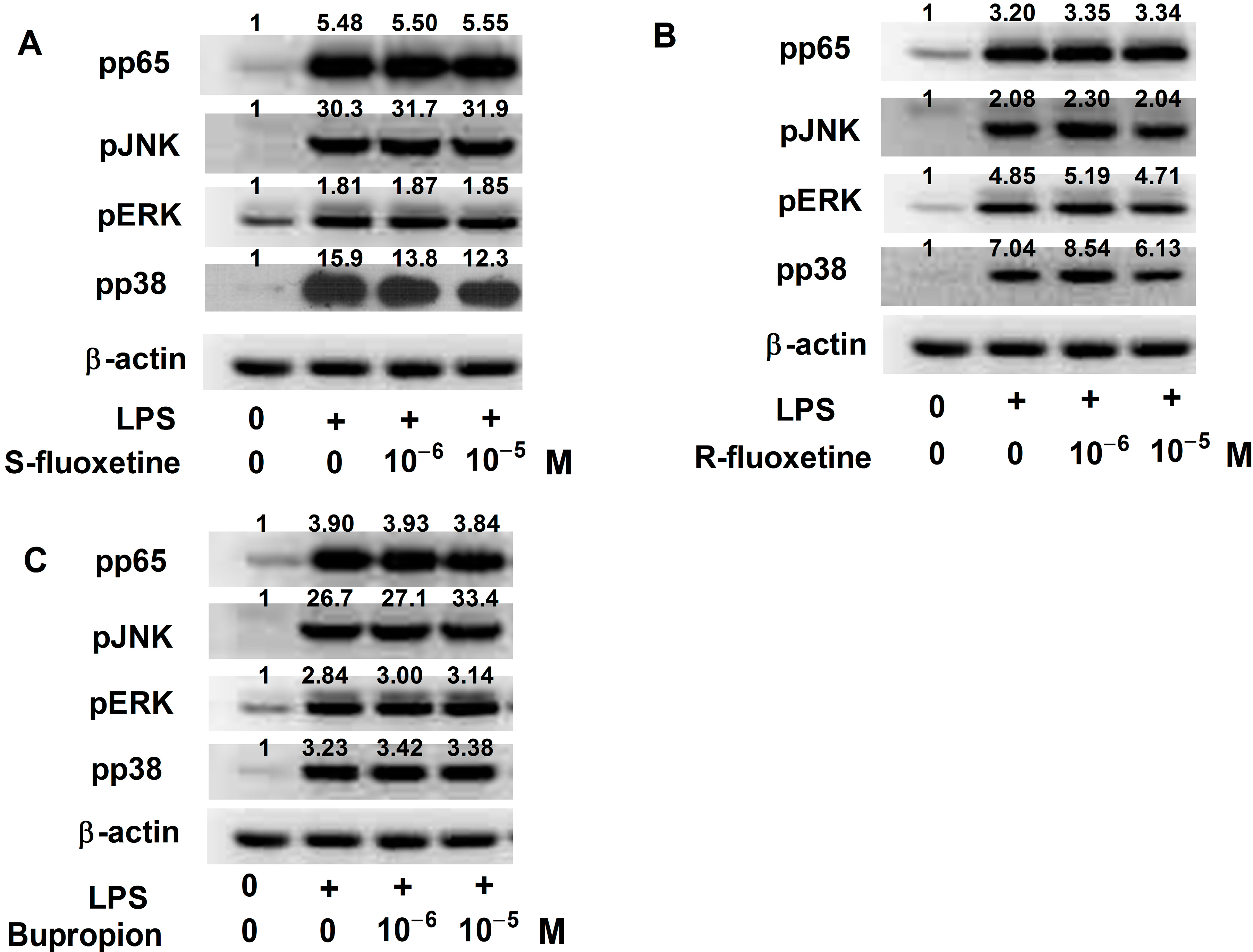
2.1.1. S-Fluoxetine Suppressed Lipopolysaccharide (LPS)-Induced Interferon-γ-Inducible Protein 10 (IP-10) Expression in THP-1 Cells

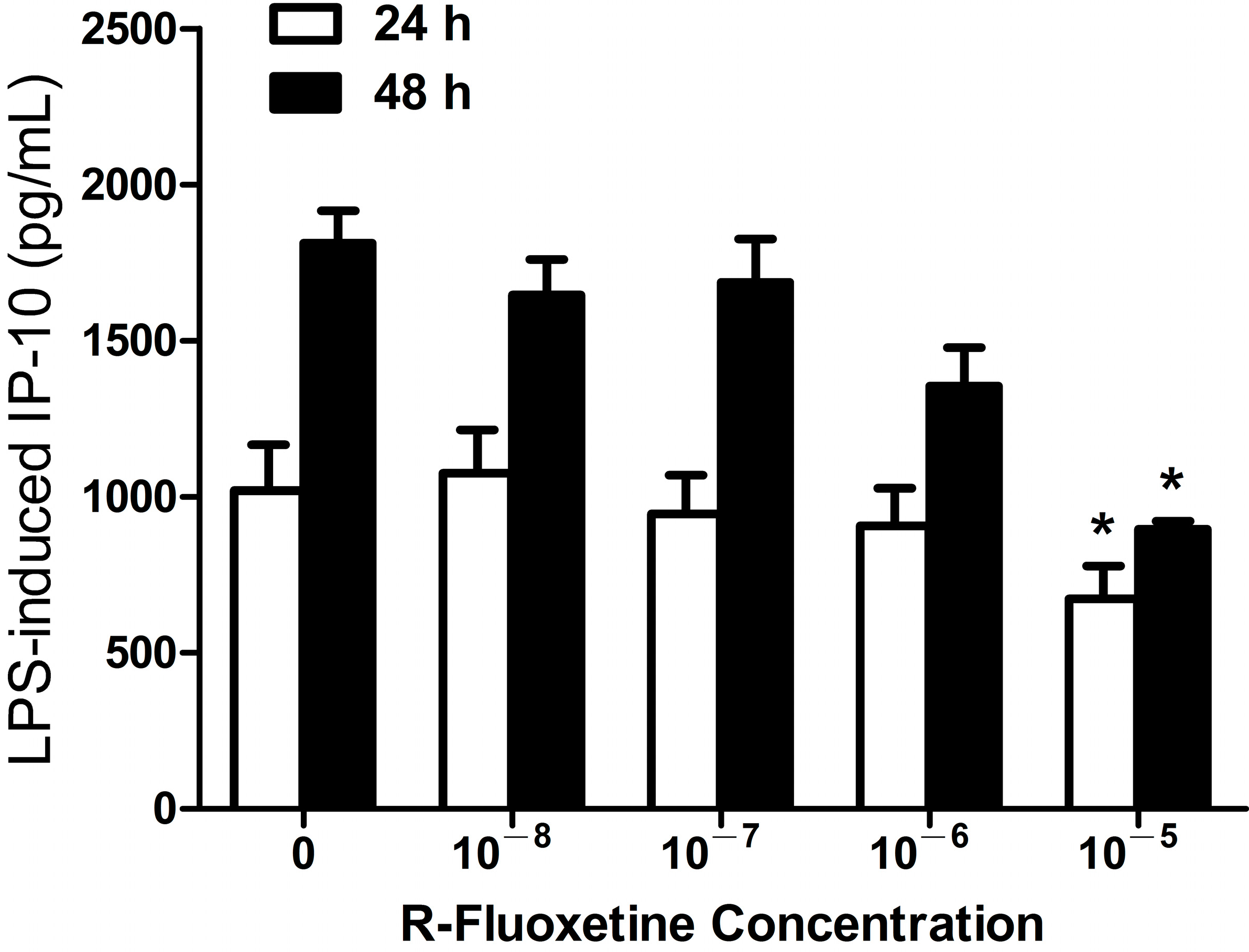
2.1.2. R-Fluoxetine also Suppressed LPS-Induced IP-10 Expression in THP-1 Cells

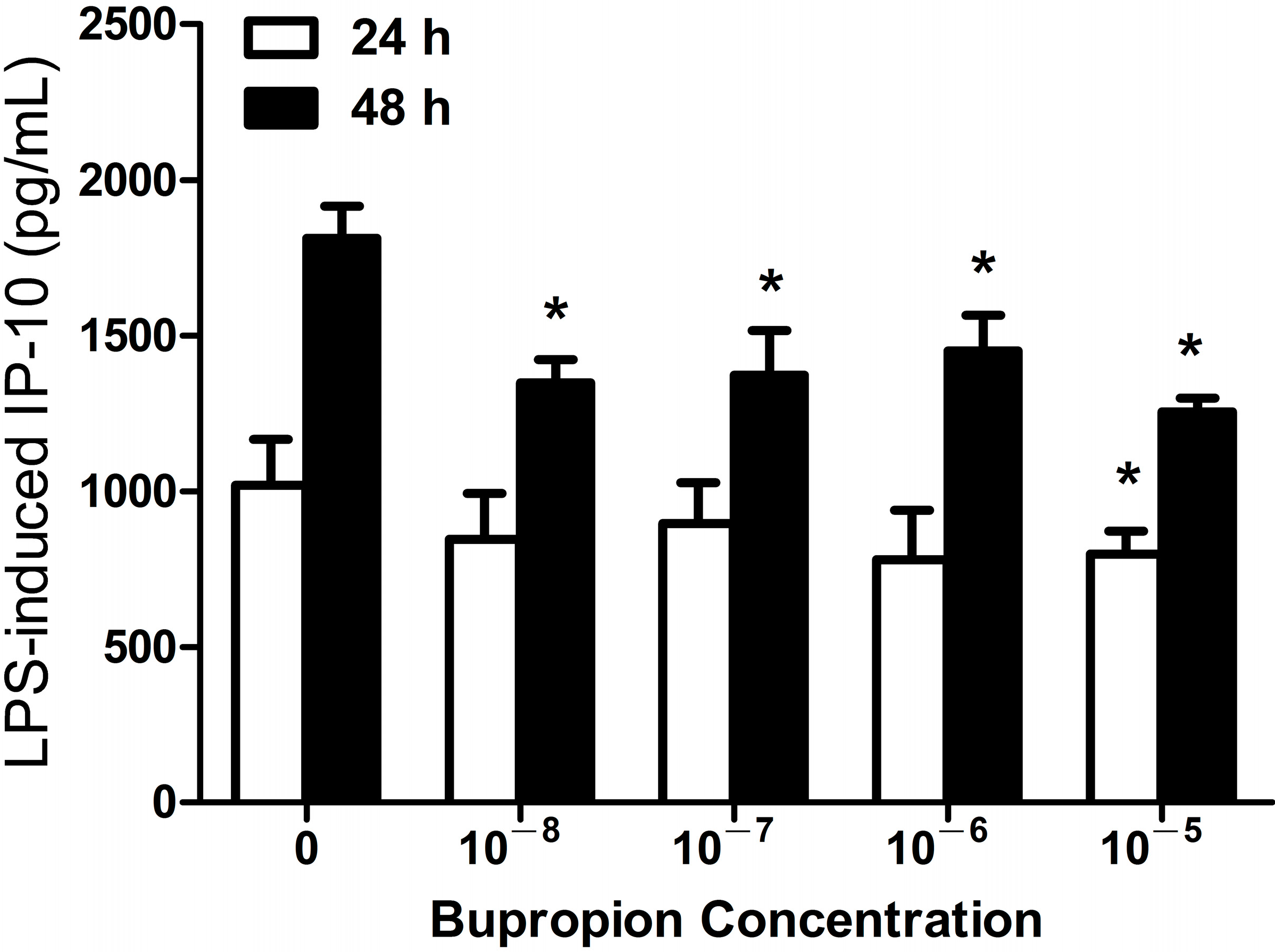
2.1.3. Bupropion Suppressed LPS-Induced IP-10 Expression in THP-1 Cells

2.1.4. Imipramine, Moclobemide, Venlafaxine and Mirtazapine Had no Effect on LPS-Induced IP-10 Expression in THP-1 Cells
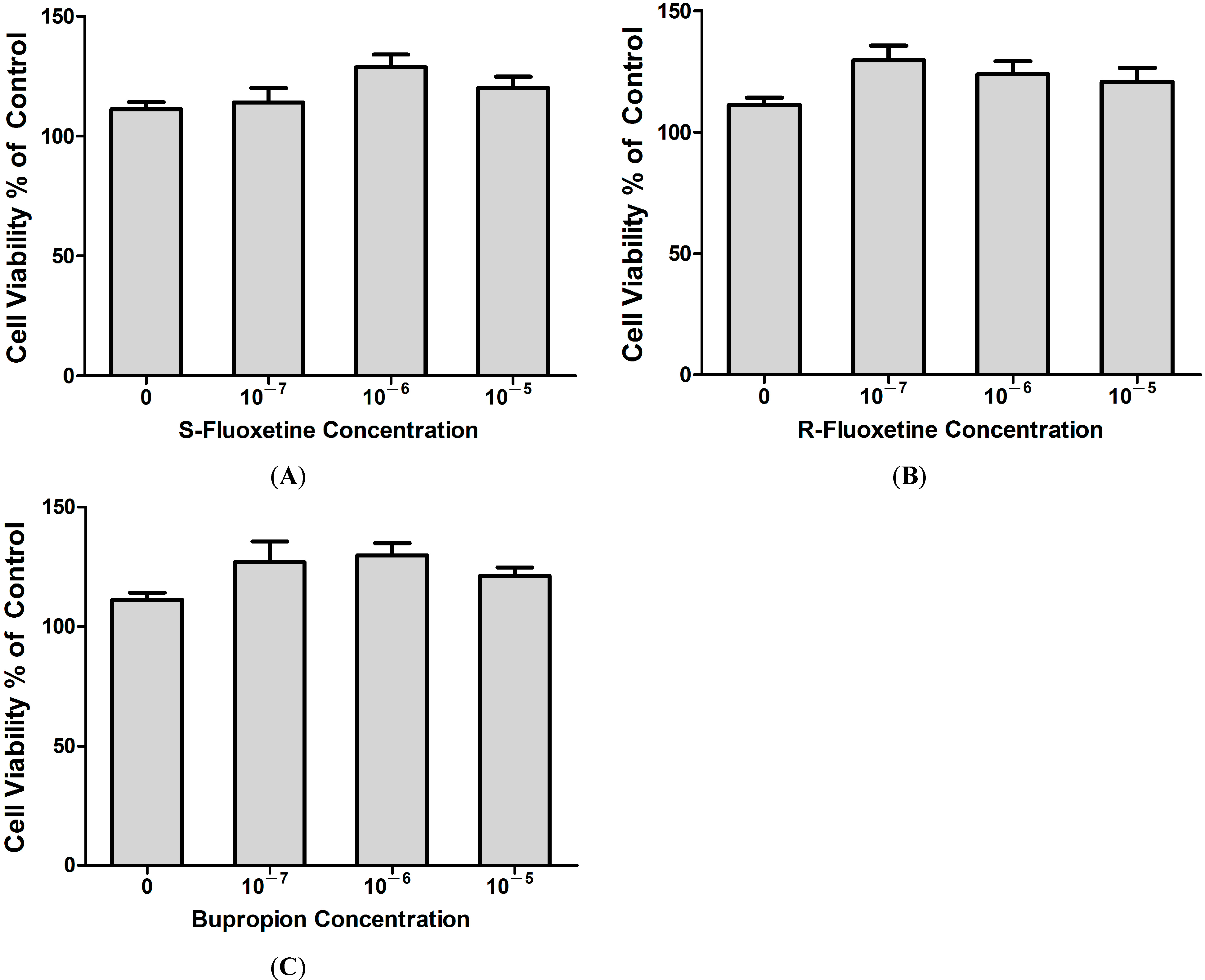
2.1.5. Fluoxetine and Bupropion Had no Cytotoxic Effect on THP-1 Cells

2.1.6. S- and R-Fluoxetine Suppressed LPS-Induced IP-10 via the Mitogen-Activated Protein Kinase (MAPK)-p38 Pathway

2.2. Discussion
3. Experimental Section
3.1. Reagents and Cell Preparation
3.2. Cell Viability Tests
3.3. Western Blotting Analysis
3.4. Enzyme-Linked Immunosorbent Assay (ELISA)
3.5. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rihmer, Z.; Angst, A. Mood disorders: Epidemiology. In Comprehensive Textbook of Psychiatry; Sadock, B.J., Sadock, V.A., Eds.; Lippincott & Williams: Baltimore, MD, USA, 2004; p. 528. [Google Scholar]
- Bostwick, J.M.; Pankratz, V.S. Affective disorders and suicide risk: A re-examination. Am. J. Psychiatry 2000, 157, 1925–1932. [Google Scholar] [CrossRef]
- Gaziano, T.; Gaziano, J.M. Global burden of cardiovascular disease, in heart disease. In A Textbook of Cardiovascular Medicine; Elsevier Saunders: Philadelphia, PA, USA, 2009. [Google Scholar]
- Glassman, A.H.; Shapiro, P.A. Depression and the course of coronary artery disease. Am. J. Psychiatry 1998, 155, 4–11. [Google Scholar]
- Jaffe, A.S.; Krumholz, H.M.; Catellier, D.J.; Freedland, K.E.; Bittner, V.; Blumenthal, J.A.; Calvin, J.E.; Norman, J.; Sequeira, R.; O’Connor, C.; et al. Prediction of medical morbidity and mortality after acute myocardial infarction in patients at increased psychosocial risk in the Enhancing Recovery in Coronary Heart Disease Patients (ENRICHD) study. Am. Heart J. 2006, 152, 126–135. [Google Scholar] [CrossRef]
- Somberg, T.C.; Arora, R.R. Depression and heart disease: Therapeutic implications. Cardiology 2008, 111, 75–81. [Google Scholar] [CrossRef]
- Wulsin, L.R.; Vaillant, G.E.; Wells, V.E. A systematic review of the mortality of depression. Psychosom. Med. 1999, 61, 6–17. [Google Scholar] [CrossRef]
- Dowlati, Y.; Herrmann, N.; Swardfager, W.; Liu, H.; Sham, L.; Reim, E.K.; Lanctôt, K.L. A meta-analysis of cytokines in major depression. Biol. Psychiatry 2010, 67, 446–457. [Google Scholar] [CrossRef]
- Howren, M.B.; Lamkin, D.M.; Suls, J. Associations of depression with C-reactive protein, IL-1, and IL-6: A meta-analysis. Psychosom. Med. 2009, 71, 171–186. [Google Scholar] [CrossRef]
- Zorilla, E.P.; Luborsky, L.; McKay, J.R.; Rosenthal, R.; Houldin, A.; Tax, A.; McCorkle, R.; Seligman, D.A.; Schmidt, K. The relationship of depression and stressors to immunological assays: A meta-analytic review. Brain Behav. Immun. 2001, 15, 199–226. [Google Scholar] [CrossRef]
- Raison, C.L.; Miller, A.H. Is depression an inflammatory disorder? Curr. Psychiatry Rep. 2011, 13, 467–475. [Google Scholar] [CrossRef]
- Pacher, P.; Kecskemeti, V. Cardiovascular side effects of new antidepressants and antipsychotics: New drugs, old concerns? Curr. Pharm. Des. 2004, 10, 2463–2475. [Google Scholar] [CrossRef]
- Wozniak, G.; Toska, A.; Saridi, M.; Mouzas, O. Serotonin reuptake inhibitor antidepressants (SSRIs) against atherosclerosis. Med. Sci. Monit. 2011, 17, RA205–RA214. [Google Scholar]
- Prather, A.A.; Rabinovitz, M.; Pollock, B.G.; Lotrich, F.E. Cytokine-induced depression during IFN-α treatment: The role of IL-6 and sleep quality. Brain Behav. Immun. 2009, 23, 1109–1116. [Google Scholar] [CrossRef]
- Raison, C.L.; Borisov, A.S.; Woolwine, B.J.; Massung, B.; Vogt, G.; Miller, A.H. Interferon-α effects on diurnal hypothalamic pituitary-adrenal axis activity: Relationship with proinflammatory cytokines and behavior. Mol. Psychiatry 2010, 15, 535–547. [Google Scholar]
- Eisenberger, N.I.; Berkman, E.T.; Inagaki, T.K.; Rameson, L.T.; Mashal, N.M.; Irwin, M.R. Inflammation-induced anhedonia: Endotoxin reduces ventral striatum responses to reward. Biol. Psychiatry 2010, 68, 748–754. [Google Scholar] [CrossRef]
- Eisenberger, N.I.; Inagaki, T.K.; Mashal, N.M.; Irwin, M.R. Inflammation and social experience: An inflammatory challenge induces feelings of social disconnection in addition to depressed mood. Brain Behav. Immun. 2010, 24, 558–563. [Google Scholar] [CrossRef]
- Janssen, D.G.; Caniato, R.N.; Verster, J.C.; Baune, B.T. A psychoneuroimmunological review on cytokines involved in antidepressant treatment response. Hum. Psychopharmacol. 2010, 25, 201–215. [Google Scholar] [CrossRef]
- Maes, M. The immunoregulatory effects of antidepressants. Hum. Psychopharmacol. 2001, 16, 95–103. [Google Scholar] [CrossRef]
- Luster, A.D. Chemokines—Chemotactic cytokines that mediate inflammation. N. Engl. J. Med. 1998, 338, 436–445. [Google Scholar] [CrossRef]
- Guha, M.; Mackman, N. LPS induction of gene expression in human monocytes. Cell. Signal. 2001, 13, 85–94. [Google Scholar] [CrossRef]
- Heller, E.A.; Liu, E.; Táger, A.M.; Yuan, Q.; Lin, A.Y.; Ahluwalia, N.; Jones, K.; Koehn, S.L.; Lok, V.M.; Aikawa, E.; et al. Chemokine CXCL10 promotes atherogenesis by modulating the local balance of effector and regulatory T cells. Circulation 2006, 113, 2301–2312. [Google Scholar] [CrossRef]
- Kawamura, A.; Miura, S.; Fujino, M.; Nishikawa, H.; Matsuo, Y.; Tanigawa, H.; Tomita, S.; Tsuchiya, Y.; Matsuo, K.; Saku, K. CXCR3 chemokine receptor-plasma IP-10 interaction in patients with coronary artery disease. Circ. J. 2003, 67, 851–854. [Google Scholar] [CrossRef]
- Rothenbacher, D.; Müller-Scholze, S.; Herder, C.; Koenig, W.; Kolb, H. Differential expression of chemokines, risk of stable coronary heart disease, and correlation with established cardiovascular risk markers. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 194–199. [Google Scholar] [CrossRef]
- Hung, C.H.; Suen, J.L.; Hua, Y.M.; Chiang, W.; Chang, H.C.; Chen, C.N.; Jong, Y.J. Suppressive effects of ketotifen on Th1- and Th2-related chemokines of monocytes. Pediatr. Allergy Immunol. 2007, 18, 378–384. [Google Scholar] [CrossRef]
- Laurin, D.; David Curb, J.; Masaki, K.H.; White, L.R.; Launer, L.J. Midlife C-reactive protein and risk of cognitive decline: A 31-year follow-up. Neurobiol. Aging 2009, 30, 1724–1727. [Google Scholar] [CrossRef]
- Pradhan, A.D.; Manson, J.E.; Rifai, N.; Buring, J.E.; Ridker, P.M. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. J. Am. Med. Assoc. 2001, 286, 327–334. [Google Scholar] [CrossRef]
- Ridker, P.M.; Hennekens, C.H.; Buring, J.E.; Rifai, N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N. Engl. J. Med. 2000, 342, 836–843. [Google Scholar] [CrossRef]
- Ridker, P.M. Inflammatory biomarkers and risks of myocardial infarction, stroke, diabetes, and total mortality: Implications for longevity. Nutr. Rev. 2007, 65, S253–S259. [Google Scholar] [CrossRef]
- Rigotti, N.A.; Thomdike, A.N.; Regan, S.; McKool, K.; Pasternak, R.C.; Chang, Y.; Swartz, S.; Torres-Finnerty, N.; Emmons, K.M.; Singer, D.E. Bupropion for smokers hospitalized with acute cardiovascular disease. Am. J. Med. 2006, 119, 1080–1087. [Google Scholar] [CrossRef]
- Taylor, D. Antidepressant drugs and cardiovascular pathology: A clinical overview of effectiveness and safety. Acta Psychiatr. Scand. 2008, 118, 434–442. [Google Scholar] [CrossRef]
- Glassman, A.H.; O’Connor, C.M.; Califf, R.M.; Swedberg, K.; Schwartz, P.; Bigger, J.T., Jr.; Krishnan, K.R.; van Zyl, L.T.; Swenson, J.R.; Finkel, M.S.; et al. Sertraline treatment of major depression in patients with acute MI or unstable angina. J. Am. Med. Assoc. 2002, 288, 701–709. [Google Scholar] [CrossRef]
- Taylor, C.B.; Youngblood, M.E.; Catellier, D.; Veith, R.C.; Carney, R.M.; Burg, M.M.; Kaufmann, P.G.; Shuster, J.; Mellman, T.; Blumenthal, J.A.; et al. Effects of antidepressant medication on morbidity and mortality in depressed patients after myocardial infarction. Arch. Gen. Psychiatry 2005, 62, 792–798. [Google Scholar]
- Schlienger, R.G.; Fischer, L.M.; Jick, H.; Meier, C.R. Current use of selective serotonin reuptake inhibitors and risk of acute myocardial infarction. Drug Saf. 2004, 27, 1157–1165. [Google Scholar] [CrossRef]
- Seino, Y.; Ikeda, U.; Sekiguchi, H.; Morita, M.; Konishi, K.; Kasahara, T.; Shimada, K. Expression of leukocyte chemotactic cytokines in myocardial tissue. Cytokine 1995, 7, 301–304. [Google Scholar] [CrossRef]
- Shioi, T.; Matsumori, A.; Kihara, Y.; Inoko, M.; Ono, K.; Iwanaga, Y.; Yamada, T.; Iwasaki, A.; Matsushima, K.; Sasayama, S. Increased expression of interleukin-1β and monocyte chemotactic and activating factor/monocyte chemoattractant protein-1 in the hypertrophied and falling heart with pressure overload. Circ. Res. 1997, 81, 664–671. [Google Scholar] [CrossRef]
- Tarzami, S.T. Chemokines and inflammation in heart disease: Adaptive or maladaptive? Int. J. Clin. Exp. Med. 2011, 4, 74–80. [Google Scholar]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Sasaguri, Y.; Tanimoto, A. Role of macrophage-derived histamine in atherosclerosis—Chronic participation in the inflammatory response. J. Atheroscler. Thromb. 2004, 11, 122–130. [Google Scholar] [CrossRef]
- Sato, T.; Takebayashi, S.; Kohchi, K. Increased subendothelial infiltration of the coronary arteries with monocytes/macrophages in patients with unstable angina. Atherosclerosis 1987, 68, 191–197. [Google Scholar] [CrossRef]
- Kumphune, S.; Chattipakorn, S.; Chattipakorn, N. Role of p38 inhibition in cardiac ischemia/reperfusion injury. Eur. J. Clin. Pharmacol. 2012, 68, 513–524. [Google Scholar] [CrossRef]
- Marber, M.S.; Rose, B.; Wang, Y. The p 38 mitogen-activated protein kinase pathway—A potential target for intervention in infarction, hypertrophy, and heart failure. J. Mol. Cell. Cardiol. 2011, 51, 485–490. [Google Scholar] [CrossRef]
- Martin, E.D.; de Nicola, G.F.; Marber, M.S. New therapeutic targets in cardiology: p38 α Mitogen-activated protein kinase for ischemic heart disease. Circulation 2012, 126, 357–368. [Google Scholar] [CrossRef]
- Pariante, C.M.; Makoff, A.; Lovestone, S.; Feroli, S.; Heyden, A.; Miller, A.H.; Kerwin, R.W. Antidepressants enhanced glucocorticoid receptor function in vitro by modulating the membrane steroid transporters. Br. J. Pharmacol. 2001, 134, 1335–1343. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tsai, J.-H.; Kuo, C.-H.; Yang, P.; Cheng, K.-H.; Wang, P.-W.; Chen, C.-C.; Hung, C.-H. Effects of Antidepressants on IP-10 Production in LPS-Activated THP-1 Human Monocytes. Int. J. Mol. Sci. 2014, 15, 13223-13235. https://doi.org/10.3390/ijms150813223
Tsai J-H, Kuo C-H, Yang P, Cheng K-H, Wang P-W, Chen C-C, Hung C-H. Effects of Antidepressants on IP-10 Production in LPS-Activated THP-1 Human Monocytes. International Journal of Molecular Sciences. 2014; 15(8):13223-13235. https://doi.org/10.3390/ijms150813223
Chicago/Turabian StyleTsai, Jui-Hsiu, Chang-Hung Kuo, Pinchen Yang, Kuang-Hung Cheng, Peng-Wei Wang, Cheng-Chung Chen, and Chih-Hsing Hung. 2014. "Effects of Antidepressants on IP-10 Production in LPS-Activated THP-1 Human Monocytes" International Journal of Molecular Sciences 15, no. 8: 13223-13235. https://doi.org/10.3390/ijms150813223