Digital Marine Bioprospecting: Mining New Neurotoxin Drug Candidates from the Transcriptomes of Cold-Water Sea Anemones

Abstract
:1. Introduction

2. Results and Discussion
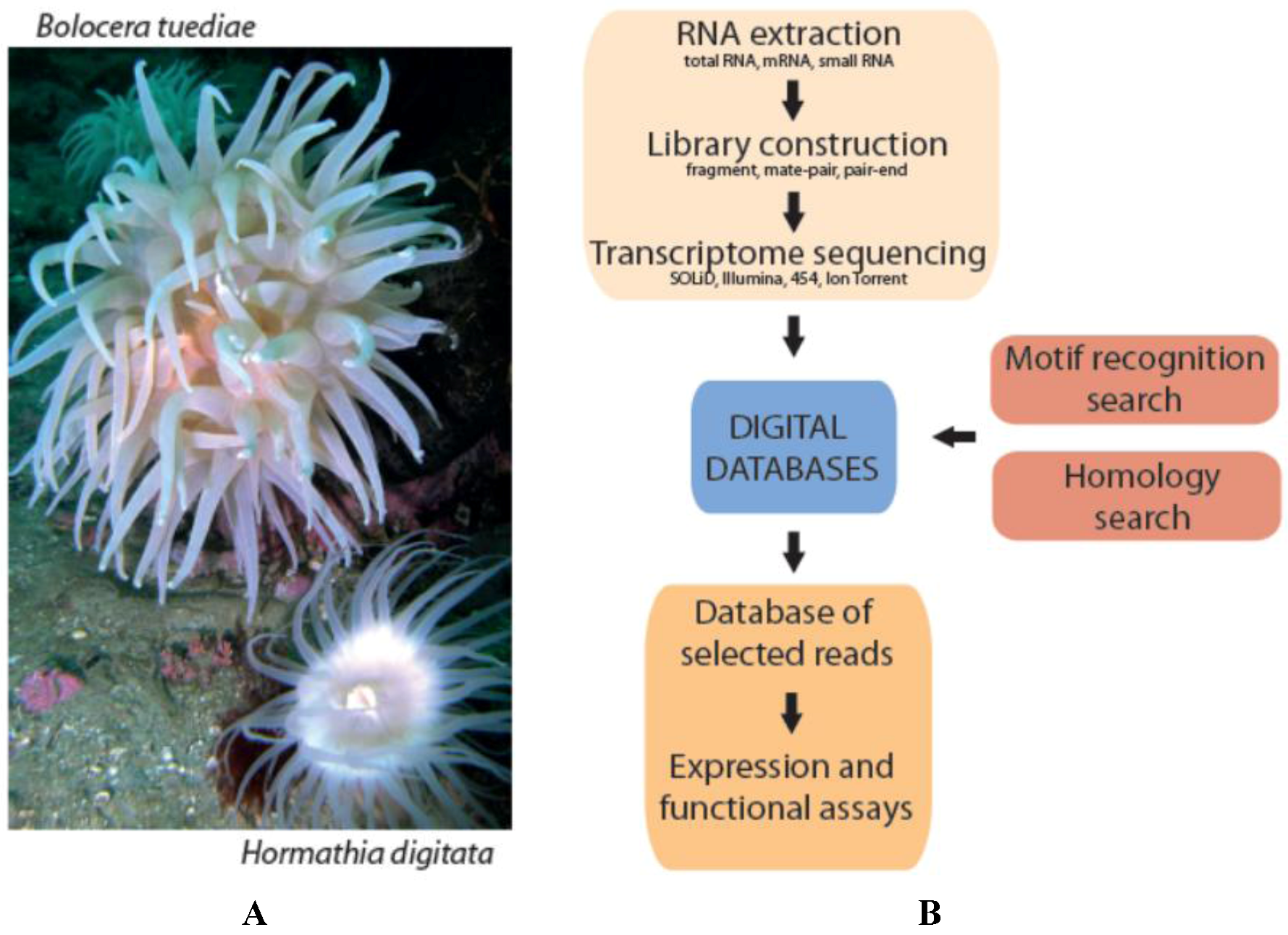
2.1. Transcriptome Sequencing and Assembly

{kind=link}
{kind=link}
{kind=link}
| Species | Reads/Contigs | Number | Average size (nt) | Total nt |
|---|---|---|---|---|
| B. tuediae | Raw reads | 547,061 | 547 | 299,232,484 |
| Trimmed reads | 546,903 | 333 | 182,128,133 | |
| All contigs | 64,442 | 591 | 38,101,858 | |
| Large contigs | 5072 | 1380 | 6,997,895 | |
| Single reads | 118,104 | 279 | 33,008,862 | |
| H. digitata | Raw reads | 546,974 | 543 | 296,833,666 |
| Trimmed reads | 546,846 | 331 | 181,169,361 | |
| All contigs | 54,293 | 613 | 33,255,104 | |
| Large contigs | 5083 | 1430 | 7,272,471 | |
| Single reads | 105,695 | 260 | 27,786,964 |
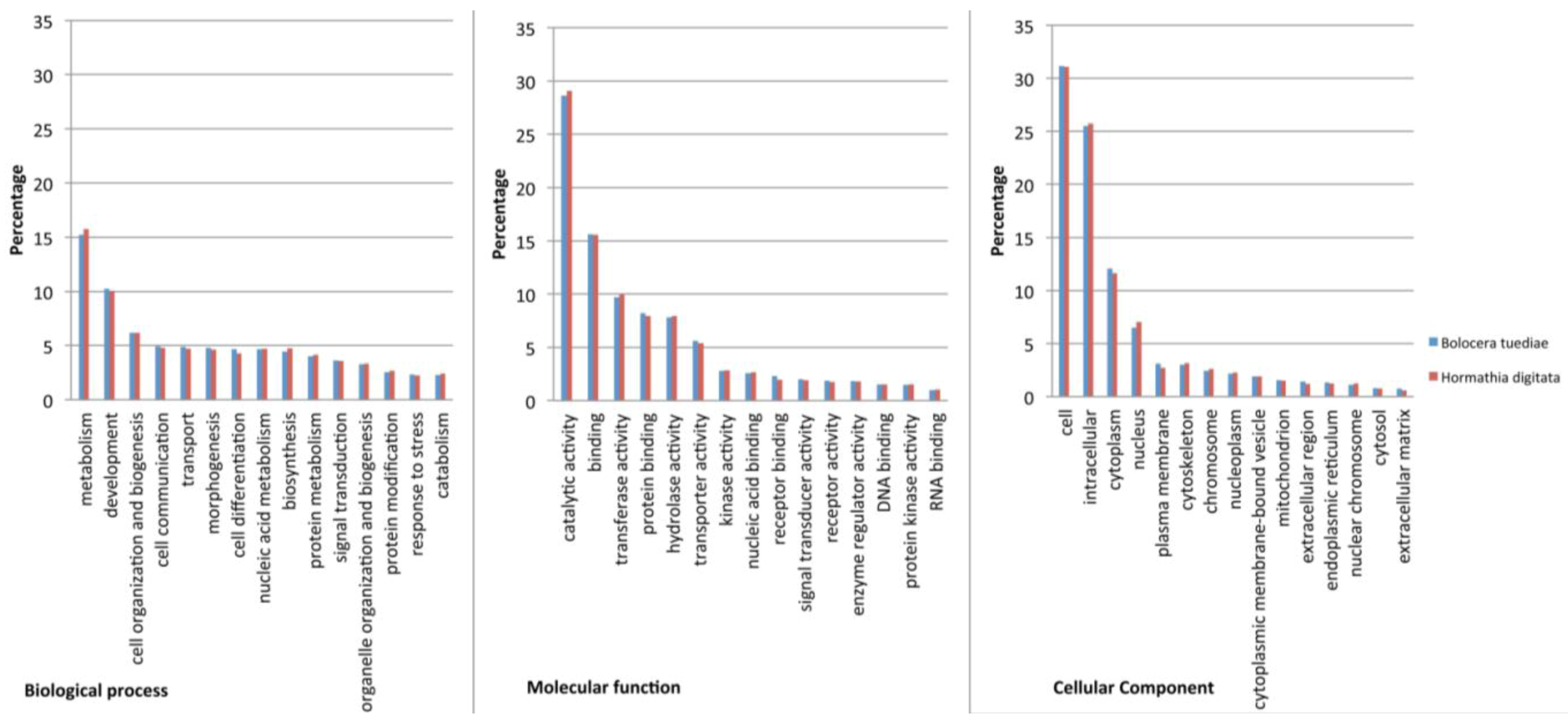
2.2. Annotation and Gene Ontology Analysis

2.3. A Protocol for Digital Marine Bioprospecting
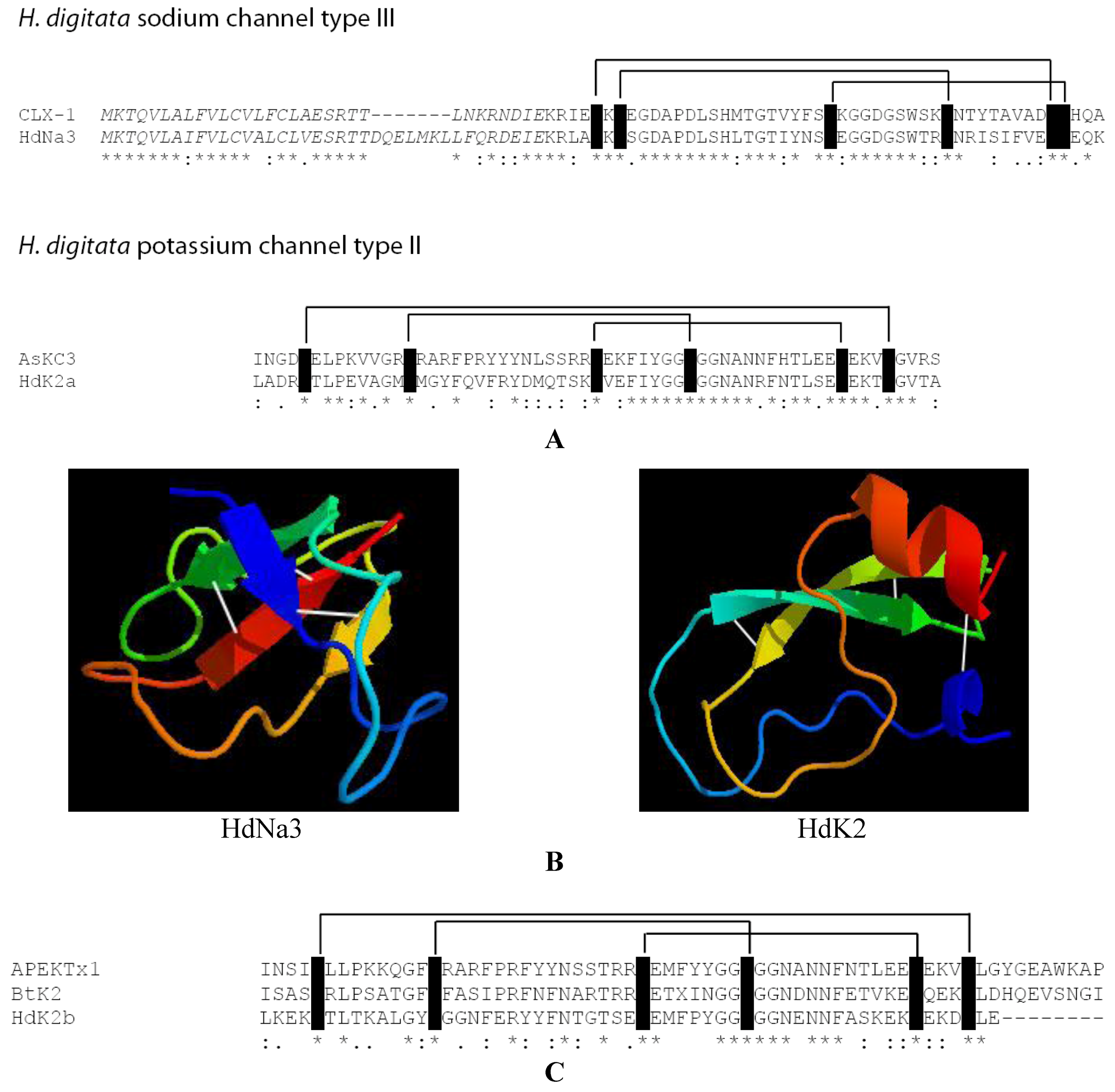
2.4. Identification of New Potential Neurotoxin Drug Candidates from Sea Anemones

| CDD, input and output a | B. tuediae | H. digitata |
|---|---|---|
| Query amino acid sequences | 864 | 1236 |
| Queries with domain hits | 131 | 229 |
| Total number of domain hits | 151 | 267 |
| Superfamilies | ||
| KU (Kunitz-type) | 135 | 211 |
| Toxin4 | - | 23 |
| KAZAL_FS | 6 | 23 |
| Antistatin | 6 | - |
| WAP | 1 | 3 |
| TY | 2 | 1 |
| ShK | - | 1 |
| VMA21-like | - | 1 |
| NTR | - | 1 |
3. Experimental Section
3.1. Sampling and RNA Extraction
3.2. Large Scale Sequencing
3.3. Assembly, Mapping and Annotation
3.4. Structure and Domain Predictions
4. Conclusion
Supplementary Files
Acknowledgments
References
- Koehn, F.E.; Carter, G.T. Rediscovering natural products as a source of new drugs. Discov. Med. 2005, 26, 159–164. [Google Scholar]
- Johansen, S.D.; Emblem, A.; Karlsen, B.O.; Okkenhaug, S.; Hansen, H.; Moum, T.; Coucheron, D.H.; Seternes, O.M. Approaching marine bioprospecting in hexacorals by RNA deep sequencing. N. Biotechnol. 2010, 27, 267–275. [Google Scholar]
- Yamaguchi, Y.; Hasegawa, Y.; Honma, T.; Nagashima, Y.; Shiomi, K. Screening and cDNA cloning of Kv1 potassium channel toxins in sea anemones. Mar. Drugs 2010, 8, 2893–2905. [Google Scholar] [CrossRef]
- Sperstad, S.V.; Haug, T.; Blencke, H.M.; Styrvold, O.B.; Li, C.; Stensvåg, K. Antimicrobial peptides from marine invertebrates: Challenges and perspectives in marine antimicrobial peptide discovery. Biotechnol. Adv. 2011, 5, 519–530. [Google Scholar]
- Vera, J.C.; Wheat, C.W.; Fescemyer, H.W.; Frilander, M.J.; Crawford, D.L.; Hanski, I.; Marden, J.H. Rapid transcriptome characterization for a nonmodel organism using 454 pyrosequencing. Mol. Ecol. 2008, 7, 1636–1647. [Google Scholar]
- Parchman, T.L.; Geist, K.S.; Grahnen, J.A.; Benkman, C.W.; Buerkle, C.A. Transcriptome sequencing in an ecologically important tree species: Assembly, annotation, and marker discovery. BMC Genomics 2010, 11, 180. [Google Scholar] [CrossRef]
- Wang, Y.; Zeng, X.; Iyer, N.J.; Bryant, D.W.; Mockler, T.C.; Mahalingam, R. Exploring the switchgrass transcriptome using second-generation sequencing technology. PLoS One 2012, 7, 3. [Google Scholar]
- Metzker, M.L. Sequencing technologies-the next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef]
- Putnam, N.H.; Srivastava, M.; Hellsten, U.; Dirks, B.; Chapman, J.; Salamov, A.; Terry, A.; Shapiro, H.; Lindquist, E.; Kapitonov, V.V.; et al. Sea Anemone Genome Reveals Ancestral Eumetazoan Gene Repertoire and Genomic Organization. Science 2007, 317, 86–94. [Google Scholar]
- Shinzato, C.; Shoguchi, E.; Kawashima, T.; Hamada, M.; Hisata, K.; Tanaka, M.; Fujie, M.; Fujiwara, M.; Koyanagi, R.; Ikuta, T.; et al. Using the Acropora digitifera genome to understand coral responses to environmental change. Nature 2011, 476, 320–323. [Google Scholar] [CrossRef]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug. Discov. 2009, 8, 69–85. [Google Scholar] [CrossRef]
- Rocha, J.; Peixe, L.; Gomes, N.C.M.; Calado, R. Cnidarians as a source of new marine bioactive compounds-an overview of the last decade and future steps for bioprospecting. Mar. Drugs 2011, 9, 1860–1886. [Google Scholar] [CrossRef]
- Meyer, E.; Aglyamova, G.V.; Wang, S.; Buchanan-Carter, J.; Abrego, D.; Colbourne, J.K.; Willis, B.L.; Matz, M.V. Sequencing and de novo analysis of a coral larval transcriptome using 454 GSFlx. BMC Genomics 2009, 10, 219. [Google Scholar]
- Polato, N.R.; Vera, J.C.; Baums, I.B. Gene discovery in the threatened elkhorn coral: 454 sequencing of the Acropora palmata transcriptome. PLoS One 2011, 6, 12. [Google Scholar]
- Traylor-Knowles, N.; Granger, B.R.; Lubinski, T.J.; Parikh, J.R.; Garamszegi, S.; Xia, Y.; Marto, J.A.; Kaufman, L.; Finnerty, J.R. Production of a reference transcriptome and transcriptomic database (PocilloporaBase) for the cauliflower coral, Pocillopora damicornis. BMC Genomics 2011, 12, 585. [Google Scholar] [CrossRef]
- Yuyama, I.; Watanabe, T.; Takei, Y. Profiling differential gene expression of symbiotic and aposymbiotic corals using a high coverage gene expression profiling (HiCEP) analysis. Mar. Biotechnol. 2011, 1, 32–40. [Google Scholar]
- Weis, V.M.; Davy, S.K.; Hoegh-Guldberg, O.; Rodriguez-Lanetty, M.; Pringle, J.R. Cell biology in model systems as the key to understanding corals. Trends Ecol. Evol. 2008, 7, 369–376. [Google Scholar]
- Sunagawa, S.; Wilson, E.C.; Thaler, M.; Smith, M.L.; Caruso, C.; Pringle, J.R.; Weis, V.M.; Medina, M.; Schwarz, J.A. Generation and analysis of transcriptomic resources for a model system on the rise: the sea anemone Aiptasia pallida and its dinoflagellate endosymbiont. BMC Genomics 2009, 10, 258. [Google Scholar] [CrossRef]
- Morgan, M.B.; Parker, C.C.; Robinson, J.W.; Pierce, E.M. Using representational difference analysis to detect changes in transcript expression of Aiptasia genes after laboratory exposure to lindane. Aquat. Toxicol. 2012, 110-111, 66–73. [Google Scholar] [CrossRef]
- Schwarz, J.A.; Brokstein, P.B.; Voolstra, C.; Terry, A.Y.; Manohar, C.F.; Miller, D.J.; Szmant, A.M.; Coffroth, M.A.; Medina, M. Coral life history and symbiosis: Functional genomic resources for two reef building Caribbean corals, Acropora palmata and Montastraea faveolata. BMC Genomics 2008, 9, 97. [Google Scholar]
- Sabourault, C.; Ganot, P.; Deleury, E.; Allemand, D.; Furla, P. Comprehensive EST analysis of the symbiotic sea anemone, Anemonia viridis. BMC Genomics 2009, 10, 333. [Google Scholar] [CrossRef]
- Norton, R.S. Structure and structure-function relationships of sea anemone proteins that interact with the sodium channel. Toxicon 1991, 29, 1051–1084. [Google Scholar] [CrossRef]
- Handbook of Neurotoxicology; Massaro, E.J. (Ed.) Humana Press: Totowa, NJ, USA, 2002; Volume I, p. 685.
- Wunderer, G.; Fritz, H.; Wachter, E.; Machleidt, W. Amino-acid sequence of a coelenterate toxin: Toxin II from Anemonia sulcata. Eur. J. Biochem. 1976, 1, 193–198. [Google Scholar]
- Anderluh, G.; Podlesek, Z.; Macek, P. A common motif in proparts of Cnidarian toxins and nematocyst collagens and its putative role. Biochim. Biophys. Acta 2000, 1476, 372–376. [Google Scholar] [CrossRef]
- Stevens, M.; Peigneur, S.; Tytgat, J. Neurotoxins and their binding areas on voltage-gated sodium channels. Front. Pharmacol. 2011, 2, 71. [Google Scholar]
- Diochot, S.; Lazdunski, M. Sea Anemone Toxins Affecting Potassium Channels. Prog. Mol. Subcell. Biol. 2009, 46, 99–122. [Google Scholar] [CrossRef]
- Lomax, J. Get ready to GO! A biologist’s guide to the gene ontology. Brief. Bioinform. 2005, 6, 298–304. [Google Scholar] [CrossRef]
- Emblem, Å. Genomic Analyses of the Cold-Water Coral Lophelia and Sea Anemones. PhD Thesis, University of Tromsø, Norway, 2011. [Google Scholar]
- St Pierre, L.; Fischer, H.; Adams, D.J.; Schenning, M.; Lavidis, N.; de Jersey, J.; Masci, P.P.; Lavin, M.F. Distinct activities of novel neurotoxins from Australian venomous snakes for nicotinic acetylcholine receptors. Cell. Mol. Life Sci. 2007, 21, 2829–2840. [Google Scholar]
- Kozlov, S.; Malyavka, A.; McCutchen, B.; Lu, A.; Schepers, E.; Herrmann, R.; Grishin, E. A novel strategy for the identification of toxinlike structures in spider venom. Proteins 2005, 59, 131–140. [Google Scholar] [CrossRef]
- Kozlov, S.; Grishin, E. The mining of toxin-like polypeptides from EST database by single residue distribution analysis. BMC Genomics 2011, 12, 88. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Anderson, J.B.; Chitsaz, F.; Derbyshire, M.K.; DeWeese-Scott, C.; Fong, J.H.; Geer, L.Y.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; et al. CDD: Specific functional annotation with the Conserved Domain Database. Nucleic Acids Res. 2009, 39, 225–229. [Google Scholar]
- Wanke, E.; Zaharenko, A.J.; Redaelli, E.; Schiavon, E. Actions of sea anemone type 1 neurotoxins on voltage-gated sodium channel isoforms. Toxicon 2009, 54, 1102–1111. [Google Scholar] [CrossRef]
- Moran, Y.; Gordon, D.; Gurevitz, M. Sea anemone toxins affecting voltage-gated sodium channels-molecular and evolutionary features. Toxicon 2009, 54, 1089–1101. [Google Scholar] [CrossRef]
- Beress, L.; Zwick, J. Purification of two crab-paralyzing polypeptides from the sea anemone Bolocera tuediae. Mar. Chem. 1980, 8, 333–338. [Google Scholar] [CrossRef]
- Dauplais, M.; Lecoq, A.; Song, J.; Cotton, J.; Jamin, N.; Gilquin, B.; Roumestand, C.; Vita, C.; de Medeiros, C.L.; Rowan, E.G. On the convergent evolution of animal toxins. Conservation of a diad of functional residues in potassium channel-blocking toxins with unrelated structures. J. Biol. Chem. 1997, 272, 4302–4309. [Google Scholar]
- Minagawa, S.; Ishida, M.; Shimakura, K.; Nagashima, Y.; Shiomi, K. Isolation and amino acid sequences of two Kunitz-typeprotease inhibitors from the sea anemone Anthopleura aff. xanthogrammica. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1997, 118, 381–386. [Google Scholar] [CrossRef]
- Strydom, D.J. Protease inhibitors as snake venom toxins. Nat. New. Biol. 1973, 243, 88–89. [Google Scholar] [CrossRef]
- Verollet, R. A major step towards efficient sample preparation with bead-beating. Biotechniques 2008, 44, 832–833. [Google Scholar]
- Conesa, A.; Götz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Hu, Z.; Bao, J.; Reecy, J.M. CateGOrizer: A Web-Based Program to Batch Analyze Gene Ontology Classification Categories. Online J. Bioinform. 2008, 9, 108–112. [Google Scholar]
- CLCbio. Available online: http://www.clcbio.com/ (accessed on 15 August 2012).
- Clustal: Multiple Sequence Alignment. Available online: http://www.clustal.org/ (accessed on 15 August 2012).
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL Workspace: A web-based environment for protein structure modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar] [CrossRef]
- SWISS-MODEL: Swiss Institute of Bioinformatics. Available online: http://swissmodel.expasy.org/ (accessed on 15 August 2012).
- PyMOL: A User-Sponsored Molecular Visualization System on an Open-Source Foundation. Available online: http://www.pymol.org/ (accessed on 15 August 2012).
- POVRAY-Persistence of Vision. Available online: http://www.povray.org/ (accessed on 15 August 2012).
- Bioinformatics Toolkit. Available online: http://toolkit.tuebingen.mpg.de/sixframe (accessed on 15 August 2012).
- Hartley, J.L.; Salehi-Ashtiani, K.; Hill, D.E. Proteome expression moves in vitro: Resources and tools for harnessing the human proteome. Nat. Methods 2008, 5, 1001–1002. [Google Scholar]
- Goshima, N.; Kawamura, Y.; Fukumoto, A.; Miura, A.; Honma, R.; Satoh, R.; Wakamatsu, A.; Yamamoto, J.; Kimura, K.; Nishikawa, T.; et al. Human protein factory for converting the transcriptome into in vitro-expressed proteome. Nat. Methods 2008, 5, 1011–1017. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Urbarova, I.; Karlsen, B.O.; Okkenhaug, S.; Seternes, O.M.; Johansen, S.D.; Emblem, Å. Digital Marine Bioprospecting: Mining New Neurotoxin Drug Candidates from the Transcriptomes of Cold-Water Sea Anemones. Mar. Drugs 2012, 10, 2265-2279. https://doi.org/10.3390/md10102265
Urbarova I, Karlsen BO, Okkenhaug S, Seternes OM, Johansen SD, Emblem Å. Digital Marine Bioprospecting: Mining New Neurotoxin Drug Candidates from the Transcriptomes of Cold-Water Sea Anemones. Marine Drugs. 2012; 10(10):2265-2279. https://doi.org/10.3390/md10102265
Chicago/Turabian StyleUrbarova, Ilona, Bård Ove Karlsen, Siri Okkenhaug, Ole Morten Seternes, Steinar D. Johansen, and Åse Emblem. 2012. "Digital Marine Bioprospecting: Mining New Neurotoxin Drug Candidates from the Transcriptomes of Cold-Water Sea Anemones" Marine Drugs 10, no. 10: 2265-2279. https://doi.org/10.3390/md10102265