Aspartate Aminotransferase (AST/GOT) and Alanine Aminotransferase (ALT/GPT) Detection Techniques

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Colorimetric Analysis
3. Spectrophotometric Measurement
4. Chemiluminescence
5. Chromatography
6. Fluorescence and UV Absorbance
7. Radiochemical Analysis
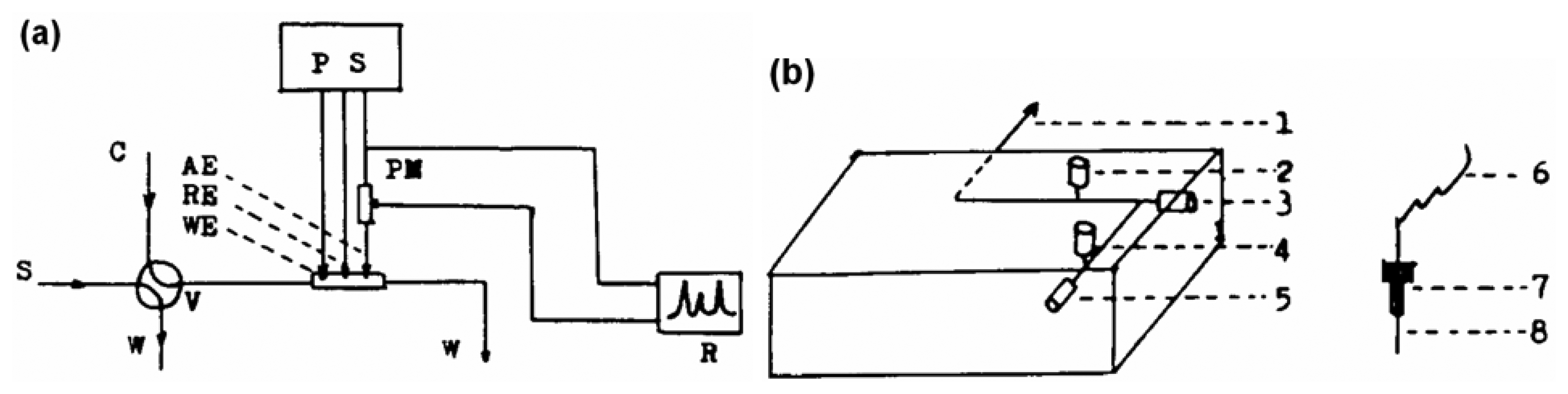
8. Electrochemical Techniques
8.1. Detection using Electrode without Modification
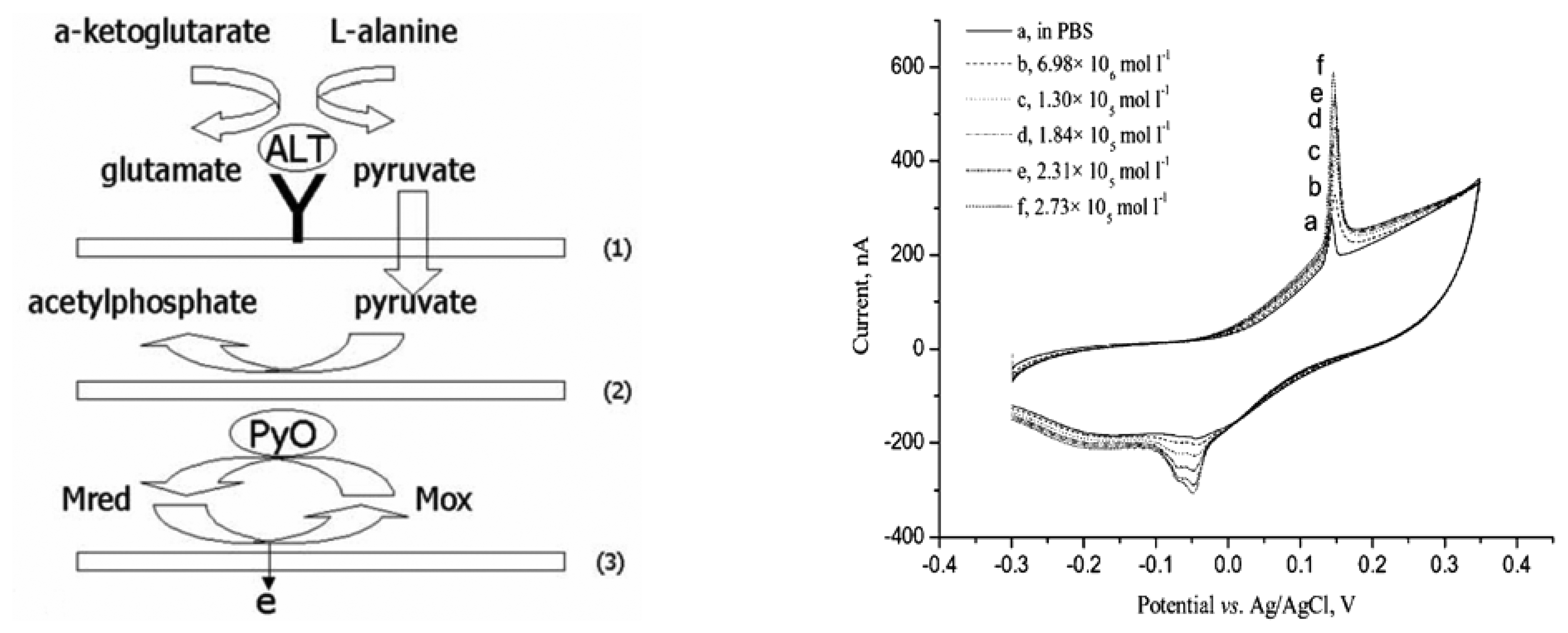
8.2. Detection using Pyruvate Oxidase Modified Electrode
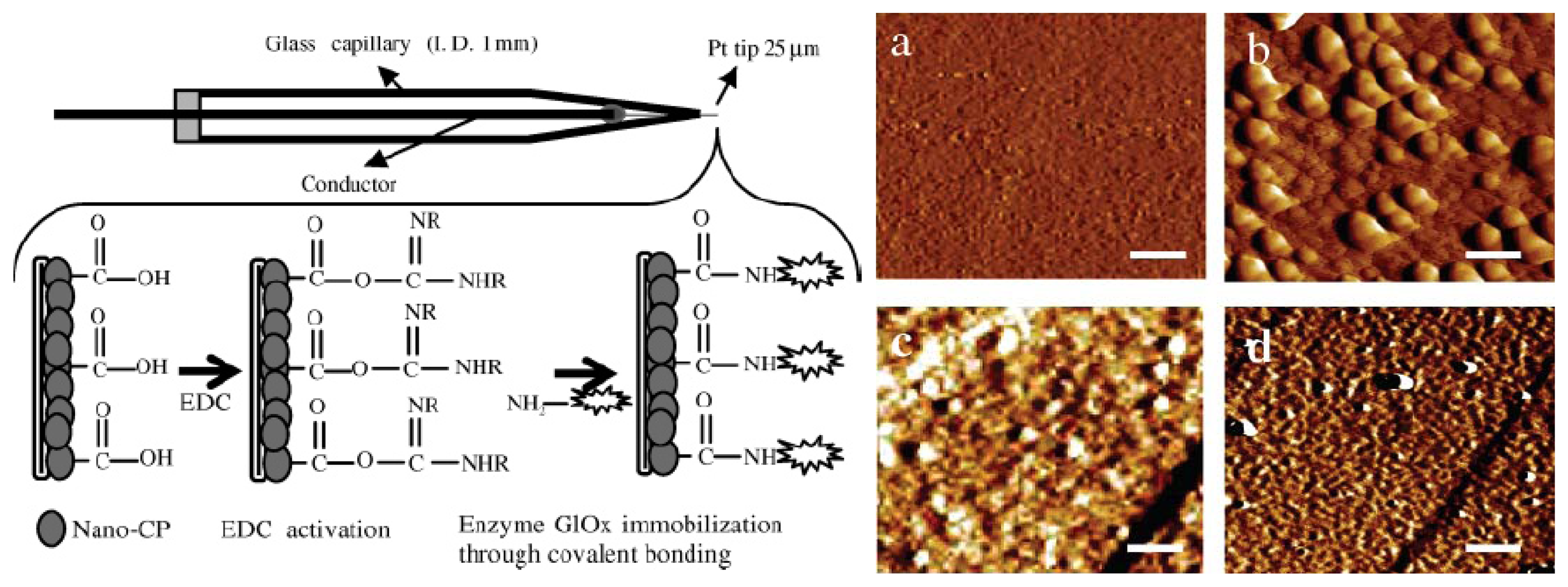
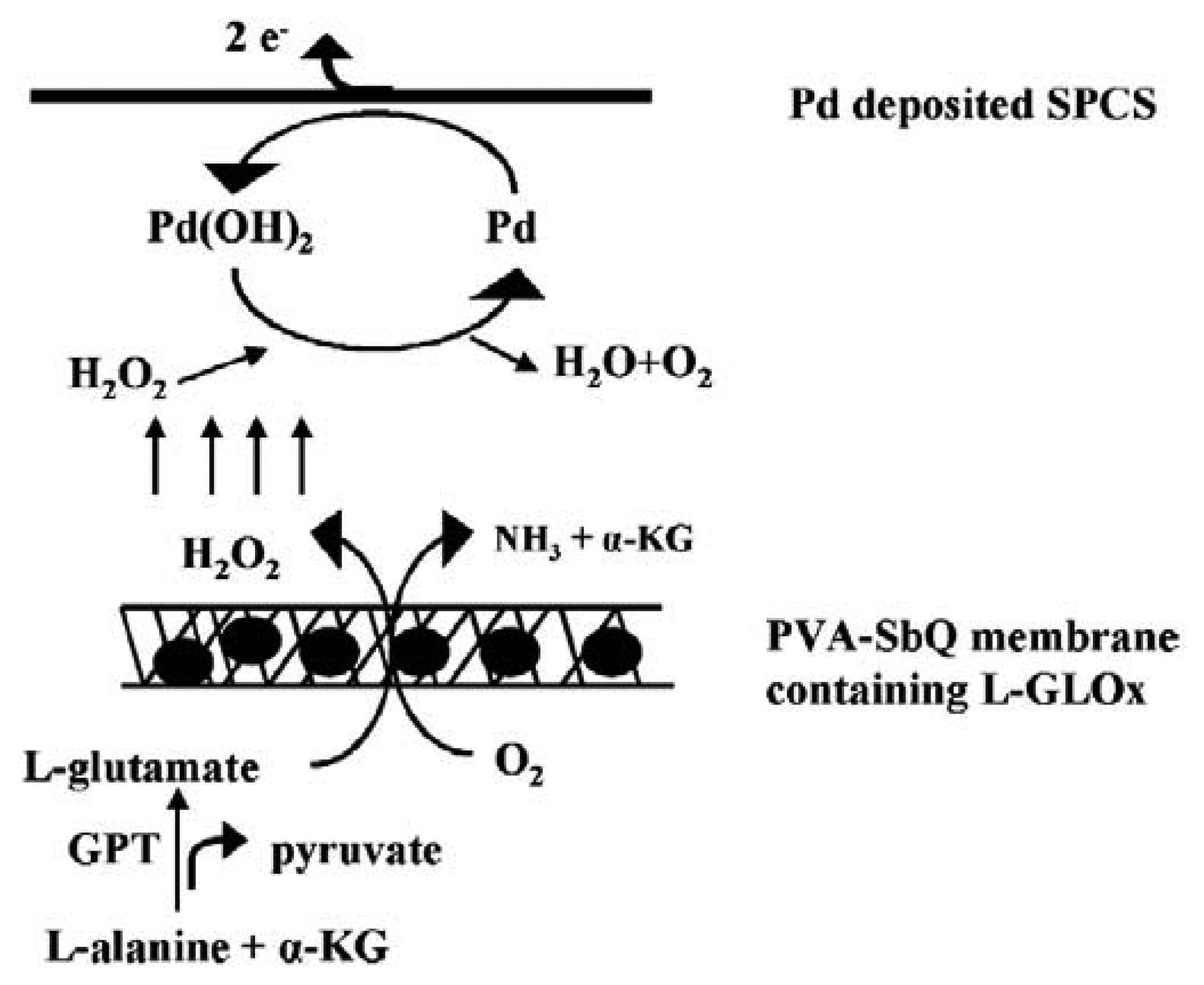
8.3. Detection using Glutamate Oxidase Modified Electrode
8.4. L-Lactate Biosensor for GOT and GPT Detection
9. Conclusions and Outlook
Acknowledgments
References and Notes
- Hafkenscheid, J.C.M.; Dijt, C.C.M. Determination of serum aminotransferases: activation by pyridoxal-5′-phosphate in relation to substrate concentration. Clin. Chem. 1979, 25, 55–59. [Google Scholar]
- Sampson, E.J.; Whitner, V.S.; Burtis, C.A.; McKneaily, S.S.; Fast, D.M.; Bayse, D.D. An interlaboratory evaluation of the IFCC methodfor aspartate aminotransferasewith use of purified enzyme materials. Clin. Chem. 1980, 26, 1156–1164. [Google Scholar]
- Jacobs, D.S. Laboratory Test Handbook., 4th ed.; Cleveland, OH; Lexi-Comp Inc., 1996. [Google Scholar]
- Purcell, G.V.; Behenna, D.B.; Walsh, P.R. Alanine aminotransferase and aspartate aminotransferase measurements with two automated analyzers, SMAC and the ABA-100, Compared. Clin.Chem. 1979, 25, 780–782. [Google Scholar]
- http://www.medfriendly.com/aspartateaminotransferase.html.
- http://www.medfriendly.com/alanineaminotransferase.html.
- Wang, J.Y. Biological Chemistry, 3rd ed.; Higher Education Press: Beijing, 2002. [Google Scholar]
- Bergmeyer, H.U.; Bernst, E. Glutamate-oxaloacetate transaminase: UV-assay manual method, in. Bergmeyer, H.U., Ed.; In Methods of Enzymatic Analysis., 2nd English Ed; Academic Press: New York, 1963; pp. 727–733. [Google Scholar]
- Bergmeyer, H.U.; Bernst, E. lutamate-pyruvate transaminase: UV-assay manual method, in. Bergmeyer, H.U., Ed.; In Methods of Enzymatic Analysis., 2nd English Ed; Academic Press: New York, 1963; pp. 752–758. [Google Scholar]
- Hanson, C.; Cleland, W. Kinetic studies of glutamic oxaloacetic transaminase isozymes. Biochem. 1964, 3, 338–345. [Google Scholar]
- Saier, M.; Jenkins, W. Alanine aminotoransferase: purification and properties. J. Biol. Chem. 1967, 242, 91–100. [Google Scholar]
- Bergmeyer, H.U.; Horder, M.; Rey, J. Approved recommendation on IFCC methods for the measurement of catalytic enzymes. Part 2: IFCC method for aspartate aminotransferase. J. Clin. Chem. Clin. Biochem. 1986a, 24, 497–510. [Google Scholar]
- Bergmeyer, H.U.; Horder, M.; Rey, J. Approved recommendation on IFCC methods for the measurement of catalytic enzymes. Part 3: IFCC method for alanine aminotransferase. J. Clin. Chem. Clin. Biochem. 1986b, 24, 481–495. [Google Scholar]
- Jung, K.; Grützmann, K.D. Comparative determinations of aminotransferase activities in serum with so-called “optimised” methods. Clin. Chim. Acta 1977, 81, 299–304. [Google Scholar]
- Gella, F.J.; Olivella, T.; Pastor, M.C.; Arenas, J.; Moreno, R.; Durban, R.; Gomez, J.A. A simple procedure for the routine determination of aspartate aminotransferase and alanine aminotransferase with pyridoxal phosphate. Clin. Chim. Acta 1985, 153, 241–247. [Google Scholar]
- Lustig, V.; Diamandis, A.P.; Goldberg, D.M. Evaluation of commercially formulated aspartate aminotransferase and alanine aminotransferase activity determinations by the scandinavian committee on enzymes and IFCC methods as modified for use with automated enzyme analysers. Clin. Biochem. 1988, 21, 283–290. [Google Scholar]
- Goldberg, D.M.; Remtulla, M.A.; Lustig, V. The Diagnostic accuracy of three recommended methods for serum aspartate aminotransferase assays in patients suspected of myocardial infarction and hepatobiliary diseases. Clin. Biochem. 1988, 21, 323–328. [Google Scholar]
- Schumann, G.; Bonora, R.; Ceriotti, F.; Férard, G.; Ferrero, C.A.; Franck, P.F.H.; Gella, F.J.; Hoelzel, W.; Jørgensen, P.J.; Kanno, T.; Kessner, A.; Klauke, R.; Kristiansen, N.; Lessinger, J.M.; J.Linsinger, T.P.; Misaki, H.; Panteghini, M.; Pauwels, J.; Schiele, F.; Schimmel, H.G. IFCC primary reference procedures for the measurement of catalytic activity concentrations of enzymes at 37 °C: Part 4. Reference procedure for the measurement of catalytic concentration of alanine-aminotransferase. Clin. Chem. Lab Med. 2002, 40, 718–724. [Google Scholar]
- Schumann, G.; Bonora, R.; Ceriotti, F.; Férard, G.; Ferrero, C.A.; Franck, P.F.H.; Gella, F.J.; Hoelzel, W.; Jørgensen, P.J.; Kanno, T.; Kessner, A.; Klauke, R.; Kristiansen, N.; Lessinger, J.M.; Linsinger, T.P.; Misaki, H.; Panteghini, M.; Pauwels, J.; Schiele, F.; Schimmel, H.G.; Weidemann, G.; Siekmann, L. IFCC primary reference procedures for the measurement of catalytic activity concentrations of enzymes at 37 °C: Part 5. Reference procedure for the measurement of catalytic concentration of aspartate-aminotransferase. Clin. Chem. Lab Med. 2002, 40, 725–733. [Google Scholar]
- Schumann, G.; Klauke, R. New IFCC reference procedures for the determination of catalytic activity concentrations of five enzymes in serum: preliminary upper reference limits obtained in hospitalized subjects. Clin. Chim. Acta 2003, 327, 69–79. [Google Scholar]
- Babson, A.L.; Shapiro, P.O.; Williams, P.A.R.; Phillips, G.E. The use of a diazonium salt for the determination of glutamic-oxalacetic transaminase in serum. Clin. Chim. Acta 1962, 7, 199–205. [Google Scholar]
- Matsuzawa, T.; Katunuma, N. Colorimetric assays for serum alanine transaminase and lactic dehydrogenase using diazonium zinc salt. Anal. Biochem. 1966, 17, 143–153. [Google Scholar]
- Lippi, U.; Guidi, G. A new colorimetric ultramicromethod for serum glutamicoxalacetic and glutamic-pyruvic transaminase determination. Clin. Chim. Acta 1970, 28, 431–437. [Google Scholar]
- Bailey, W.C.; DeRouen, T.A.; Ziskind, M.M.; Greenberg, H.B. Autoanalytic (colormetric) determinations of SGOT in isoniazid recipients are reliable. Am. Rev. Respir Dis. 1975, 111, 237–238. [Google Scholar]
- Whitaker, J.F. A general colorimetric procedure for the estimation of enzymes which are linked to the NADH/NAD+ system. Clin. Chim. Acta 1969, 24, 23–27. [Google Scholar]
- Oki, A.; Ogawa, H.; Takamura, Y.; Horiike, Y. Biochip which examines hepatic function by employing colorimetric method. Jpn. J. Appl. Phys. 2003, 42, L342–L345. [Google Scholar]
- Karmen, A. A note on the spectrometric assay of glutamic-oxalacetic transaminase in human blood serum. J. Clin. Invest. 1955, 34, 131–133. [Google Scholar]
- Giusti, G.; Ruggiero, G.; Cacciatore, L. A comparative study of some spectrophotometric and colorimetric procedures for the determination of serum glutamic-oxaloacetic and glutamic-pyruvic transaminase in hepatic diseases. Enzymol. Biol. Clin. 1969, 10, 17–38. [Google Scholar]
- Itoh, H.; Srere, P.A. A new assay for glutamate-oxaloacetate transaminase. Anal. Biochem. 1970, 35, 405–410. [Google Scholar]
- Rodgerson, D.O.; Osberg, I.M. Sources of error in spectrophotometric measurement of aspartate aminotransferase and alanine aminotransferaseActivities in serum. Clin.Chem. 1974, 20, 43–50. [Google Scholar]
- Amador, E.; Wacker, W.B.C. Serum glutamic-oxaloacetic transaminase activity: A new modification and an analytical assessment of current assay technics. Clin. Chem. 1962, 8, 343–350. [Google Scholar]
- Henry, R.J.; Chiamori, N.; Golub, O.J.; Berkman, S. Revised spectrophotometric methods for the determination of glutamic-oxalacetic transaminase, glutainic-pyruvic transaminase, and lactic dehydrogenase. Am. J. Clin. Pathol. 1960, 34, 318–322. [Google Scholar]
- Pearson, J.R.; Pinfield, B.R.; Cooper, D. A system for computer analysis of kinetic enzyme data from a modified Beckman DSA-560. Clin. Chem. 1972, 18, 775–777. [Google Scholar]
- Hamada, H.; Ohkura, Y. A new photometric method for the determination of serum glutamate pyruvate transaminase activity using pyruvate and glutamate as substrates. Chem. Pharm. Bull 1976, 24, 1865–1869. [Google Scholar]
- Yagi, T.; Kagamiyama, H.; Ohtawara, S.; Soda, K.; Nozaki, M. A new assay for L-aspartate: 2-oxoglutarate aminotransferase. Anal. Biochem. 1979, 100, 20–24. [Google Scholar]
- Phillip, M.A.; Graham, R.P. Designing a coupled assay system for aspartate aminotransferase. Biochem. Edu. 1995, 23, 40–41. [Google Scholar]
- Fernández-Romero, J.M.; Luque de Castro, M.D.; Valcfircel, M. Determination of alanine aminotransferase in human serum in an open-closed flow injection configuration. J. Biotechnol. 1990, 14, 43–52. [Google Scholar]
- Valero, E.; Garcia-Carmona, F. A Continuous spectrophotometric method based on enzymatic cycling for determining l-glutamate. Anal. Biochem. 1998, 259, 265–271. [Google Scholar]
- Mayer, D.; Spielman, D.M. Detection of glutamate in the human brain at 3 T using optimized constant time point resolved spectroscopy. Magn. Reson. Med. 2005, 54, 439–442. [Google Scholar]
- Chang, K.S.; Hsu, W.L.; Chen, H.Y.; Chang, C.K.; Chen, C.Y. Determination of glutamate pyruvate transaminase activity in clinical specimens using a biosensor composed of immobilized l-glutamate oxidase in a photo-crosslinkable polymer membrane on a palladium-deposited screen-printed carbon electrode. Anal. Chim. Acta 2003, 481, 199–208. [Google Scholar]
- He, Y.N.; Chen, H.Y. The kinetics-based electrochemical determination of serum glutamate pyruvate transaminase activity with a gold microelectrode. Anal. Chim. Acta 1997, 353, 319–323. [Google Scholar]
- Catz, S.D.; Carreras, M.C.; Poderoso, J.J. Nitric oxide synthase inhibitors decrease human polymorphonuclear leukocyte luminol-dependent chemiluminescence. Free Radic. Biol. Med. 1995, 19, 741–748. [Google Scholar]
- Janasek, D.; Spohn, U. Chemiluminometric flow injection analysis procedures for the enzymatic determination of l-alanine, α-ketoglutarate and lglutamate. Biosens. Bioelectron. 1999, 14, 123–129. [Google Scholar]
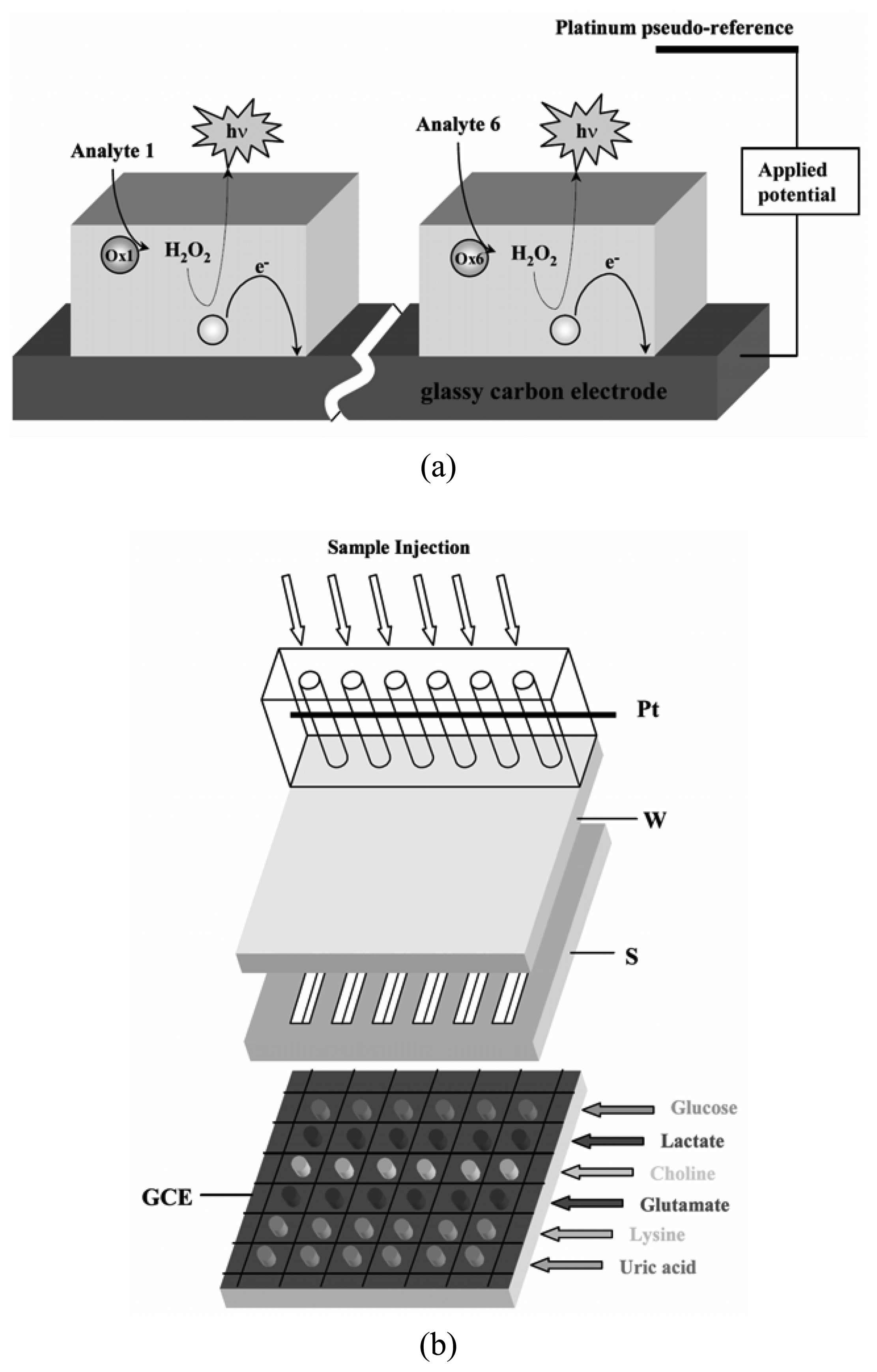
- Marquette, C.A.; Degiuli, A.; Blum, L.J. Electrochemiluminescent biosensors array for the concomitant detection of choline, glucose, glutamate, lactate, lysine and urate. Biosens. Bioelectron. 2003, 19, 433–439. [Google Scholar]
- Canepari, S.; Carunchio, V.; Girelli, A.M.; Messina, A. Determination of aspartate aminotransferase activity by high-performance liquid chromatography. J. Chromatogr. B 1994, 656, 191–195. [Google Scholar]
- Carunchio, V.; Castellano, R.; Girelli, A.M.; Messina, A. Simultaneous assay for aspartate aminotransferase and guanase in human serum by high-performance liquid chromatography. J. Chromatogr. B 1997, 689, 305–311. [Google Scholar]
- Ohmori, S.; Tanaka, Y.; Ikeda, M.; Hirota, K. A Sensitive determination of α-keto acids by gas-liquid chromatography and its application to the assay of l-glutamate Dehydrogenase and Aminotransferases. Anal. Biochem. 1981, 112, 204–212. [Google Scholar]
- Zhuang, X.W.; Ha, T.J.; Kim, H.D.; Centner, T.; Labeit, S.; Chu, S. Fluorescence quenching: A tool for single-molecule protein-folding study. PNAS 2000, 97, 14241–14244. [Google Scholar]
- Takeuchi, T.; Sumida, J. Indirect detection of halide ions via fluorescence quenching of quinine sulfate in microcoluman ion chromatograph. Anal. Sci. 2004, 20, 983–985. [Google Scholar]
- Rietz, B.; Guilbault, G.G. Fluorimetric assay of serum glutamate oxaloacetate transaminase, glutamate pyruvate transaminase and α-hydroxybutyrate dehydrogenàse by solution and solid surface fluorescent methods. Anal. Chim. Acta 1975, 77, 191–198. [Google Scholar]
- Khampha, W.; Yakovleva, J.; Isarangkul, D.; Wiyakrutta, S.; Meevootisom, V.; Emnéus, J. Specific detection of l-glutamate in food using flow-injection analysis and enzymatic recycling of substrate. Anal. Chim. Acta 2004, 518, 127–135. [Google Scholar]
- Bonizzoni, M.; Fabbrizzi, L.; Piovani, G.; Taglietti, A. Fluorescent detection of glutamate with a dicopper(II) polyamine cage. Tetrahedron 2004, 60, 11159–11162. [Google Scholar]
- Okumoto, S.; Looger, L.L.; Micheva, K.D.; Reimer, R.J.; Smith, S.J.; Frommer, W.B. Detection of glutamate release from neurons by genetically encoded surface-displayed FRET nanosensors. PNAS 2005, 102, 8740–8745. [Google Scholar]
- Parvin, R.; Caramancion, M.N.V.; Pande, S.V. Convenient rapid determination of picomole amounts of oxaloacetate and aspartate. Anal. Biochem. 1980, 104, 296–299. [Google Scholar]
- Garrison, D.; Beattie, J.; Namboodiri, M.A.A. A radiochemical microassay for aspartate aminotransferase activity in the nervous system. Anal. Biochem. 1988, 172, 96–103. [Google Scholar]
- Perales, M.A.; Sener, A.; Malaisse, W.J. Radioisotopic assay of aspartate and alanine aminotransferase. Clin. Biochem. 1992, 25, 105–107. [Google Scholar]
- He, Y.N.; Chen, H.Y. An improved ELISA for the determination of thyroglobulin with differential pulse voltammetry. Anal. Lett. 1996, 29, 2463–2474. [Google Scholar]
- You, T.Y.; Niwa, O.; Kurita, R.; Iwasaki, Y.; Hayashi, K.; Suzuki, K.; Hirono, S. Reductive H2O2 detection at nanoparticle iridium/carbon film flectrode and its application as l-glutamate enzyme sensor. Electroanalysis 2004, 16, 54–59. [Google Scholar]
- Peguin, S.; Coulet, P.R.; Bardeletti, G. Pyruvate oxidase and oxaloacetate decarboxylase enzyme electrodes Simultaneous Determination of Transaminases with a Two-electrode-based Analyzer. Anal. Chim. Acta 1989, 222, 83–93. [Google Scholar]
- Kihara, K.; Yasukawa, E.; Hayashi, M.; Hirose, S. Determination of glutamate-pyruvate transaminase activity in blood serum with a pyruvate oxidase/poly(vinyl chloride) membrane sensor. Anal. Chim. Acta 1984, 159, 81–86. [Google Scholar]
- Kihara, K.; Yasukawa, E.; Hirose, S. Sequential Determination of Glutamate oxalacetate transaminase and glutamate pyruvate transaminase activities in serum using an immobilized bienzyme-poly(viny1 chloride) membrane electrode. Anal. Chem. 1984, 56, 1876–1880. [Google Scholar]
- Xuan, G.S.; Oh, S.W.; Choi, E.Y. Development of an electrochemical immunosensor for alanine aminotransferase. Biosens. Bioelectron. 2003, 19, 365–371. [Google Scholar]
- Pan, S.; Arnold, M.A. Selectivity enhancement for glutamate with a Nafion/glutamate oxidase biosensor. Talanta 1996, 43, 1157–1162. [Google Scholar]
- Ye, B.C.; Li, Q.S.; Li, Y.R.; Li, X.B.; Yu, J.T. l-glutamate biosensor using a novel l-glutamate oxidase and its application to flow injection analysis system. J. Biotechnol. 1995, 42, 45–52. [Google Scholar]
- Oldenziel, W.H.; Westerink, B.H.C. Improving glutamate Microsensors by optimizing the composition of the redox hydrogel. Anal. Chem. 2005, 77, 5520–5528. [Google Scholar]
- Rahman, M.A.; Kwon, N.H.; Won, M.S.; Choe, E.S.; Shim, Y.B. Functionalized conducting polymer as an enzyme-immobilizing substrate: an amperometric glutamate microbiosensor for in vivo measurements. Anal. Chem. 2005, 77, 4854–4860. [Google Scholar]
- Castillo, J.; Blöchl, A.; Dennison, S.; Schuhmann, W.; Csöregi, E. Glutamate detection from nerve cells using a planar electrodes array integrated in a microtiter plate. Biosens. Bioelectron. 2005, 20, 2116–2119. [Google Scholar]
- Castillo, J.; Isik, S.; Blöchl, A.; Pereira-Rodrigues, N.; Bedioui, F.; Csöregi, E.; Schuhmann, W.; Oni, J. Simultaneous detection of the release of glutamate and nitric oxide from adherently growing cells using an array of glutamate and nitric oxide selective electrodes. Biosens. Bioelectron. 2005, 20, 1559–1565. [Google Scholar]
- O'Neill, R.D.; Chang, S.C.; Lowry, J.P.; McNeil, C.J. Comparisons of platinum, gold, palladium and glassy carbon as electrode materials in the design of biosensors for glutamate. Biosens. Bioelectron. 2004, 19, 1521–1528. [Google Scholar]
- Mikeladze, E.; Schulte, A.; Mosbach, M.; Blöchl, A.; Csöregi, E.; Solomonia, R.; Schuhmann, W. Redox hydrogel-based bienzyme microelectrodes for amperometric monitoring of l-glutamate. Electroanalysis 2002, 14, 393–399. [Google Scholar]
- Compagnone, D.; Federici, G.; Massoud, R.; Santoro, L.; Anichini, M.; Palleschi, G. Analysis for transaminases in serum with an amperometric glutamate electrode. Clin. Chem. 1992, 38, 2306–2310. [Google Scholar]
- Cooper, J.M.; McNeil, C.J.; Spoors, J.A. Amperometric enzyme electrode for the determination of aspartate aminotransferase and alanine aminotransferase in serum. Anal. Chim. Acta 1991, 245, 57–62. [Google Scholar]
- Mizutani, F.; Yabuki, S.; Hirata, Y. Amperometric L-lactate sensing electrode based on polyion complex layer containing lactate oxidase: application to serum and milk samples. Anal. Chim. Acta 1995, 314, 233–239. [Google Scholar]
- Mizutani, F.; Yabuki, S.; Hirata, Y. Amperometric biosensors using poly-L-lysine: poly (4-styrenesulfonate) membranes with immobilized enzymes. Denki Kagaku 1995, 63, 1100–1105. [Google Scholar]
- Mizutani, F.; Yabuki, S.; Hirata, Y. Flow injection analysis of L-lactic acid using an enzyme-polyion complex-coated electrode as the detector. Talanta 1996, 43, 1815–1820. [Google Scholar]
- Mizutani, F.; Yabuki, S.; Iijima, S. Amperometric alcohol-sensing electrode based on a polyion complex membrane containing alcohol oxidase. Anal. Sci. 1997, 13, 83–87. [Google Scholar]
- Mizutani, F.; Sato, Y.; Hirata, Y.; Sawaguchi, T.; Yabuki, S. Glucose oxidase–polyion complex-bilayer membrane for elimination of electroactive interferents in amperometric glucose sensor. Anal. Chim. Acta 1998, 364, 173–179. [Google Scholar]
- Mizutani, F.; Sato, Y.; Sawaguchi, T.; Yabuki, S.; Iijima, S. Rapid measurement of transaminase activities using an amperometric l-glutamate-sensing electrode based on a glutamate oxidase-polyion complex-bilayer membrane. Sens. Actuators B 1998, 52, 23–29. [Google Scholar]
- Sohn, B.K.; Cho, B.W.; Kim, C.S.; Kwon, D.H. ISFET glucose and sucrose sensors by using platinum electrode and photo-crosslinkable polymers. Sens. Actuators B 1997, 41, 7–11. [Google Scholar]
- Jaffrezic-Renault, N.; Senillou, A.; Martelet, C.; Wan, K.; Chovelon, J.M. ISFET microsensors for the detection of pollutants in liquid media. Sens. Actuators B 1999, 59, 154–164. [Google Scholar]
- Renneberg, R.; Sonomoto, K.; Katoh, S.; Tanaka, A. Oxygen diffusivity of synthetic gels derived from polymers. Appl. Microbiol. Biotechnol. 1988, 28, 1–7. [Google Scholar]
- Rouillon, R.; Tocabensand, M.; Marty, J.L. Stabilization of chloroplasts by entrapment in polyvinylalcohol bearing styrylpyridinium groups. Anal. Lett. 1994, 27, 2239–2248. [Google Scholar]
- Perdomo, J.; Sundermeier, C.; Hinkers, H.; Martínez Morell, O.; Seifert, W.; Knoll, M. Containment sensors for the determination of L-lactate and glucose. Biosens. Bioelectron. 1999, 14, 27–32. [Google Scholar]
- Chi, Q.J.; Dong, S.J. Flow-injection analysis of glucose at an amperometric glucose sensor based on electrochemical deposition of palladium and glucose oxidase on a glassy carbon electrode. Anal. Chim. Acta 1993, 278, 17–23. [Google Scholar]
- Dong, S.J.; Deng, Q.; Cheng, G.J. Cholesterol sensor based on electrodeposition of catalytic palladium particles. Anal. Chim. Acta 1993, 279, 235–240. [Google Scholar]
- Moser, I.; Jobst, G.; Aschauer, E.; Svasek, P.; Varahram, M.; Urban, G. Miniaturized thin film glutamate and glutamine biosensors. Biosens. Bioelectron. 1995, 10, 527–532. [Google Scholar]
- Moser, I.; Jobst, G.; Svasek, P.; Varahram, M.; Urban, G. Rapid liver enzyme assay with miniaturized liquid handling system comprising thin film biosensor array. Sens. Actuators B 1997, 44, 377–380. [Google Scholar]
- Suman, S.; Singhal, R.; Sharma, A.L.; Malthotra, B.D.; Pundir, C.S. Development of a lactate biosensor based on conducting copolymer bound lactate oxidase. Sens. Actuators B 2005, 107, 768–772. [Google Scholar]
- Kwan, R.C.H.; Hon, P.Y.T.; Mak, K.K.W.; Renneberg, R. Amperometric determination of lactate with novel trienzyme/poly(carbamoyl) sulfonate hydrogel-based sensor. Biosens. Bioelectron. 2004, 19, 1745–1752. [Google Scholar]
- Erickson, D.; Li, D.Q. Integrated microfluidic devices. Anal. Chim. Acta 2004, 507, 11–26. [Google Scholar]
- Erickson, D.; Liu, X.Z.; Krull, U.; Li, D.Q. Electrokinetically controlled DNA hybridization microfluidic chip enabling rapid target analysis. Anal. Chem. 2004, 76, 7269–7277. [Google Scholar]
- Yamaguchi, A.; Jin, P.; Tsuchiyama, H.; Masuda, T.; Sun, K.; Matsuo, S.; Misawa, H. Rapid fabrication of electrochemical enzyme sensor chip using polydimethylsiloxane microfluidic channel. Anal. Chim. Acta 2002, 468, 143–152. [Google Scholar]
- Lv, Y.; Zhang, Z.J.; Chen, F.N. Chemiluminescence microfluidic system sensor on a chip for determination of glucose in human serum with immobilized reagents. Talanta 2003, 59, 571–576. [Google Scholar]
- Yun, K.S.; Lee, D.H.; Kim, M.S.; Kim, H.S.; Lee, G.M.; Yoon, E. High throughput bio-molecule detection using microbead-based assay with quantum dot fluorescence in a microfluidic chip. Proceedings of International Conference on Miniaturized Systems for Chemistry and Life Sciences (micro TAS'04), Malmo, Sweden, September. 2004; pp. 222–224.
- Kim, J.H.; Kim, B.G.; Nam, H.; Park, D.E.; Yun, K.S.; Yoon, J.B.; You, J.; Yoon, E. A disposable DNA sample preparation microfluidic chip for nucleic acid probe assay. IEEE International MEMS Conference 2002, Las Vegas, USA, Jan. 2002; pp. 133–136.
- Im, M.S.; Cho, I.J.; Yun, K.S.; Yoon, E. An electronmagnetically actuated polymer micro-pen for picoliter biological assay patterning. 13th International Conference on Solid-State Sensors and Actuators (Transducers'05), Digest of Technical Papers Seoul, Korea, June. 2005; pp. 1588–1591.
- Yun, K.S.; Yoon, E. Micro/Nanofluidic device for single-cell-based assay. Biomed. Microdevices 2005, 7, 35–40. [Google Scholar]











© 2006 by MDPI ( http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Huang, X.-J.; Choi, Y.-K.; Im, H.-S.; Yarimaga, O.; Yoon, E.; Kim, H.-S. Aspartate Aminotransferase (AST/GOT) and Alanine Aminotransferase (ALT/GPT) Detection Techniques. Sensors 2006, 6, 756-782. https://doi.org/10.3390/s6070756
Huang X-J, Choi Y-K, Im H-S, Yarimaga O, Yoon E, Kim H-S. Aspartate Aminotransferase (AST/GOT) and Alanine Aminotransferase (ALT/GPT) Detection Techniques. Sensors. 2006; 6(7):756-782. https://doi.org/10.3390/s6070756
Chicago/Turabian StyleHuang, Xing-Jiu, Yang-Kyu Choi, Hyung-Soon Im, Oktay Yarimaga, Euisik Yoon, and Hak-Sung Kim. 2006. "Aspartate Aminotransferase (AST/GOT) and Alanine Aminotransferase (ALT/GPT) Detection Techniques" Sensors 6, no. 7: 756-782. https://doi.org/10.3390/s6070756