Nucleic Acids for Ultra-Sensitive Protein Detection

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- DNA and RNA both adhere to simple complementarity rules, which cater the easy design of specific interactions between different probes and affinity reagents
- Oligonucleotides can be easily synthesized and chemically modified at scale, affording flexibility in assay design
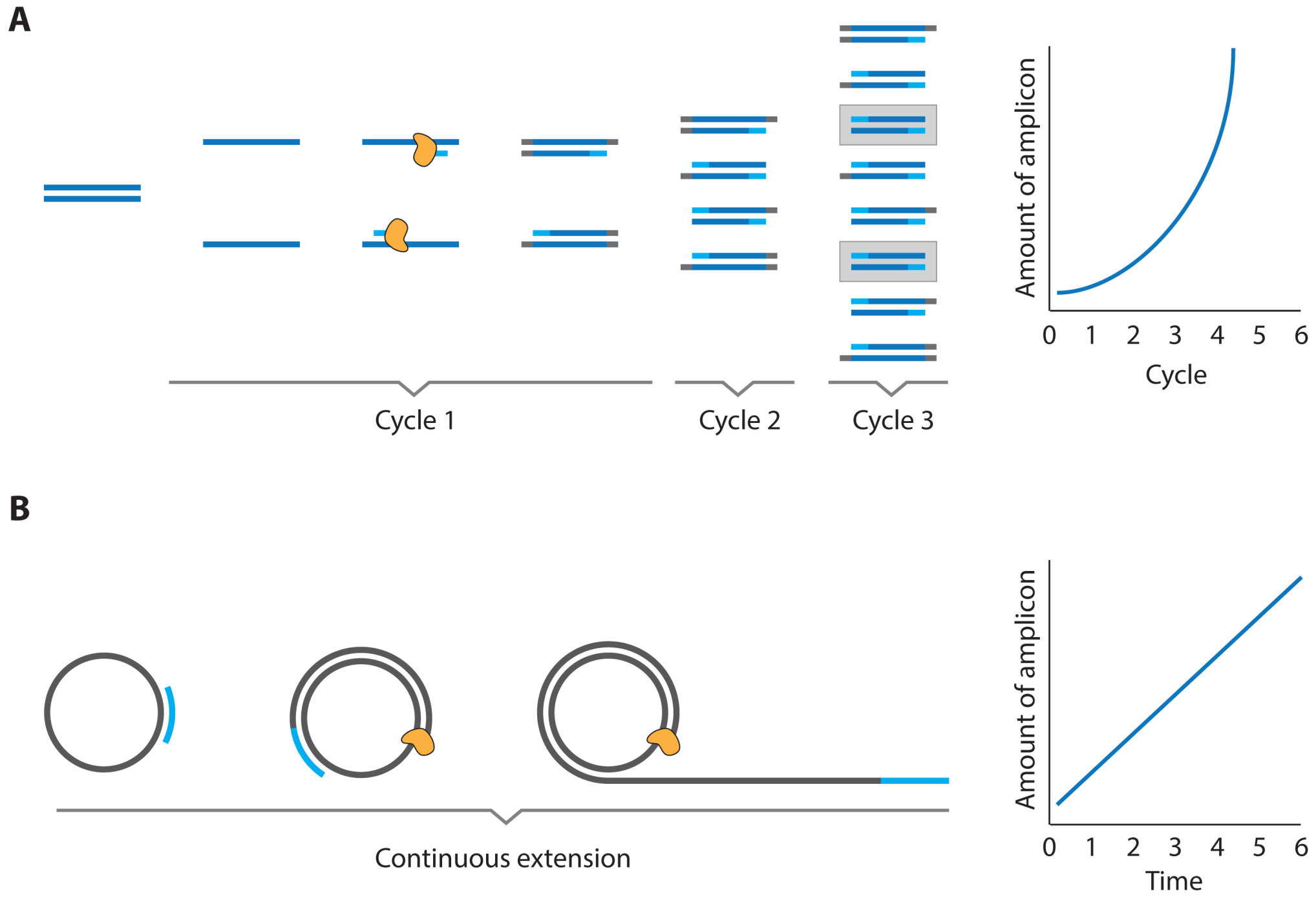
- Oligonucleotides can be processed with a large and versatile enzymatic “toolbox” that enables the facile amplification of oligonucleotide material
- Specific sequences inherently carry information that can be used for direct “barcoding” of the reagents and/or analytes to which they are coupled
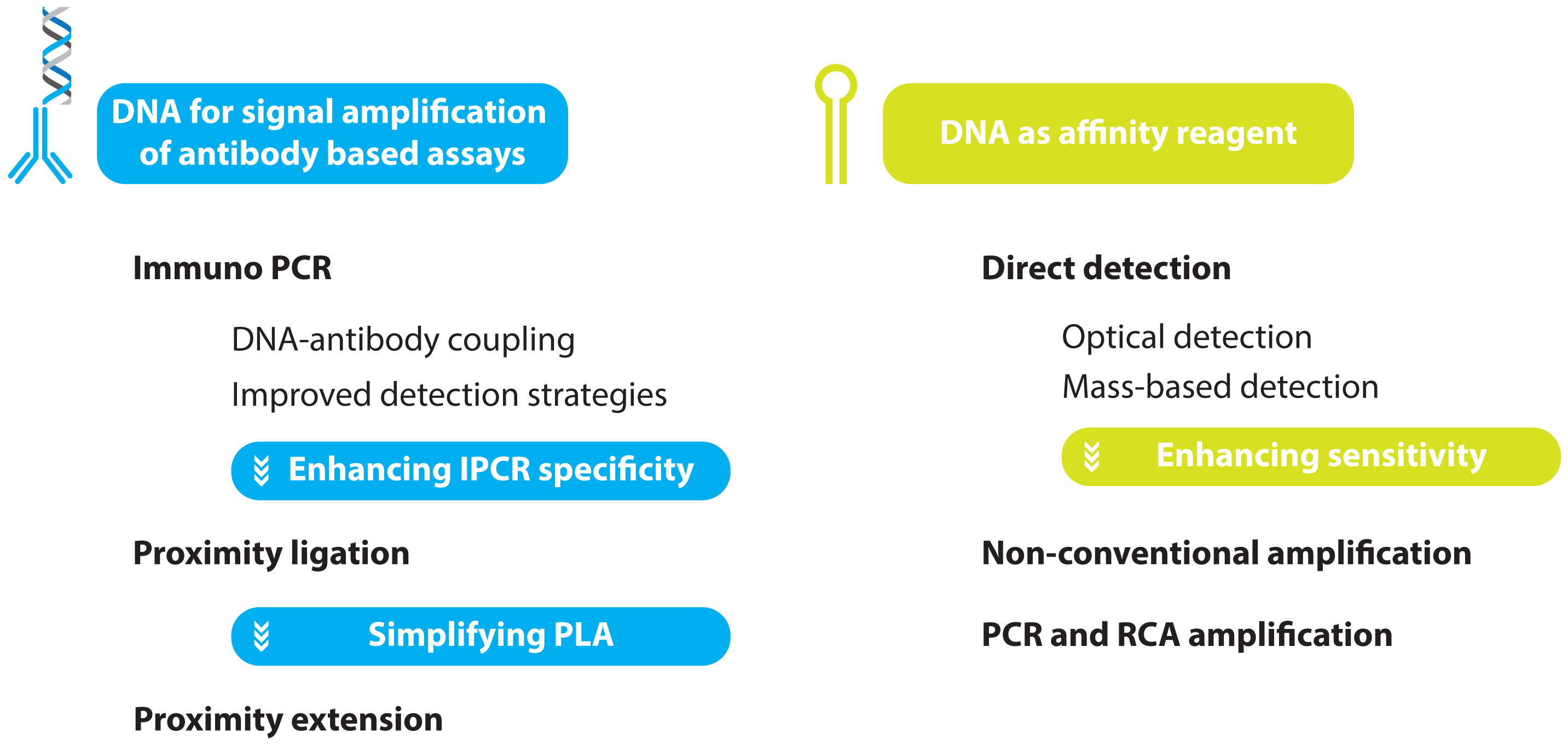
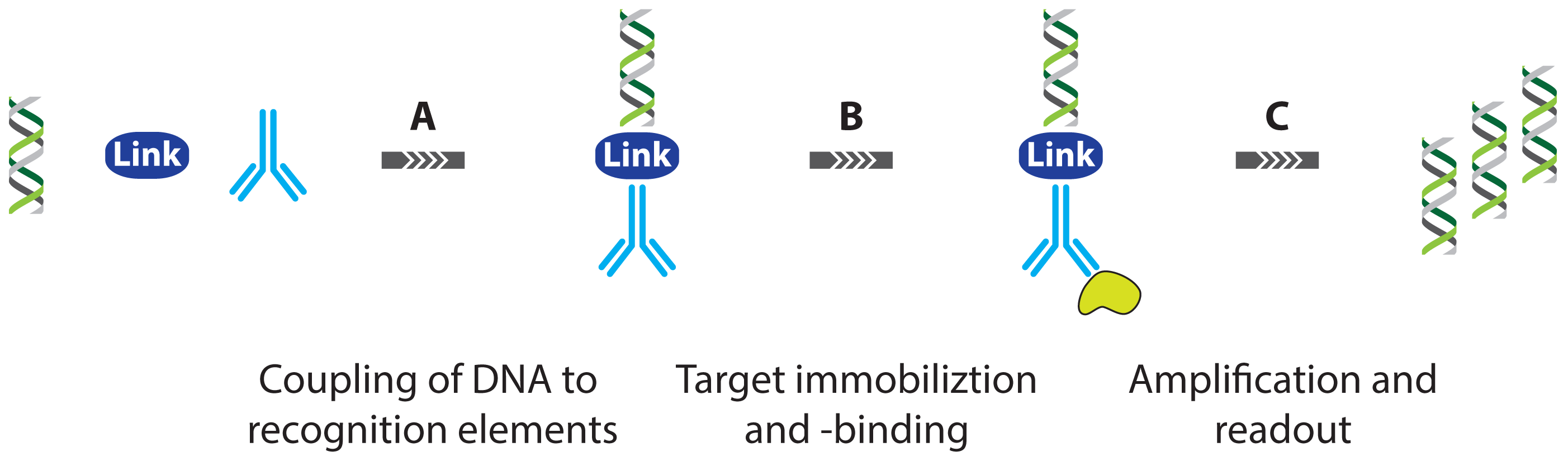
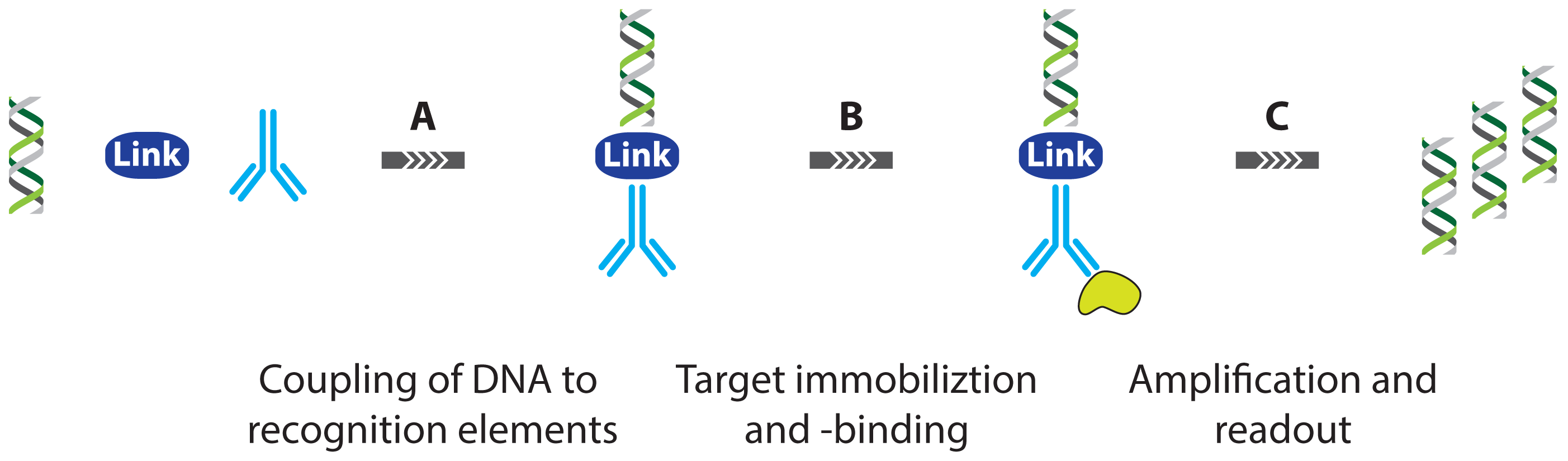
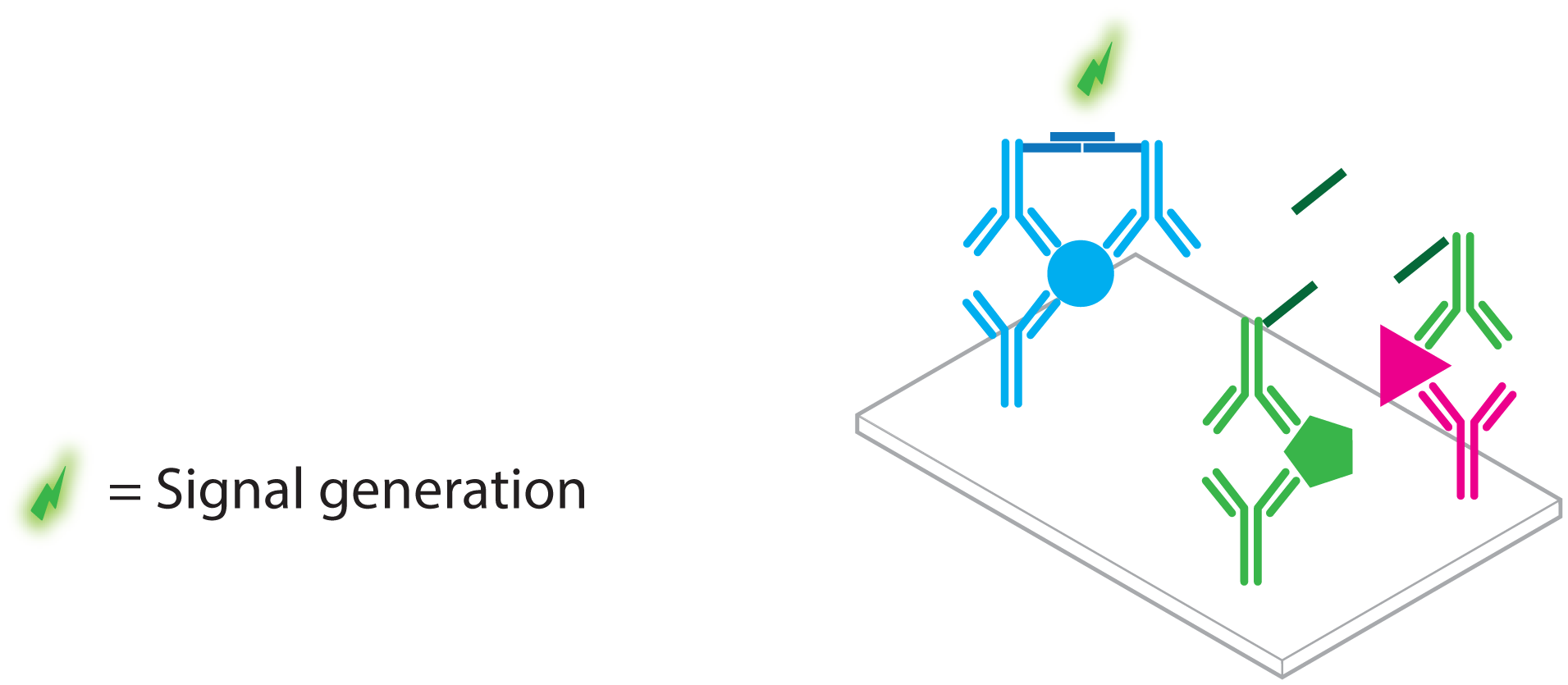
2. Protein Detection Using DNA Labels
- immuno PCR (IPCR)
- proximity ligation assay (PLA)
- proximity extension assay (PEA)
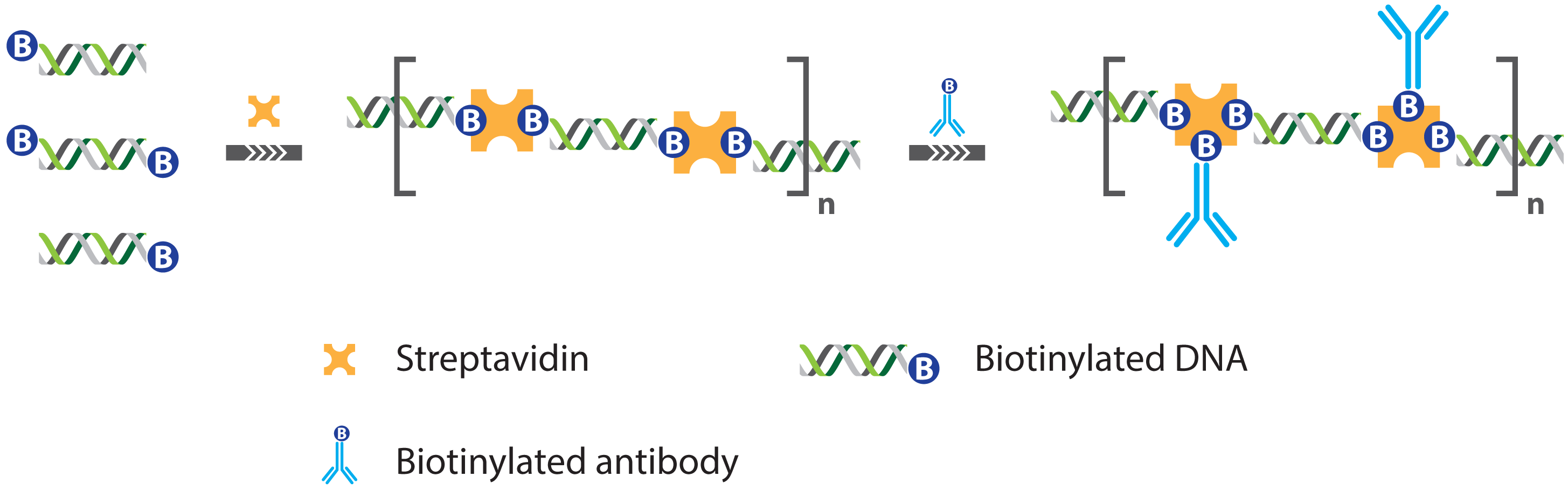
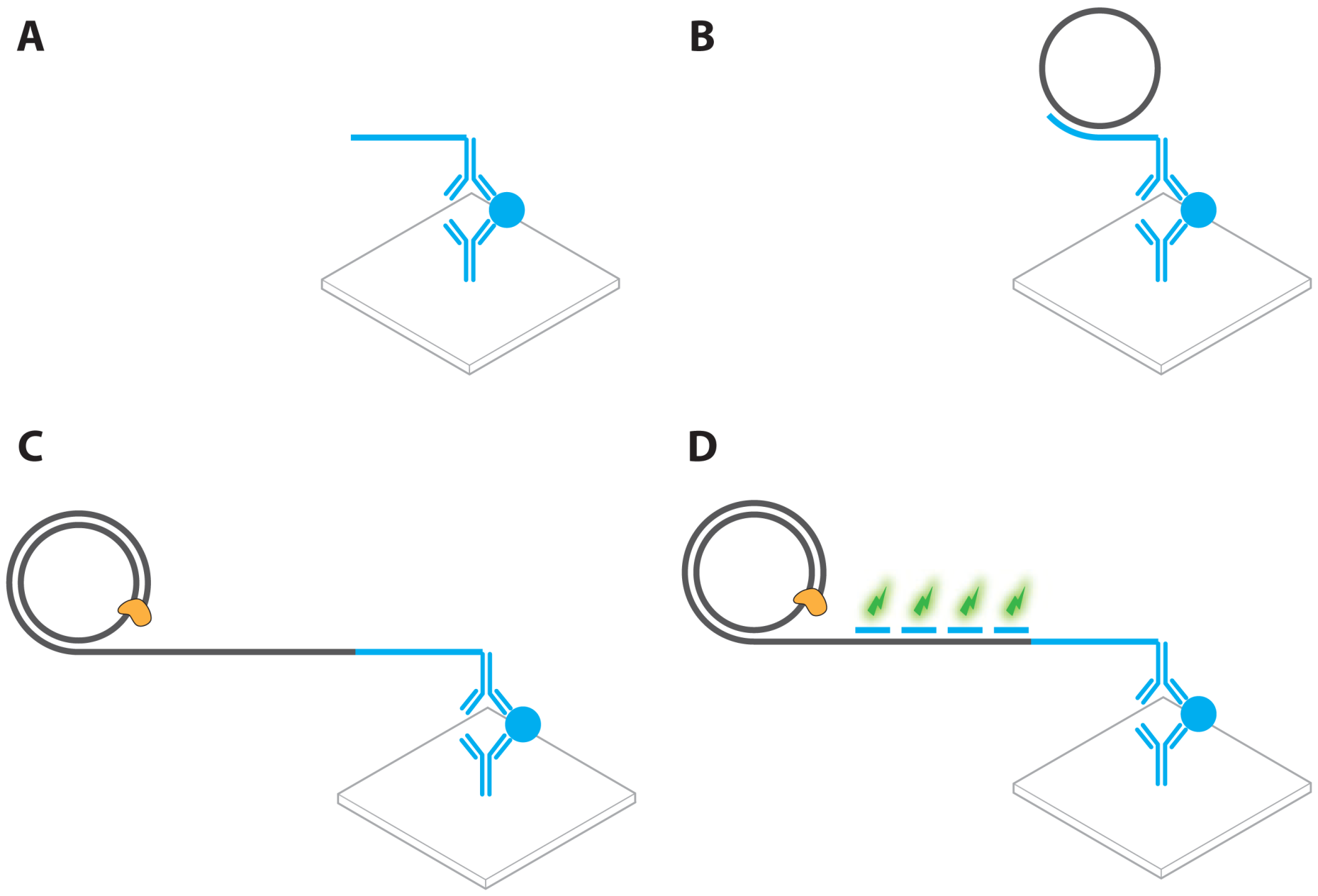
2.1. Immuno PCR and -RCA
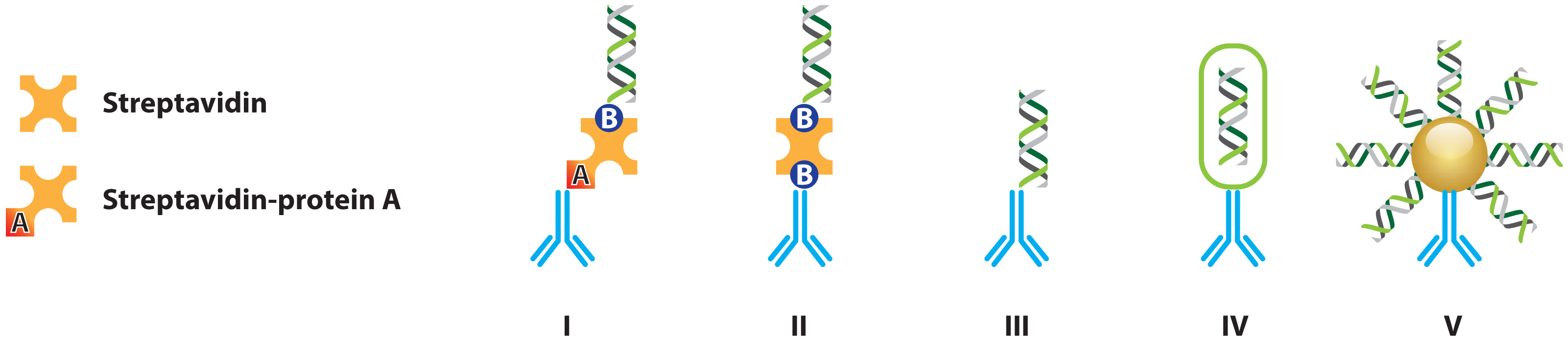
2.1.1. Improved Methods for DNA-Antibody Coupling in IPCR
2.1.2. Improved Detection Strategies in IPCR
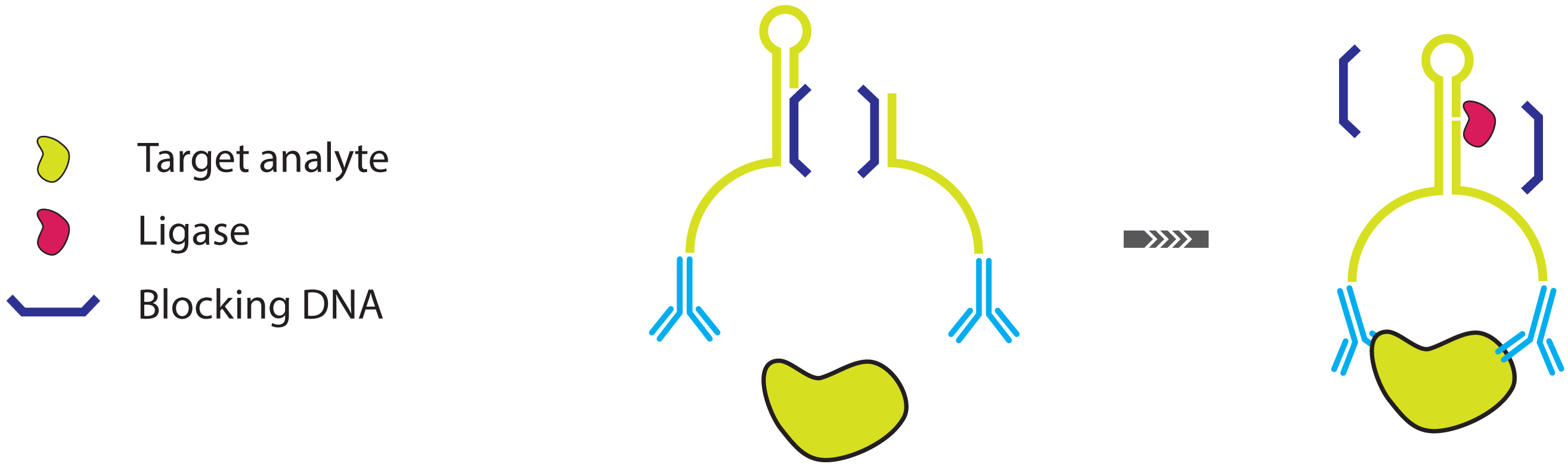
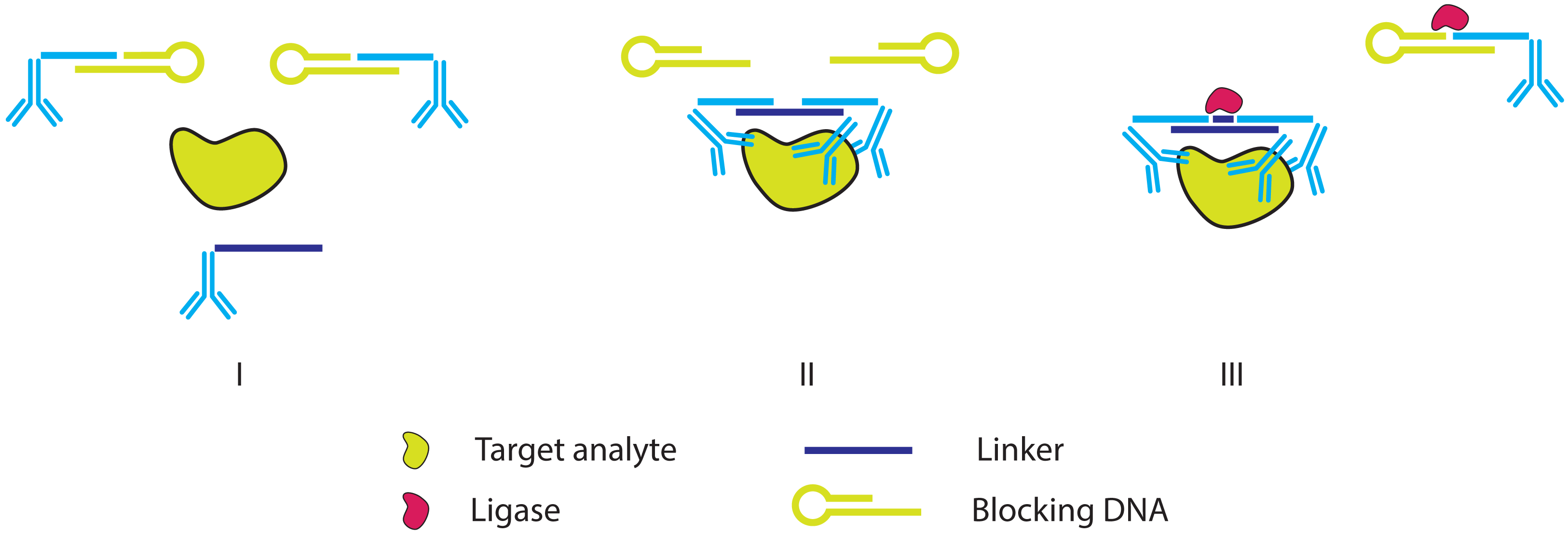
2.2. Improving the Specificity of IPCR: The Proximity Ligation Assay
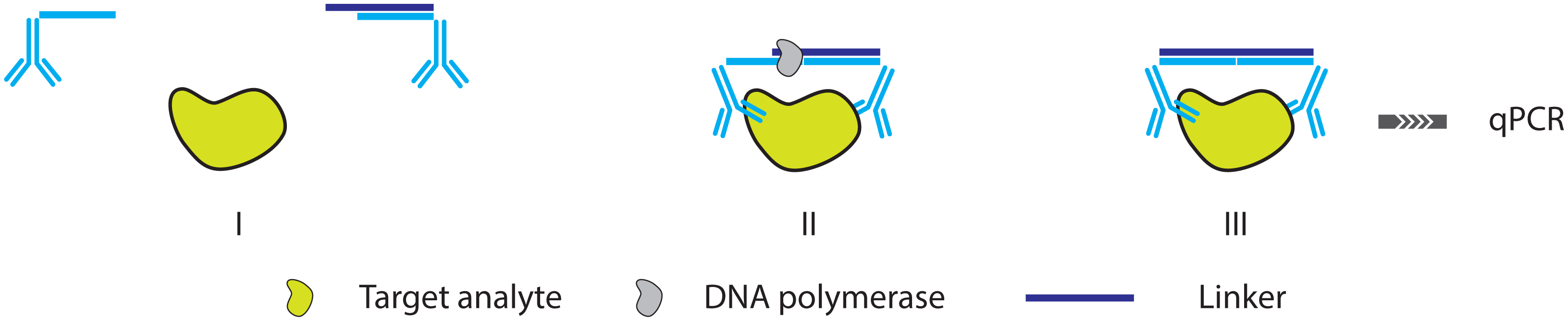
2.3. Further Simplifying IPCR and PLA: The Proximity Extension Assay
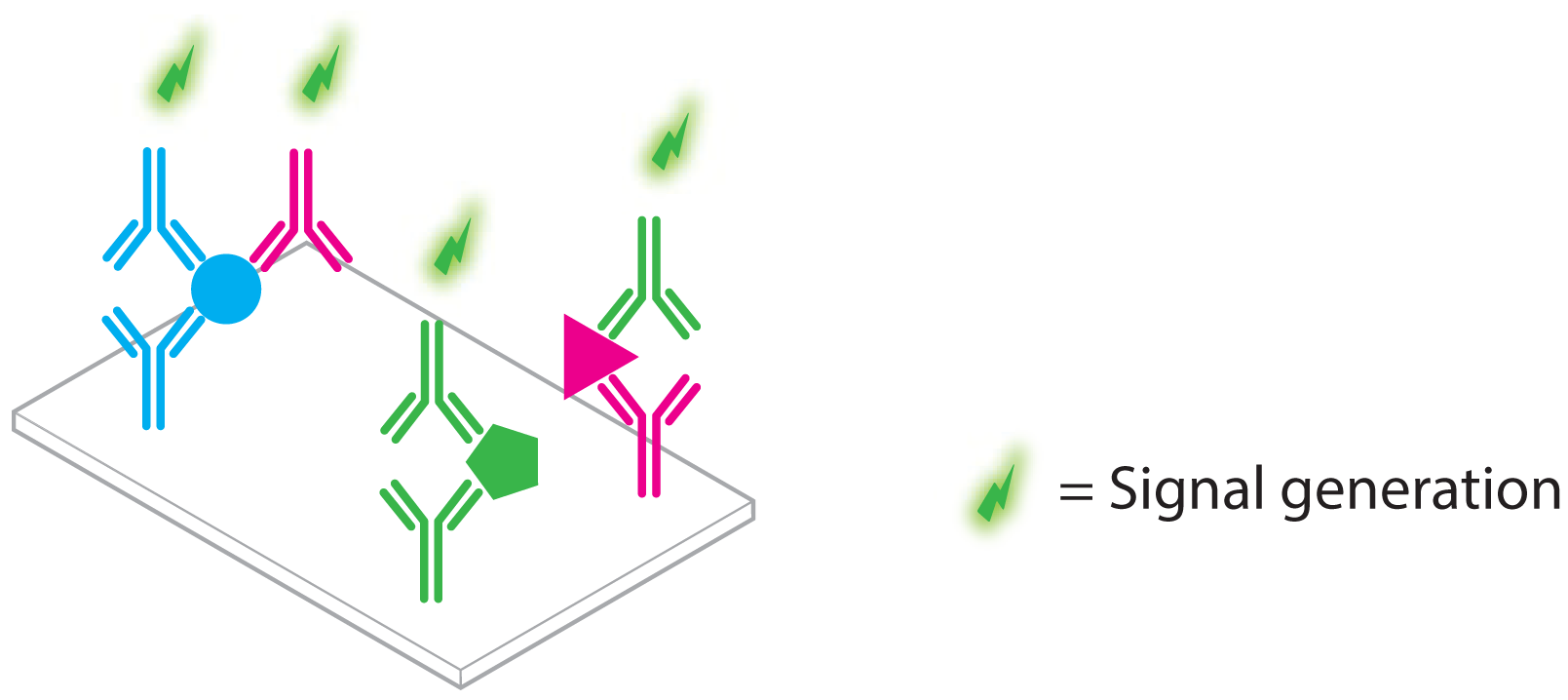
3. Aptamer Based Ultra-Sensitive Protein Asays
3.1. Non-Amplified Systems
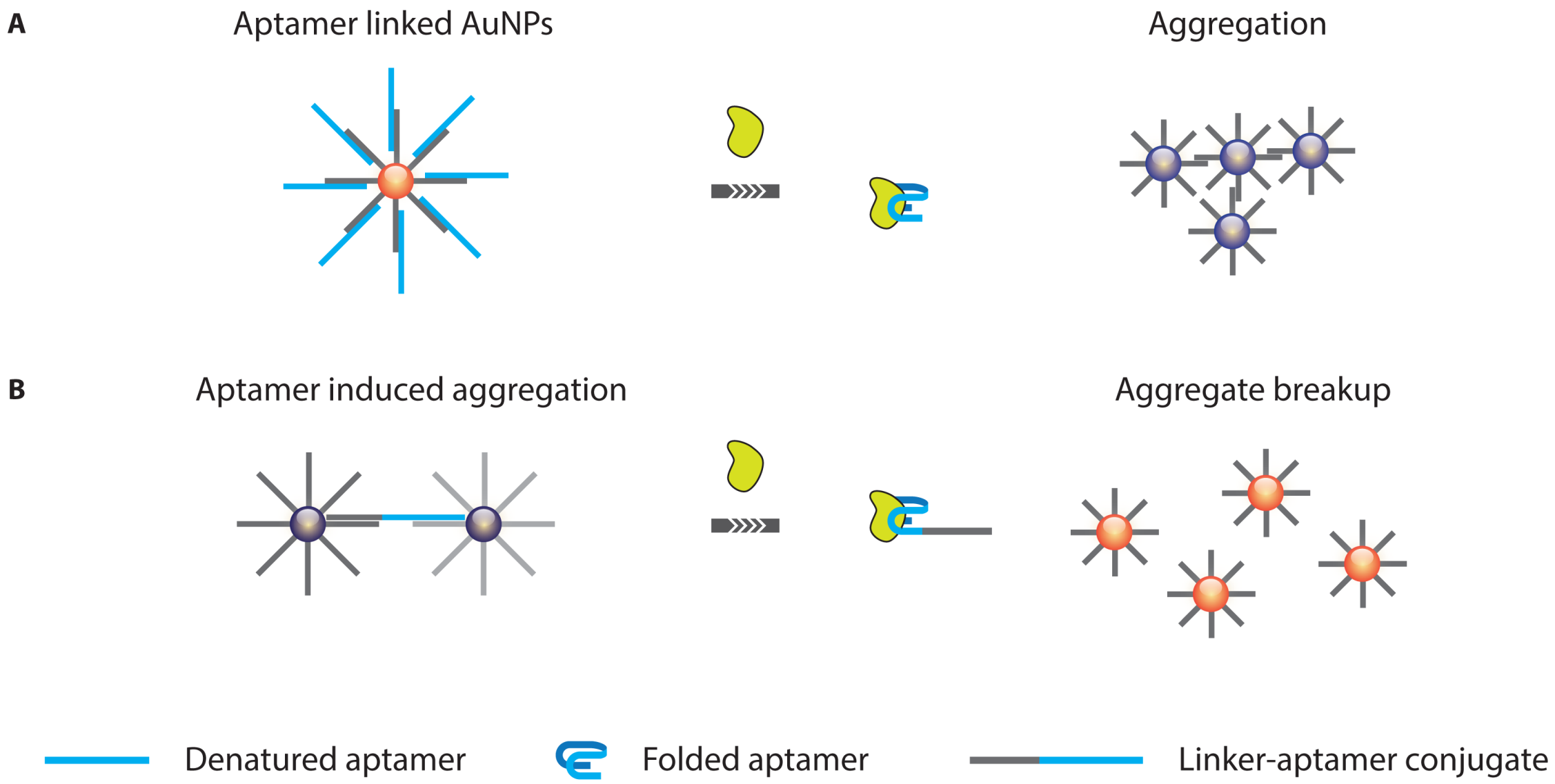
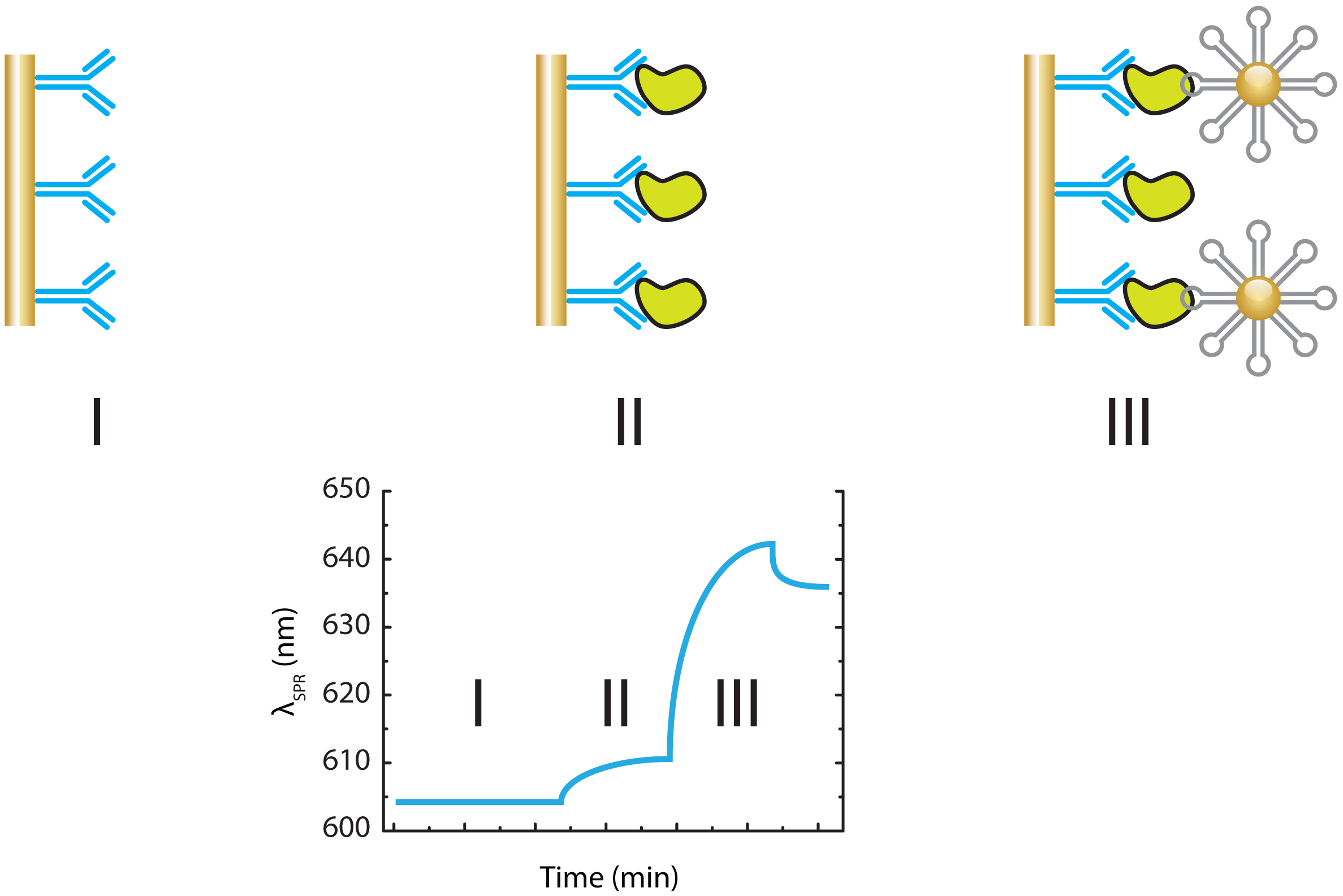
3.1.1. Optical Aptasensing Assays
3.1.2. Mass Based Aptasensing Assays
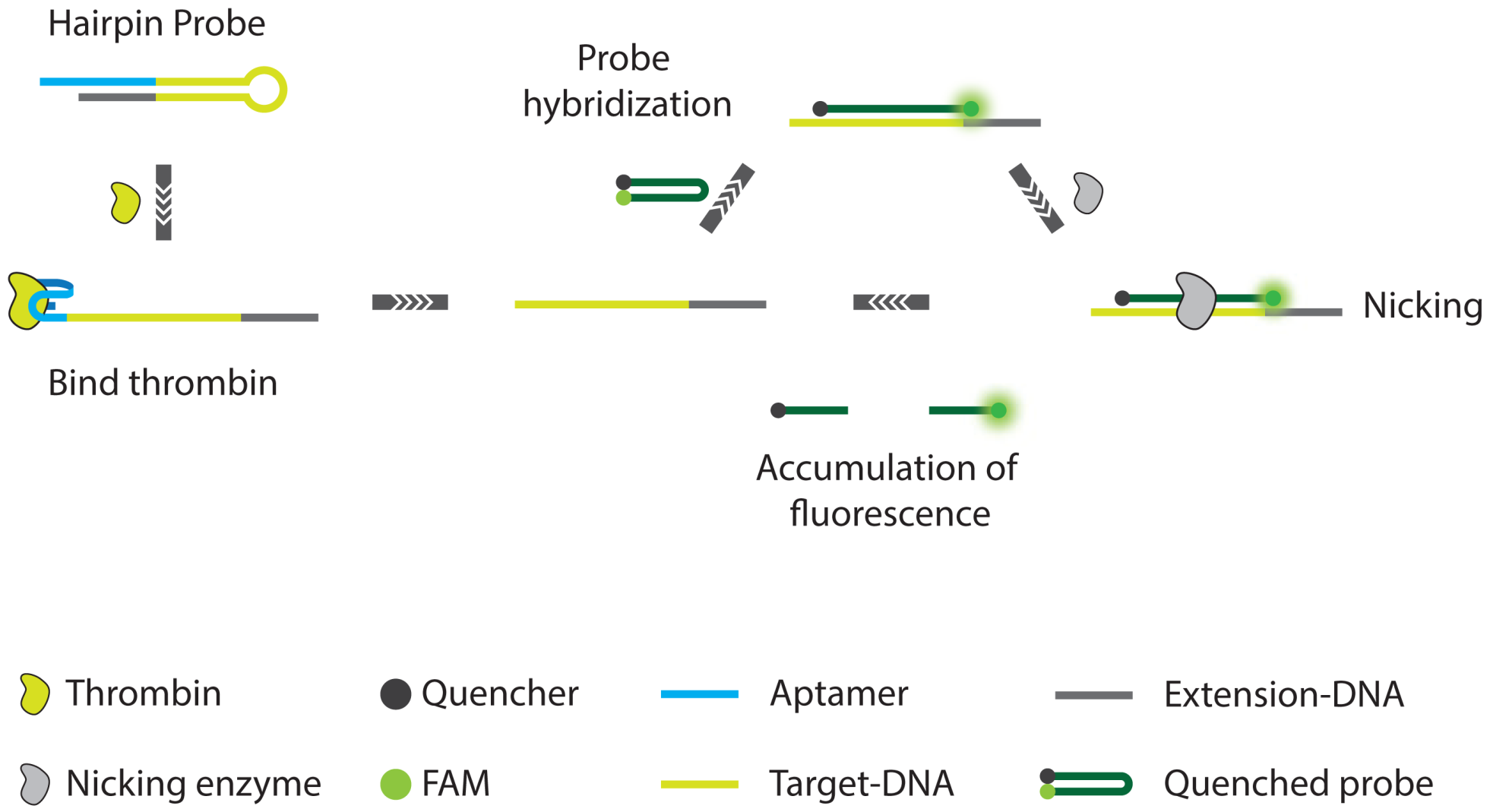
3.2. Non-PCR Signal Amplification
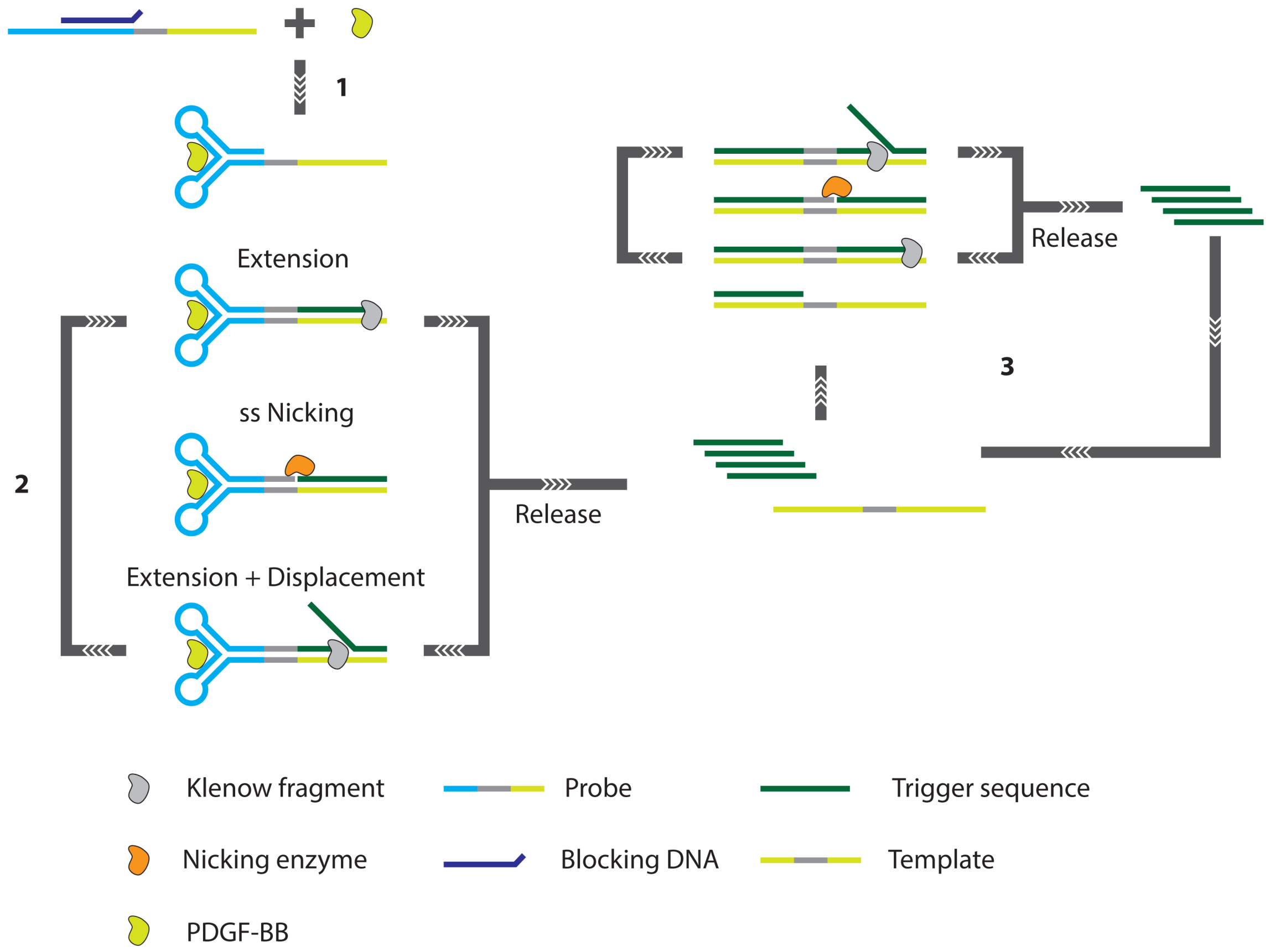
3.3. PCR and RCA Enhanced Detection Aptamer Based Protein Detection
4. Conclusions
Acknowledgments
References
- Crick, F. Central dogma of molecular biology. Nature 1970, 227, 561–563. [Google Scholar]
- Snyder, M.; Weissman, S.; Gerstein, M. Personal phenotypes to go with personal genomes. Mol. Syst. Biol. 2009, 5. [Google Scholar] [CrossRef]
- Ansong, C.; Purvine, S.O.; Adkins, J.N.; Lipton, M.S.; Smith, R.D. Proteogenomics: Needs and roles to be filled by proteomics in genome annotation. Briefings Funct. Genom. Proteom. 2008, 7, 50–62. [Google Scholar]
- Picotti, P.; Bodenmiller, B.; Mueller, L.N.; Domon, B.; Aebersold, R. Full dynamic range proteome analysis of S. cerevisiae by targeted proteomics. Cell 2009, 138, 795–806. [Google Scholar]
- Anderson, N.L. The Human plasma proteome: History, character, and diagnostic prospects. Mol. Cell. Proteomics 2002, 1, 845–867. [Google Scholar]
- Yalow, R.S.; Berson, S.A. Immunoassay of endogenous plasma insulin in man. J. Clin. Investig. 1960, 39, 1157–1175. [Google Scholar]
- Engvall, E.; Perlmann, P. Enzyme-linked immunosorbent assay (ELISA). Quantitative assay of immunoglobulin G. Immunochemistry 1971, 8, 871–874. [Google Scholar]
- Van Weemen, B.K. The rise of EIA/ELISA. Clin. Chem. 2005, 51. [Google Scholar] [CrossRef]
- Lequin, R.M. Enzyme immunoassay (EIA)/enzyme-linked immunosorbent assay (ELISA). Clin. Chem. 2005, 51, 2415–2418. [Google Scholar]
- Yates, J.R.; Ruse, C.I.; Nakorchevsky, A. Proteomics by mass spectrometry: Approaches, advances, and applications. Ann. Rev. Biomed. Eng. 2009, 11, 49–79. [Google Scholar]
- Gstaiger, M.; Aebersold, R. Applying mass spectrometry-based proteomics to genetics, genomics and network biology. Nat. Rev. Genet. 2009, 10, 617–627. [Google Scholar]
- Nong, R.Y.; Gu, J.; Darmanis, S.; Kamali-Moghaddam, M.; Landegren, U. DNA-assisted protein detection technologies. Exp. Rev. Proteom. 2012, 9, 21–32. [Google Scholar]
- Saiki, R.K.; Scharf, S.; Faloona, F.; Mullis, K.B.; Horn, G.T.; Erlich, H.A.; Arnheim, N. Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 1985, 230, 1350–1354. [Google Scholar]
- Saiki, R.K.; Gelfand, D.H.; Stoffel, S.; Scharf, S.J.; Higuchi, R.; Horn, G.T.; Mullis, K.B.; Erlich, H.A. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 1988, 239, 487–491. [Google Scholar]
- Rittié, L.; Perbal, B. Enzymes used in molecular biology: A useful guide. J. Cell Commun. Signal. 2008, 2, 25–45. [Google Scholar]
- Fire, A.; Xu, S.Q. Rolling replication of short DNA circles. Proc. Natl. Acad. Sci. USA 1995, 92, 4641–4645. [Google Scholar]
- Banér, J.; Nilsson, M.; Mendel-Hartvig, M.; Landegren, U. Signal amplification of padlock probes by rolling circle replication. Nucleic Acids Res. 1998, 26, 5073–5078. [Google Scholar]
- Sano, T.; Smith, C.L.; Cantor, C.R. Immuno-PCR: Very sensitive antigen detection by means of specific antibody-DNA conjugates. Science 1992, 258, 120–122. [Google Scholar]
- Niemeyer, C.M.; Adler, M.; Wacker, R. Immuno-PCR: High sensitivity detection of proteins by nucleic acid amplification. Trends Biotechnol. 2005, 23, 208–216. [Google Scholar]
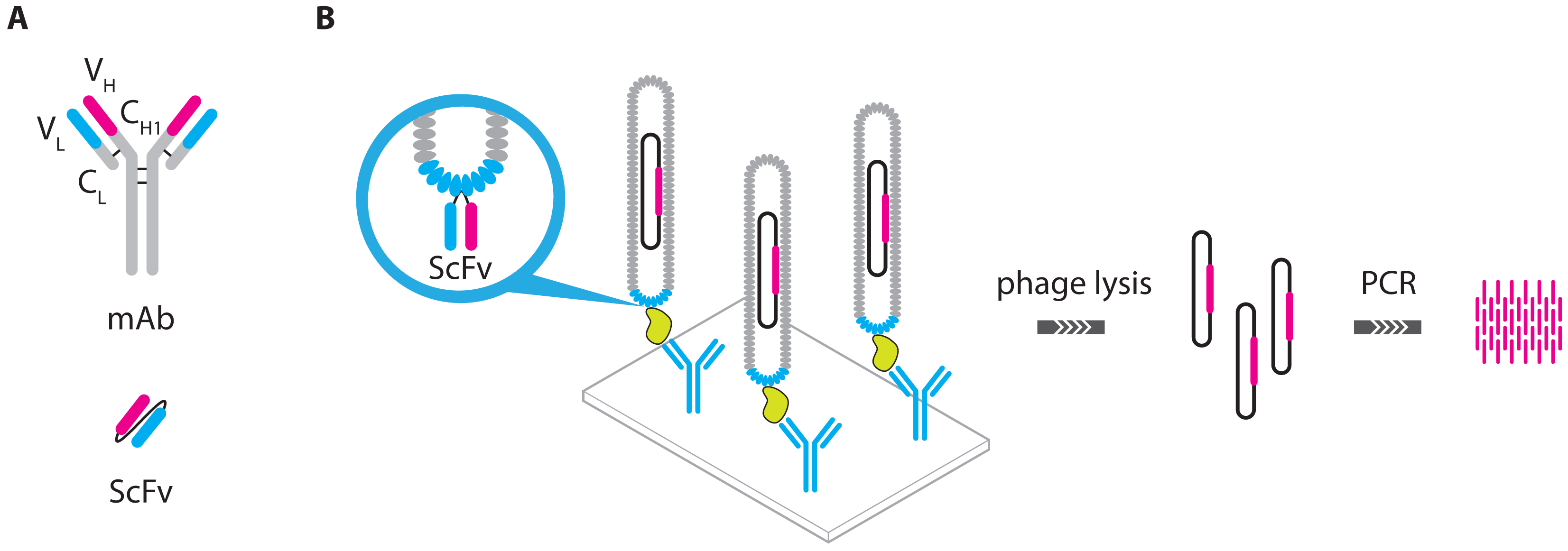
- Ren, J.; Ge, L.; Li, Y.; Bai, J.; Liu, W.C.; Si, X.M. Detection of circulating CEA molecules in human sera and leukopheresis of peripheral blood stem cells with E. coli expressed bispecific CEAScFv-streptavidin fusion protein-based immuno-PCR technique. Ann. N. Y. Acad. Sci. 2001, 945, 116–118. [Google Scholar]
- Zhou, H.; Fisher, R.J.; Papas, T.S. Universal immuno-PCR for ultra-sensitive target protein detection. Nucleic Acids Res. 1993, 21, 6038–6039. [Google Scholar]
- Verschoor, M.L.; Wilson, L.A.; Verschoor, C.P.; Singh, G. Ets-1 regulates energy metabolism in cancer cells. PLoS one 2010, 5, e13565. [Google Scholar]
- Malou, N.; Raoult, D. Immuno-PCR: A promising ultrasensitive diagnostic method to detect antigens and antibodies. Trends Microbiol. 2011, 19, 295–302. [Google Scholar]
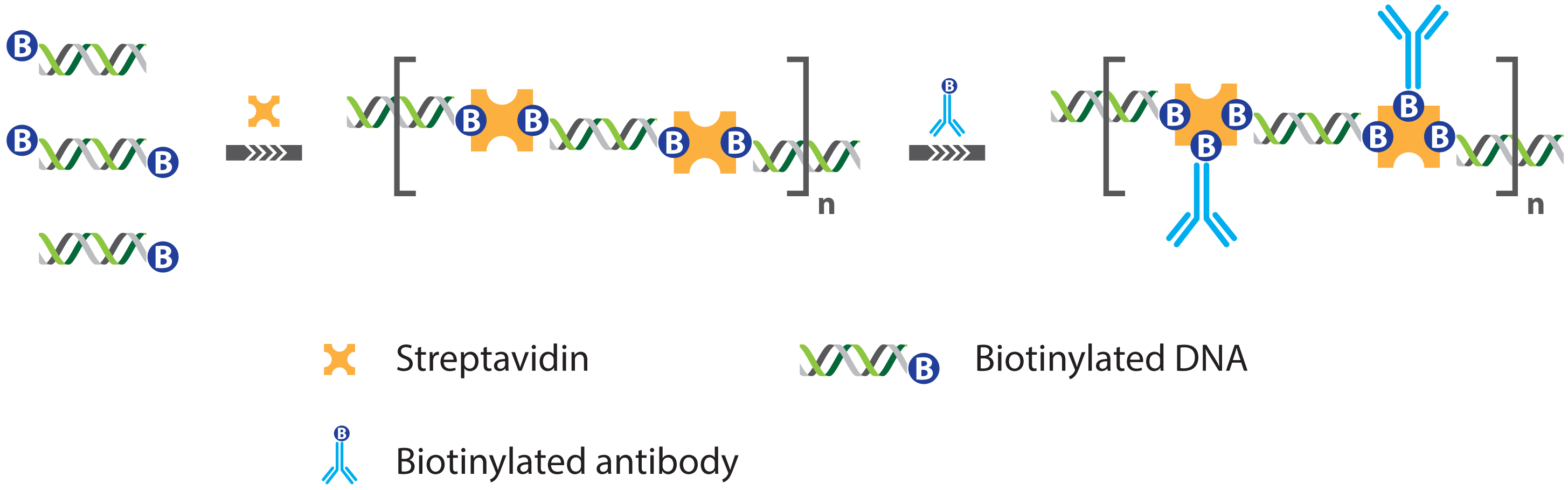
- Niemeyer, C.M.; Adler, M.; Pignataro, B.; Lenhert, S.; Gao, S.; Chi, L.; Fuchs, H.; Blohm, D. Self-assembly of DNA-streptavidin nanostructures and their use as reagents in immuno-PCR. Nucleic Acids Res. 1999, 27, 4553–4561. [Google Scholar]
- Hendrickson, E.R.; Truby, T.M.; Joerger, R.D.; Majarian, W.R.; Ebersole, R.C. High sensitivity multianalyte immunoassay using covalent DNA-labeled antibodies and polymerase chain reaction. Nucleic Acids Res. 1995, 23, 522–529. [Google Scholar]
- Sims, P.W.; Vasser, M.; Wong, W.L.; Williams, P.M.; Meng, Y.G. Immunopolymerase chain reaction using real-time polymerase chain reaction for detection. Anal. Biochem. 2000, 281, 230–232. [Google Scholar]
- Mason, J.T.; Xu, L.; Sheng, Z.m.; O'Leary, T.J. A liposome-PCR assay for the ultrasensitive detection of biological toxins. Nat. Biotechnol. 2006, 24, 555–557. [Google Scholar]
- Guo, Y.C.; Zhou, Y.F.; Zhang, X.E.; Zhang, Z.P.; Qiao, Y.M.; Bi, L.J.; Wen, J.K.; Liang, M.F.; Zhang, J.B. Phage display mediated immuno-PCR. Nucleic Acids Res. 2006, 34. [Google Scholar] [CrossRef]
- Mao, C.; Liu, A.; Cao, B. Virus-based chemical and biological sensing. Angew. Chem. 2009, 48, 6790–6810. [Google Scholar]
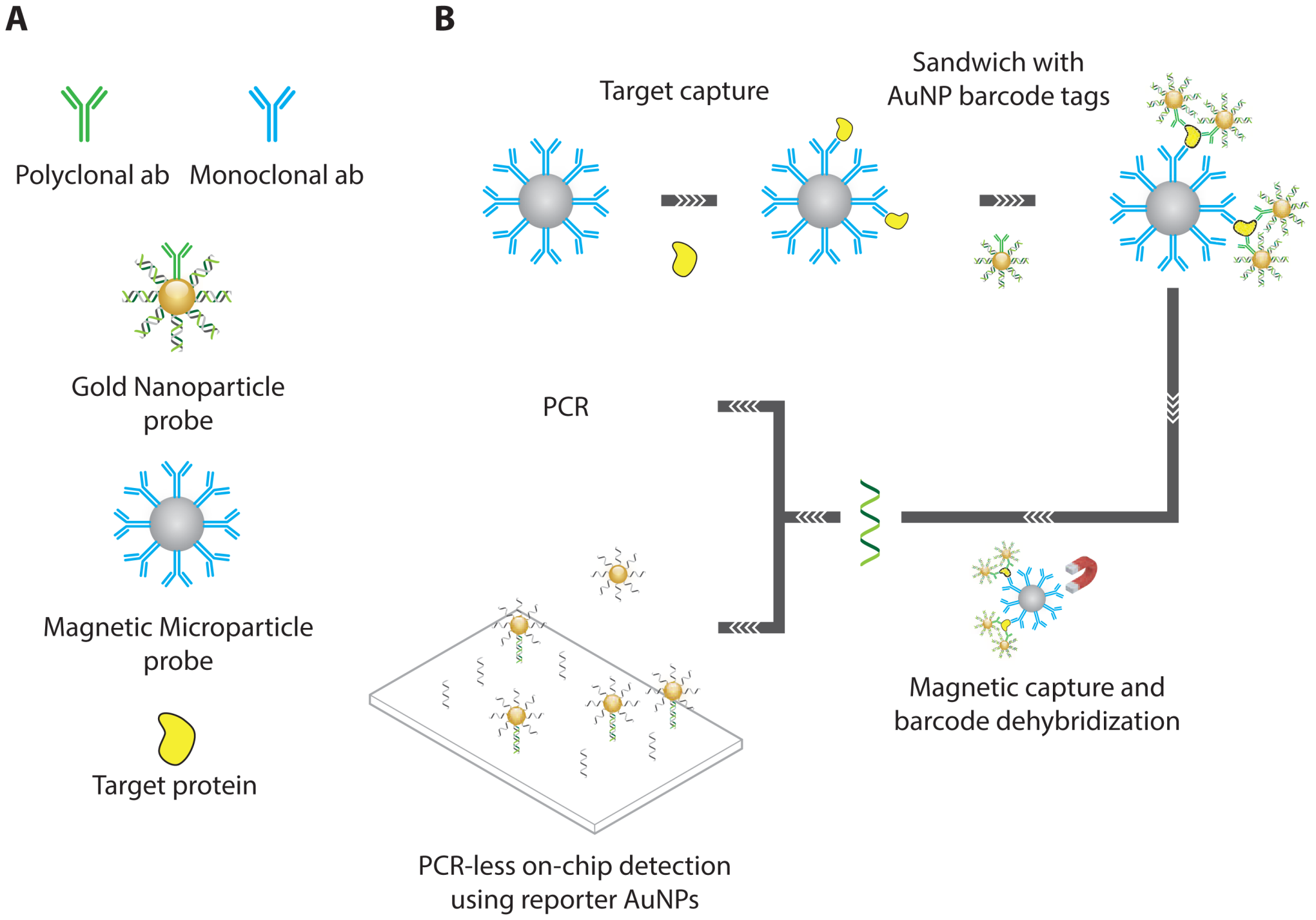
- Nam, J.M.; Thaxton, C.S.; Mirkin, C.A. Nanoparticle-based bio-bar codes for the ultrasensitive detection of proteins. Science 2003, 301, 1884–1886. [Google Scholar]
- Stoeva, S.I.; Lee, J.S.; Smith, J.E.; Rosen, S.T.; Mirkin, C.A. Multiplexed detection of protein cancer markers with biobarcoded nanoparticle probes. J. Am. Chem. Soc. 2006, 128, 8378–8379. [Google Scholar]
- Goluch, E.D.; Nam, J.M.; Georganopoulou, D.G.; Chiesl, T.N.; Shaikh, K.A.; Ryu, K.S.; Barron, A.E.; Mirkin, C.A.; Liu, C. A bio-barcode assay for on-chip attomolar-sensitivity protein detection. Lab Chip 2006, 6, 1293–1299. [Google Scholar]
- Schweitzer, B.; Wiltshire, S.; Lambert, J.; O'Malley, S.; Kukanskis, K.; Zhu, Z.; Kingsmore, S.F.; Lizardi, P.M.; Ward, D.C. Immunoassays with rolling circle DNA amplification: A versatile platform for ultrasensitive antigen detection. Proc. Natl. Acad. Sci. USA 2000, 97, 10113–10119. [Google Scholar]
- Akter, F.; Mie, M.; Kobatake, E. Immuno-rolling circle amplification using a multibinding fusion protein. Anal. Biochem. 2011, 416, 174–179. [Google Scholar]
- Gusev, Y.; Sparkowski, J.; Raghunathan, A.; Ferguson, H.; Montano, J.; Bogdan, N.; Schweitzer, B.; Wiltshire, S.; Kingsmore, S.F.; Maltzman, W.; Wheeler, V. Rolling circle amplification: A new approach to increase sensitivity for immunohistochemistry and flow cytometry. Am. J. Pathol. 2001, 159, 63–69. [Google Scholar]
- Schweitzer, B.; Roberts, S.; Grimwade, B.; Shao, W.; Wang, M.; Fu, Q.; Shu, Q.; Laroche, I.; Zhou, Z.; Tchernev, V.T.; Christiansen, J.; Velleca, M.; Kingsmore, S.F. Multiplexed protein profiling on microarrays by rolling-circle amplification. Nat. Biotechnol. 2002, 20, 359–365. [Google Scholar]
- Wiltshire, S.; O'Malley, S.; Lambert, J.; Kukanski, K.; Edgar, D.; Kingsmore, S.F.; Schweitzer, B. Detection of multiple allergen-specific IgEs on microarrays by immunoassay with rolling circle amplification. Clin. Chem. 2000, 46, 1990–1993. [Google Scholar]
- Shao, W.; Zhou, Z.; Laroche, I.; Lu, H.; Zong, Q.; Patel, D.D.; Kingsmore, S.; Piccoli, S.P. Optimization of rolling-circle amplified protein microarrays for multiplexed protein profiling. J. Biomed. Biotechnol. 2003, 2003, 299–307. [Google Scholar]
- Zhou, H.; Bouwman, K.; Schotanus, M.; Verweij, C.; Marrero, J.a.; Dillon, D.; Costa, J.; Lizardi, P.; Haab, B.B. Two-color, rolling-circle amplification on antibody microarrays for sensitive, multiplexed serum-protein measurements. Genome Biol. 2004, 5. [Google Scholar] [CrossRef]
- Ceyhan, B.; Alhorn, P.; Lang, C.; Schüler, D.; Niemeyer, C.M. Semisynthetic biogenic magnetosome nanoparticles for the detection of proteins and nucleic acids. Small 2006, 2, 1251–1255. [Google Scholar]
- Wacker, R.; Ceyhan, B.; Alhorn, P.; Schueler, D.; Lang, C.; Niemeyer, C.M. Magneto immuno-PCR: A novel immunoassay based on biogenic magnetosome nanoparticles. Biochem. Biophys. Res. Commun. 2007, 357, 391–396. [Google Scholar]
- Fredriksson, S.; Gullberg, M.; Jarvius, J.; Olsson, C.; Pietras, K.; Gústafsdóttir, S.M.; Ostman, A.; Landegren, U. Protein detection using proximity-dependent DNA ligation assays. Nat. Biotechnol. 2002, 20, 473–477. [Google Scholar]
- Gullberg, M.; Gústafsdóttir, S.M.; Schallmeiner, E.; Jarvius, J.; Bjarnegard, M.; Betsholtz, C.; Landegren, U.; Fredriksson, S. Cytokine detection by antibody-based proximity ligation. Proc. Natl. Acad. Sci. USA 2004, 101, 8420–8424. [Google Scholar]
- Zhang, H.; Li, X.F.F.; Le, X.C. Binding-induced DNA assembly and its application to yoctomole detection of proteins. Anal. Chem. 2012, 84, 877–884. [Google Scholar]
- SantaLucia, J.; Hicks, D. The thermodynamics of DNA structural motifs. Ann. Rev. Biophys. Biomol. Struct. 2004, 33, 415–440. [Google Scholar]
- Gullberg, M.; Fredriksson, S.; Taussig, M.; Jarvius, J.; Gustafsdottir, S.; Landegren, U. A sense of closeness: Protein detection by proximity ligation. Curr. Opin. Biotechnol. 2003, 14, 82–86. [Google Scholar]
- Fredriksson, S.; Dixon, W.; Ji, H.; Koong, A.C.; Mindrinos, M.; Davis, R.W. Multiplexed protein detection by proximity ligation for cancer biomarker validation. Nat. Meth. 2007, 4, 327–329. [Google Scholar]
- Darmanis, S.; Nong, R.Y.; Hammond, M.; Gu, J.; Alderborn, A.; Vänelid, J.; Siegbahn, A.; Gustafsdottir, S.; Ericsson, O.; Landegren, U.; Kamali-Moghaddam, M. Sensitive plasma protein analysis by microparticle-based proximity ligation assays. Mol. Cell. Proteom. 2010, 9, 327–335. [Google Scholar]
- Kamali-moghaddam, M.; Pettersson, F.E.; Wu, D.; Englund, H.; Darmanis, S.; Lord, A.; Tavoosidana, G.; Sehlin, D.; Gustafsdottir, S.; Nilsson, L.N.G.; Lannfelt, L.; Landegren, U. Sensitive detection of Ab protofibrils by proximity ligation-relevance for Alzheimers disease. BMC Neurosci. 2010, 11, 1–7. [Google Scholar]
- Schallmeiner, E.; Oksanen, E.; Ericsson, O. Sensitive protein detection via triple-binder proximity ligation assays. Nat. Meth. 2007, 4, 135–137. [Google Scholar]
- Darmanis, S.; Nong, R.Y.; Vänelid, J.; Siegbahn, A.; Ericsson, O.; Fredriksson, S.; Bäcklin, C.; Gut, M.; Heath, S.; Gut, I.G.; Wallentin, L.; Gustafsson, M.G.; Kamali-Moghaddam, M.; Landegren, U. ProteinSeq: High-performance proteomic analyses by proximity ligation and next generation sequencing. PLoS ONE 2011, 6, e25583. [Google Scholar]
- Göransson, J.; Ke, R.; Nong, R.Y.; Howell, W.M.; Karman, A.; Grawé, J.; Stenberg, J.; Granberg, M.; Elgh, M.; Herthnek, D.; Wikström, P.; Jarvius, J.; Nilsson, M. Rapid identification of bio-molecules applied for detection of biosecurity agents using rolling circle amplification. PLoS ONE 2012, 7, e31068. [Google Scholar]
- Lundberg, M.; Eriksson, A.; Tran, B.; Assarsson, E.; Fredriksson, S. Homogeneous antibody-based proximity extension assays provide sensitive and specific detection of low-abundant proteins in human blood. Nucleic Acids Res. 2011, 39. [Google Scholar] [CrossRef]
- Di Giusto, D.A.; Wlassoff, W.A.; Gooding, J.J.; Messerle, B.A.; King, G.C. Proximity extension of circular DNA aptamers with real-time protein detection. Nucleic Acids Res. 2005, 33. [Google Scholar] [CrossRef]
- McGregor, L.M.; Gorin, D.J.; Dumelin, C.E.; Liu, D.R. Interaction-dependent PCR: Identification of ligand-target pairs from libraries of ligands and libraries of targets in a single solution-phase experiment. J. Am. Chem. Soc. 2010, 132, 15522–15524. [Google Scholar]
- Cho, E.J.; Lee, J.W.; Ellington, A.D. Applications of aptamers as sensors. Ann. Rev. Anal. Chem. 2009, 2, 241–264. [Google Scholar]
- Sassolas, A.; Blum, L.J.; Leca-Bouvier, B.D. Homogeneous assays using aptamers. Analyst 2011, 136, 257–274. [Google Scholar]
- Hermann, T. Adaptive recognition by nucleic acid aptamers. Science 2000, 287, 820–825. [Google Scholar]
- Liu, J.; Lu, Y. Fast colorimetric sensing of adenosine and cocaine based on a general sensor design involving aptamers and nanoparticles. Angew. Chem. 2005, 45, 90–94. [Google Scholar]
- Huang, C.C.; Huang, Y.F.; Cao, Z.; Tan, W.; Chang, H.T. Aptamer-modified gold nanoparticles for colorimetric determination of platelet-derived growth factors and their receptors. Anal. Chem. 2005, 77, 5735–5741. [Google Scholar]
- Liu, J.; Mazumdar, D.; Lu, Y. A simple and sensitive “dipstick” test in serum based on lateral flow separation of aptamer-linked nanostructures. Angew. Chem. 2006, 45, 7955–7959. [Google Scholar]
- Wei, H.; Li, B.; Li, J.; Wang, E.; Dong, S. Simple and sensitive aptamer-based colorimetric sensing of protein using unmodified gold nanoparticle probes. Chem. Commun. 2007, 3735–3737. [Google Scholar]
- Saha, K.; Agasti, S.S.; Kim, C.; Li, X.; Rotello, V.M. Gold nanoparticles in chemical and biological sensing. Chem. Rev. 2012, 112, 2739–2779. [Google Scholar]
- Zhang, H.; Li, X.F.F.; Le, X.C. Tunable aptamer capillary electrophoresis and its application to protein analysis. J. Am. Chem. Soc. 2008, 130, 34–35. [Google Scholar]
- Gokulrangan, G.; Unruh, J.R.; Holub, D.F.; Ingram, B.; Johnson, C.K.; Wilson, G.S. DNA aptamer-based bioanalysis of IgE by fluorescence anisotropy. Anal. Chem. 2005, 77, 1963–1970. [Google Scholar]
- Jhaveri, S.; Rajendran, M.; Ellington, A.D. In vitro selection of signaling aptamers. Nat. Biotechnol. 2000, 18, 1293–1297. [Google Scholar]
- Zhou, X.; Duan, R.; Xing, D. Highly sensitive detection of protein and small molecules based on aptamer-modified electrochemiluminescence nanoprobe. Analyst 2012, 137, 1963–1969. [Google Scholar]
- Pollet, J.; Strych, U.; Willson, R.C. A peroxidase-active aptazyme as an isothermally amplifiable label in an aptazyme-linked oligonucleotide assay for low-picomolar IgE detection. Analyst 2012, 137, 5710–5712. [Google Scholar]
- Sassolas, A.; Blum, L.J.; Leca-Bouvier, B.D. Optical detection systems using immobilized aptamers. Biosens. Bioelectron. 2011, 26, 3725–3736. [Google Scholar]
- Liss, M.; Petersen, B.; Wolf, H.; Prohaska, E. An aptamer-based quartz crystal protein biosensor. Anal. Chem. 2002, 74, 4488–95. [Google Scholar]
- Minunni, M.; Tombelli, S.; Gullotto, a.; Luzi, E.; Mascini, M. Development of biosensors with aptamers as bio-recognition element: The case of HIV-1 Tat protein. Biosens. Bioelectron. 2004, 20, 1149–1156. [Google Scholar]
- Tombelli, S.; Minunni, M.; Luzi, E.; Mascini, M. Aptamer-based biosensors for the detection of HIV-1 Tat protein. Bioelectrochemistry 2005, 67, 135–141. [Google Scholar]
- Nakamura, C.; Kobayashi, T. Usage of a DNA aptamer as a ligand targeting microcystin. Mol. Cryst. Liq. Cryst. Sci. Tech. A 2001, 371, 369–374. [Google Scholar]
- Hasegawa, H.; Taira, K.I.; Sode, K.; Ikebukuro, K.; Science, L. Improvement of aptamer affinity by dimerization. Sensors 2008, 8, 1090–1098. [Google Scholar]
- Pollet, J.; Delport, F.; Janssen, K.P.F.; Jans, K.; Maes, G.; Pfeiffer, H.; Wevers, M.; Lammertyn, J. Fiber optic SPR biosensing of DNA hybridization and DNAprotein interactions. Biosens. Bioelectron. 2009, 25, 864–869. [Google Scholar]
- Tran, D.T.; Vermeeren, V.; Grieten, L.; Wenmackers, S.; Wagner, P.; Pollet, J.; Janssen, K.P.F.; Michiels, L.; Lammertyn, J. Nanocrystalline diamond impedimetric aptasensor for the label-free detection of human IgE. Biosens. Bioelectron. 2011, 26, 2987–1793. [Google Scholar]
- Bini, A.; Minunni, M.; Tombelli, S.; Centi, S.; Mascini, M. Analytical performances of aptamer-based sensing for thrombin detection. Anal. Chem. 2007, 79, 3016–3019. [Google Scholar]
- Strehlitz, B.; Nikolaus, N.; Stoltenburg, R. Protein detection with aptamer biosensors. Sensors 2008, 8, 4296–4307. [Google Scholar]
- Lee, S.J.; Youn, B.S.; Park, J.W.; Niazi, J.H.; Kim, Y.S.; Gu, M.B. ssDNA aptamer-based surface plasmon resonance biosensor for the detection of retinol binding protein 4 for the early diagnosis of type 2 diabetes. Anal. Chem. 2008, 80, 2867–2873. [Google Scholar]
- Wang, J.; Munir, A.; Li, Z.; Zhou, H.S. Aptamer-Au NPs conjugates-enhanced SPR sensing for the ultrasensitive sandwich immunoassay. Biosens. Bioelectron. 2009, 25, 124–129. [Google Scholar]
- Li, J.; Fu, H.E.; Wu, L.J.; Zheng, A.X.; Chen, G.N.; Yang, H.H. General colorimetric detection of proteins and small molecules based on cyclic enzymatic signal amplification and hairpin aptamer probe. Anal. Chem. 2012, 84, 5309–5315. [Google Scholar]
- Xue, L.; Zhou, X.; Xing, D. Highly sensitive protein detection based on aptamer probe and isothermal nicking enzyme assisted fluorescence signal amplification. Chem. Commun. 2010, 46, 7373–7375. [Google Scholar]
- Xue, L.; Zhou, X.; Xing, D. Sensitive and homogeneous protein detection based on target-triggered aptamer hairpin switch and nicking enzyme assisted fluorescence signal amplification. Anal. Chem. 2012, 84, 3507–3513. [Google Scholar]
- Zhang, Z.Z.; Zhang, C.Y. Highly sensitive detection of protein with aptamer-based target-triggering two-stage amplification. Anal. Chem. 2012, 84, 1623–1629. [Google Scholar]
- Yoshida, Y.; Horii, K.; Sakai, N.; Masuda, H.; Furuichi, M.; Waga, I. Antibody-specific aptamer-based PCR analysis for sensitive protein detection. Anal. Bioanal. Chem. 2009, 395, 1089–1096. [Google Scholar]
- Liao, S.; Liu, Y.; Zeng, J.; Li, X.; Shao, N.; Mao, A.; Wang, L.; Ma, J.; Cen, H.; Wang, Y.; Zhang, X.; Zhang, R.; Wei, Z.; Wang, X. Aptamer-based sensitive detection of target molecules via RT-PCR signal amplification. Bioconjug. Chem. 2010, 21, 2183–2189. [Google Scholar]
- Fischer, N.O.; Tarasow, T.M.; Tok, J.B.H. Protein detection via direct enzymatic amplification of short DNA aptamers. Anal. Biochem. 2008, 373, 121–128. [Google Scholar]
- Pinto, A.; Lennarz, S.; Rodrigues-Correia, A.; Heckel, A.; O'Sullivan, C.K.; Mayer, G. Functional detection of proteins by caged aptamers. ACS Chem. Biol. 2012, 7, 360–366. [Google Scholar]
- Yang, L.; Ellington, A.D. Real-time PCR detection of protein analytes with conformation switching aptamers. Anal. Biochem. 2008, 380, 164–173. [Google Scholar]
- Wu, Z.S.; Zhang, S.; Zhou, H.; Shen, G.L.; Yu, R. Universal aptameric system for highly sensitive detection of protein based on structure-switching-triggered rolling circle amplification. Anal. Chem. 2010, 82, 2221–2227. [Google Scholar]
- Zhang, H.; Wang, Z.; Li, X.F.; Le, X.C. Ultrasensitive detection of proteins by amplification of affinity aptamers. Angew. Chem. 2006, 45, 1576–1580. [Google Scholar]
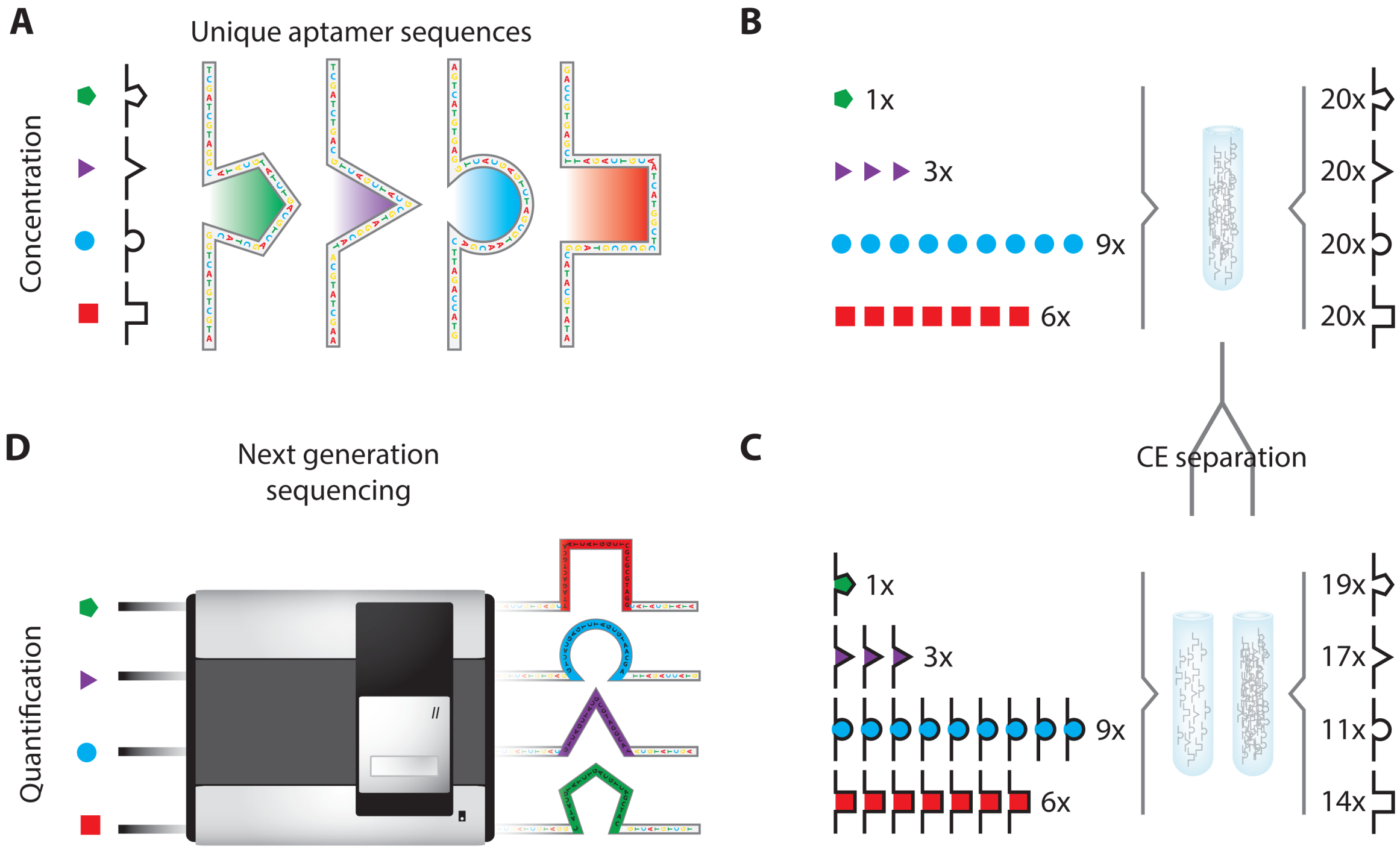
- Janssen, K.P.F.; Knez, K.; Pollet, J.; Roberts, S.J.; Schrooten, J.; Lammertyn, J. Assay design considerations for use of affinity aptamer amplification in ultra-sensitive protein assays using capillary electrophoresis. Anal. Meth. 2011, 3, 2156–2159. [Google Scholar]
- Janssen, K.P.F.; Knez, K.; Spasic, D.; Schrooten, J.; Lammertyn, J. Multiplexed protein detection using an affinity aptamer amplification assay. Anal. Bioanal. Chem. 2012, 404, 2073–2081. [Google Scholar]
- Turner, D.J.D.J.; Tuytten, R.; Janssen, K.P.F.; Lammertyn, J.; Wuyts, J.; Pollet, J.; Eyckerman, S.; Brown, C.; Kas, K. Toward clinical proteomics on a next-generation sequencing platform. Anal. Chem. 2011, 83, 666–670. [Google Scholar]
- Wang, X.L.; Li, F.; Su, Y.H.; Sun, X.; Li, X.B.; Schluesener, H.J.; Tang, F.; Xu, S.Q. Ultrasensitive detection of protein using an aptamer-based exonuclease protection assay. Anal. Chem. 2004, 76, 5605–5610. [Google Scholar]
- Zheng, D.; Zou, R.; Lou, X. Label-free fluorescent detection of ions, proteins, and small molecules using structure-switching aptamers, SYBR Gold, and exonuclease I. Anal. Chem. 2012, 84, 3554–3560. [Google Scholar]
- Qiu, L.P.; Wu, Z.S.; Shen, G.L.; Yu, R.Q. Highly sensitive and selective bifunctional oligonucleotide probe for homogeneous parallel fluorescence detection of protein and nucleotide sequence. Anal. Chem. 2011, 83, 3050–3057. [Google Scholar]
- Di Giusto, D.A.; King, G.C. Construction, stability, and activity of multivalent circular anticoagulant aptamers. J. Biol. Chem. 2004, 279, 46483–46489. [Google Scholar]
- Cheglakov, Z.; Weizmann, Y.; Basnar, B.; Willner, I. Diagnosing viruses by the rolling circle amplified synthesis of DNAzymes. Org. Biomol. Chem. 2007, 5, 223–225. [Google Scholar]
- Cheglakov, Z.; Weizmann, Y.; Braunschweig, A.B.; Wilner, O.I.; Willner, I. Increasing the complexity of periodic protein nanostructures by the rolling-circle-amplified synthesis of aptamers. Angew. Chem. 2008, 47, 126–30. [Google Scholar]
- Furukawa, K.; Abe, H.; Abe, N.; Harada, M.; Tsuneda, S.; Ito, Y. Fluorescence generation from tandem repeats of a malachite green RNA aptamer using rolling circle transcription. Bioorg. Med. Chem. Lett. 2008, 18, 4562–4565. [Google Scholar]
- Baird, G.S. Where are all the aptamers? Am. J. Clin. Pathol. 2010, 134, 529–531. [Google Scholar]
- Gold, L.; Ayers, D.; Bertino, J.; Bock, C.; Bock, A.; Brody, E.N.; Carter, J.; Dalby, A.B.; Eaton, B.E.; Fitzwater, T.; et al. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS ONE 2010, 5, e15004. [Google Scholar]
- Vaught, J.D.; Bock, C.; Carter, J.; Fitzwater, T.; Otis, M.; Schneider, D.; Rolando, J.; Waugh, S.; Wilcox, S.K.; Eaton, B.E. Expanding the chemistry of DNA for in vitro selection. J. Am. Chem. Soc. 2010, 132, 4141–4151. [Google Scholar]
- Cox, J.C.; Ellington, A.D. Automated selection of anti-protein aptamers. Bioorg. Med. Chem. 2001, 9, 2525–2531. [Google Scholar]
- Sooter, L.J.; Riedel, T.; Davidson, E.A.; Levy, M.; Cox, J.C.; Ellington, A.D. Toward automated nucleic acid enzyme selection. Biol. Chem. 2001, 382, 1327–1334. [Google Scholar]
- Keeney, T.R.; Bock, C.; Gold, L.; Kraemer, S.; Lollo, B.; Nikrad, M.; Stanton, M.; Stewart, A.; Vaught, J.D.; Walker, J.J. Automation of the somaLogic proteomics assay: A platform for biomarker discovery. J. Assoc. Lab. Autom. 2009, 14, 360–366. [Google Scholar]
- Cho, M.; Xiao, Y.; Nie, J.; Stewart, R.; Csordas, A.T.; Oh, S.S.; Thomson, J.A.; Soh, H.T. Quantitative selection of DNA aptamers through microfluidic selection and high-throughput sequencing. Proc. Natl. Acad. Sci. USA 2010, 107, 15373–15378. [Google Scholar]
- Famulok, M.; Mayer, G. Aptamer modules as sensors and detectors. Accounts Chem. Res. 2011, 44, 1349–1358. [Google Scholar]
- Ostroff, R.M.; Bigbee, W.L.; Franklin, W.; Gold, L.; Mehan, M.; Miller, Y.E.; Pass, H.I.; Rom, W.N.; Siegfried, J.M.; Stewart, A.; et al. Unlocking biomarker discovery: Large scale application of aptamer proteomic technology for early detection of lung cancer. PLoS ONE 2010, 5, e15003. [Google Scholar]
- Diamandis, E.P. Cancer biomarkers: Can we turn recent failures into success? J.Natl. Cancer Inst. 2010, 102, 1462–1467. [Google Scholar]
- Brody, E.; Gold, L.; Mehan, M.; Ostroff, R.; Rohloff, J.; Walker, J.; Zichi, D. Life's simple measures: Unlocking the proteome. J. Mol. Biol. 2012, 422, 595–606. [Google Scholar]
- Mehan, M.; Ostroff, R.; Wilcox, S.; Steele, F.; Schneider, D.; Jarvis, T.; Baird, G.; Gold, L.; Janjic, N. Highly multiplexed proteomic platform for biomarker discovery, diagnostics, and therapeutics. In Complement Therapeutics SE-20; Lambris, J.D., Holers, V.M., Ricklin, D., Eds.; Springer: New York, NY, USA, 2011; Volume 734, pp. 283–300. [Google Scholar]
- Landegren, U.; Vanelid, J.; Hammond, M.; Nong, R.Y.; Wu, D.; Ulleras, E.; Kamali-Moghaddam, M. Opportunities for sensitive plasma proteome analysis. Anal. Chem. 2012, 84, 1824–1830. [Google Scholar]
- Zichi, D.; Eaton, B.; Singer, B.; Gold, L. Proteomics and diagnostics: Let's get specific, again. Curr. Opin. Chem. Biol. 2008, 12, 78–85. [Google Scholar]




© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Janssen, K.P.F.; Knez, K.; Spasic, D.; Lammertyn, J. Nucleic Acids for Ultra-Sensitive Protein Detection. Sensors 2013, 13, 1353-1384. https://doi.org/10.3390/s130101353
Janssen KPF, Knez K, Spasic D, Lammertyn J. Nucleic Acids for Ultra-Sensitive Protein Detection. Sensors. 2013; 13(1):1353-1384. https://doi.org/10.3390/s130101353
Chicago/Turabian StyleJanssen, Kris P. F., Karel Knez, Dragana Spasic, and Jeroen Lammertyn. 2013. "Nucleic Acids for Ultra-Sensitive Protein Detection" Sensors 13, no. 1: 1353-1384. https://doi.org/10.3390/s130101353