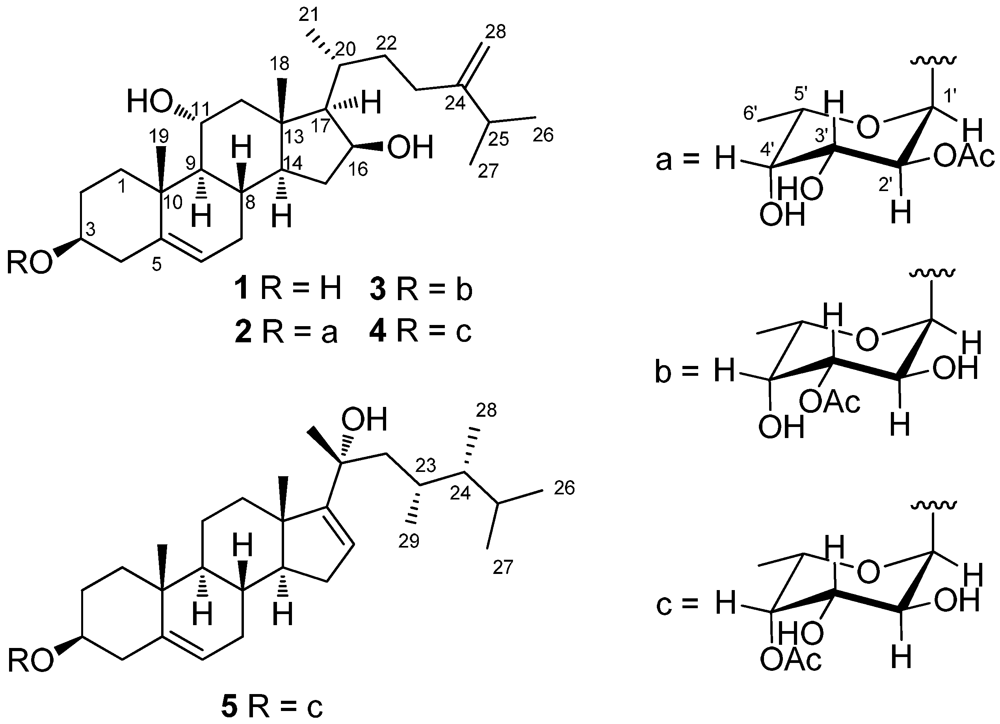
2. Results and Discussion
The HRESIMS of crassarosterol A (
1) exhibited a pseudomolecular ion peak at
m/z 453.3342 [M + Na]
+, consistent with a molecular formula of C
28H
46O
3 and requiring six degrees of unsaturation. The
13C NMR and DEPT spectroscopic data (
Table 1) displayed 28 carbon signals, including five methyls, nine methylenes, ten methines, and four quaternary carbons. The IR spectrum revealed the presence of hydroxy groups (3389 cm
–1). The carbon resonances at
δ 141.2 (C), 121.3 (CH), 156.9 (C), and 106.3 (CH
2) suggested the presence of two double bonds. The above data coupled with the characteristic
1H NMR signals for methyl groups at
δ 0.92 (3H, s), 1.18 (3H, s), 1.04 (3H, d,
J = 6.8 Hz), and 1.03 (6H, d,
J = 7.2 Hz) and signals for olefinic protons at
δH 5.41 (1H, d,
J = 5.6 Hz), 4.76 (1H, s), and 4.70 (1H, s) (
Table 2) suggested
1 to be a member of the 24-methylenecholesterol class [
12,
13]. The
1H−
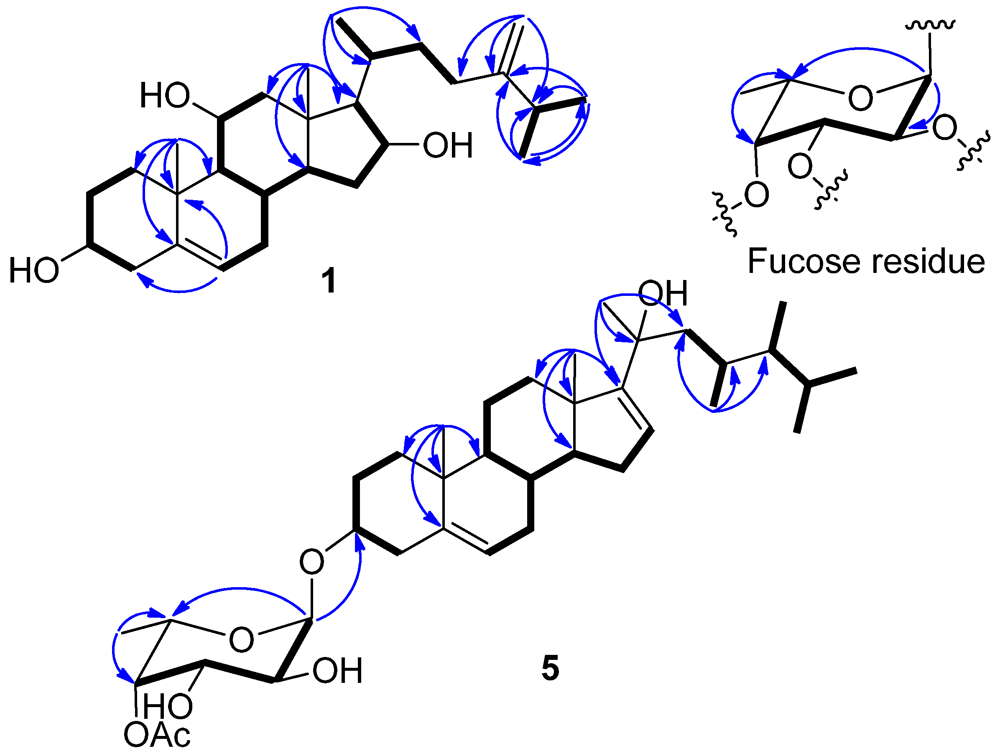
1H COSY correlations allowed the assignment of four separated spin systems (
Figure 2). The presence of an sp
2 methylene substituent at C-24 was further confirmed by the HMBC correlations from H
2-28 to C-23, C-24, and C-25 (
Figure 2). Likewise, the steroidal nucleus was confirmed by the HMBC correlations from H
3-18 to C-12, C-13, C-14, and C-17 and H
3-19 to C-1, C-5, C-9, and C-10. The NOE correlations between H
3-19/H-1β, H-3/H-1α, and H
3-19/H-11 suggested the α and β orientations for H-3 and H-11, respectively. The absence of an NOE correlation between H
3-18/H-17 and the presence of the correlation between H-17/H-16, H-16/H-14, and H-14/H-9 suggested the α orientation for H-9, H-14, H-16, and H-17. Moreover, the β orientation for H-20 was evidenced from the NOE correlations between H
3-18/H-20, H
3-18/H-12a, and H
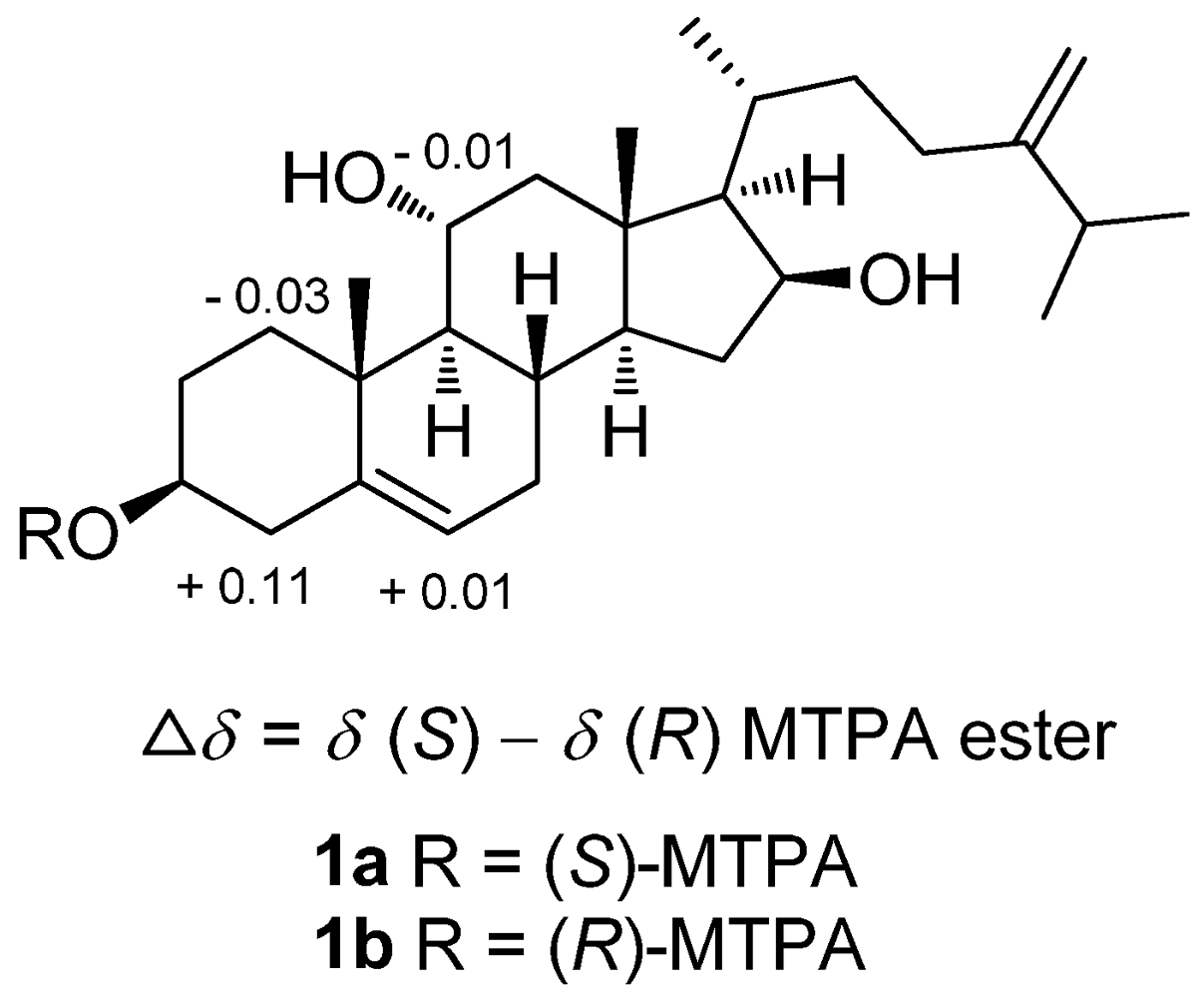
3-21/H-12a. The absolute configuration of
1 was determined by the application of Mosher’s method [
14]. Analysis of the
1H NMR data of esters
1a and
1b resulted in the determination of a 3
S configuration (
Figure 3).
Table 1.
13C NMR spectroscopic data of compounds 1−5.
Table 1.
13C NMR spectroscopic data of compounds 1−5.
| position | 1a | 2b | 3b | 4a | 5a |
|---|
| 1 | 39.1, CH2 | 39.2, CH2 | 39.2, CH2 | 39.2, CH2 | 37.1, CH2 |
| 2 | 31.8, CH2 | 29.7, CH2 | 29.5, CH2 | 29.7, CH2 | 29.6, CH2 |
| 3 | 71.8, CH | 78.2, CH | 77.8, CH | 78.2, CH | 78.3, CH |
| 4 | 42.7, CH2 | 39.1, CH2 | 39.0, CH2 | 39.1, CH2 | 38.8, CH2 |
| 5 | 141.2, C | 140.6, C | 140.7, C | 140.5, C | 140.3, C |
| 6 | 121.3, CH | 121.7, CH | 121.6, CH | 121.8, CH | 122.1, CH |
| 7 | 31.9, CH2 | 31.8, CH2 | 31.8, CH2 | 31.8, CH2 | 31.5, CH2 |
| 8 | 31.4, CH | 31.3, CH | 31.3, CH | 31.3, CH | 30.4, CH |
| 9 | 56.9, CH | 56.8, CH | 56.9, CH | 56.8, CH | 50.3, CH |
| 10 | 38.1, C | 38.3, C | 38.3, C | 38.3, C | 36.9, C |
| 11 | 68.9, CH | 68.9, CH | 68.9, CH | 68.9, CH | 21.0, CH2 |
| 12 | 51.4, CH2 | 51.4, CH2 | 51.4, CH2 | 51.4, CH2 | 36.2, CH2 |
| 13 | 42.9, C | 42.9, C | 42.9, C | 42.9, C | 47.4, C |
| 14 | 53.7, CH | 53.6, CH | 53.6, CH | 53.6, CH | 57.9, CH |
| 15 | 36.3, CH2 | 36.3, CH2 | 36.3, CH2 | 36.3, CH2 | 31.0, CH2 |
| 16 | 72.5, CH | 72.5, CH | 72.5, CH | 72.5, CH | 123.8, CH |
| 17 | 61.2, CH | 61.2, CH | 61.2, CH | 61.2, CH | 160.9, CH |
| 18 | 14.0, CH3 | 14.1, CH3 | 14.1, CH3 | 14.1, CH3 | 18.1, CH3 |
| 19 | 19.1, CH3 | 19.0, CH3 | 19.0, CH3 | 19.0, CH3 | 19.3, CH3 |
| 20 | 29.6, CH | 29.6, CH | 29.6, CH | 29.6, CH | 76.0, C |
| 21 | 18.2, CH3 | 18.2, CH3 | 18.2, CH3 | 18.2, CH3 | 29.6, CH3 |
| 22 | 34.8, CH2 | 34.8, CH2 | 34.7, CH2 | 34.7, CH2 | 49.1, CH2 |
| 23 | 31.2, CH2 | 31.2, CH2 | 31.2, CH2 | 31.2, CH2 | 29.6, CH |
| 24 | 156.9, C | 156.9, C | 156.9, C | 156.9, C | 45.5, CH |
| 25 | 34.1, CH | 34.0, CH | 34.0, CH | 34.0, CH | 30.9, CH |
| 26 | 21.8, CH3 | 21.8, CH3 | 21.8, CH3 | 21.8, CH3 | 20.9, CH3 |
| 27 | 21.9, CH3 | 21.9, CH3 | 21.9, CH3 | 21.9, CH3 | 21.5, CH3 |
| 28 | 106.3, CH2 | 106.3, CH2 | 106.3, CH2 | 106.4, CH2 | 11.6, CH3 |
| 29 | | | | | 15.7, CH3 |
| 1’ | | 97.4, CH | 94.7, CH | 97.2, CH | 97.2, CH |
| 2’ | | 74.0, CH | 68.6, CH | 70.1, CH | 70.1, CH |
| 3’ | | 66.9, CH | 72.1, CH | 69.5, CH | 69.5, CH |
| 4’ | | 70.9, CH | 72.4, CH | 73.0, CH | 73.0, CH |
| 5’ | | 65.8, CH | 65.3, CH | 65.2, CH | 65.2, CH |
| 6’ | | 16.0, CH3 | 16.1, CH3 | 16.2, CH3 | 16.2, CH3 |
| OAc | | 170.9, C | 171.5, C | 171.3, C | 171.3, C |
| | | 21.2, CH3 | 21.1, CH3 | 20.8, CH3 | 20.8, CH3 |
Figure 2.
Selected 1H−1H COSY (▬) and HMBC (→) correlations of 1 and 5 and the fucose residue in 1–5.
Figure 2.
Selected 1H−1H COSY (▬) and HMBC (→) correlations of 1 and 5 and the fucose residue in 1–5.
Analysis of the HRESIMS and
13C NMR spectroscopic data of crassarosteroside A (
2) suggested a molecular formula of C
36H
58O
8. The IR spectrum of
2 showed the presence of hydroxy (3461 cm
−1) and carbonyl (1741 cm
−1) groups. The latter was identified as an acetoxy group according to the carbon resonances at
δ 170.9 (C) and 21.2 (CH
3) (
Table 1). The
1H NMR spectroscopic data of
2 showed characteristic methyl signals at
δ 0.92 (3H, s), 1.18 (3H, s), 1.04 (3H, d,
J = 6.8 Hz), 1.03 (6H, d,
J = 7.0 Hz), 5.41 (1H, d,
J = 5.6 Hz), 4.76 (1H, s), and 4.70 (1H, s) (
Table 2), revealing that
2 has the same 24-methylenecholesterol skeleton as that of
1. By excluding the steroidal moiety and the acetoxy group, the remaining six carbons [δ 97.4 (CH), 74.0 (CH), 66.9 (CH), 70.9 (CH), 65.8 (CH), and 16.0 (CH
3)] were ascribed to the presence of a 6’-deoxyhexose residue. The sugar residue was deduced as an α-fucopyranose on the basis of 2D NMR analysis (
Figure 2) and the coupling constants of
3JH-1’,H-2’ (4.0 Hz),
3JH-2’,H-3’ (10.0 Hz),
3JH-3’,H-4’ (3.5 Hz), and
3JH-4’,H-5’ (<1 Hz) (
Table 2) [
12,
15,
16]. The acetoxy group attached at C-2’ of the α-fucose residue was evidenced from the downfield chemical shift of H-2’ (
δ 5.07), which was confirmed by the HMBC correlations from this proton to the acetate carbonyl carbon. The HMBC correlation from H-1’ to C-3 revealed that the α-fucose residue was attached at C-3 of the steroidal aglycone. The absolute configuration of the sugar moiety in
2 was determined by reversed phase HPLC analysis of its
o-tolylthiocarbamate [
17]. The liberated fucose from acid hydrolysis of
2 was treated with
L-cysteine methyl ester followed by reaction with
o-tolylisothiocyanate to afford the corresponding
o-tolylthiocarbamate derivative. The retention time of the liberated sugar derivative by HPLC analysis was found to be consistent with that of a standard
L-fucose derivative.
Crassarosteroside B (
3) gave the same molecular formula as that of
2 based on the analysis of the HRESIMS and
13C NMR spectroscopic data (
Table 1). The NMR spectroscopic data of
3 were similar to those of
2, with some exceptions for those of the sugar residue. An HMBC correlation from the anomeric proton at
δ 5.12 (H-1’) to the carbon signal at
δ 77.8 (C-3) connected the fucose residue to C-3 of the steroidal aglycone. The downfield proton chemical shift at
δ 4.87 (1H, dd,
J = 10.5, 4.0 Hz) was ascribed to the presence of an acetoxy group at C-3’ (
Table 2). The detailed 2D NMR analysis confirmed the above elucidation. Likewise, the
L-fucose residue was deduced according to RP HPLC analysis of the corresponding
o-tolylthiocarbamate as described above.
Figure 3.
1H NMR chemical shift differences of MTPA esters of 1.
Figure 3.
1H NMR chemical shift differences of MTPA esters of 1.
Crassarosteroside C (
4) was assigned the same molecular formula as those of
2 and
3. A comparison of NMR spectroscopic data of
4 with those of
2 and
3 revealed that an acetoxy group should be located at C-4’ of the fucose residue (
Table 1 and
Table 2). This was evidenced by the
1H NMR shift of H-4’ at 5.20 (1H, d,
J = 2.8 Hz). In the same manner, RP HPLC anslysis of the corresponding
o-tolylthiocarbamate derived from the hydrolyte of
4 allowed the determination of the
L-fucose moiety.
The HRESIMS and
13C NMR spectroscopic data of crassarosteroside D (
5) established a molecular formula of C
37H
60O
7. The presence of an acetoxy group was evidenced by the
1H NMR signal at δ 2.17 (3H, s) (
Table 1) and
13C NMR signals at δ 171.3 (C) and 20.8 (CH
3) (
Table 2) as well as the IR absorption band at 1737 cm
−1. The NMR spectroscopic data for the sugar moiety of
5 were quite similar to those of
4, suggesting that they shared the same 4’-
O-acetylfucose residue. Except for the sugar moiety, the remaining 29 carbon signals as well as the characteristic methyl signals at
δ 1.00 (3H, s), 1.05 (3H, s), 1.38 (3H, s), 0.86 (3H, d,
J = 6.4 Hz), 0.89 (3H, d,
J = 6.4 Hz), 0.76 (3H, d,
J = 6.8 Hz), and 0.78 (3H, d,
J = 6.4 Hz) (
Table 2) revealed that the aglycone of
5 should have a C
29 steroidal skeleton [
18]. The 23,24-dimethyl-20-hydroxy side chain was deduced by the
1H−
1H COSY correlations from H
2-22 to H
3-29 through H-23, from H-24 to both H
3-26 and H
3-27, and from H-24 to H
3-28 as well as the HMBC correlations from H
3-21 to C-17, C-20, and C-22 and H
3-29 to C-22, C-23, and C-24. This rare steroidal side chain is the same as that of sarcophytosterol isolated previously by us from the soft coral
Lobophytum sarcophytoides [
18]. The NMR spectroscopic data for the aglycone moiety of
5 are almost the same as those of sarcophytosterol, except for some minor variations in
1H and
13C chemical shifts at C-2, C-3, and C-4 between both compounds. This is due to the attachment of the sugar residue at C-3 of the steroidal aglycone. Similarly, HPLC analysis of the relevant
o-tolylthiocarbamate derived from the hydrolysis of
5 suggested the presence of
L-fucose.
Table 2.
1H NMR spectroscopic data of compounds 1−5.
Table 2.
1H NMR spectroscopic data of compounds 1−5.
| # | 1, δH (J in Hz)a | 2, δH (J in Hz)b | 3, δH (J in Hz)b | 4, δH (J in Hz)a | 5, δH (J in Hz)a |
|---|
| 1 | a: 2.55, dt | a: 2.58, dt | a: 2.56, dt | a: 2.58, dt | a: 1.86, m |
| | (13.6, 3.6) | (13.5, 3.5) | (13.5, 3.5) | (14.0, 3.6) |
| | b: 1.16, m | b: 1.16, m | b: 1.16, m | b: 1.16, m | b: 1.10, m |
| 2 | a: 1.81, m | a: 1.85, m | a: 1.79, m | a: 1.81, m | a: 1.89, m |
| | b: 1.58, m | b: 1.65, m | b: 1.66, m | b: 1.64, m | b: 1.60, m |
| 3 | 3.53, m | 3.51, m | 3.43, m | 3.49, m | 3.49, m |
| 4 | a: 2.30, m | a: 2.36, m | 2.24, m | a: 2.36, m | a: 2.36, m |
| | b: 2.26, m | b: 2.26, m | | b: 2.26, m | b: 2.24, m |
| 6 | 5.41, d (5.6) | 5.41, d (5.5) | 5.40, d (5.5) | 5.41, d (5.6) | 5.38, br d (3.2) |
| 7 | a: 1.99, m | a: 1.99, m | a: 1.98, m | a: 1.99, m | a: 2.01, m |
| | b: 1.54, m | b: 1.56, m | b: 1.54, m | b: 1.54, m | b: 1.61, m |
| 8 | 1.50, m | 1.50, m | 1.49, m | 1.50, m | 1.66, m |
| 9 | 0.99, m | 0.99, m | 0.97, m | 0.98, m | 1.01, m |
| 11 | 4.07, td | 4.07, m | 4.06, m | 4.07, td | 1.59, m |
| | (10.8, 4.8) | (10.8, 4.4) |
| 12 | a: 2.31, m | a: 2.31, m | a: 2.31, m | a: 2.31, m | a: 2.10, m |
| | b: 1.18, m | b: 1.18, m | b: 1.18, m | b: 1.18, m | b: 1.59, m |
| 14 | 0.98, m | 0.98, m | 0.98, m | 0.98, m | 1.41, m |
| 15 | a: 2.24, m | a: 2.24, m | a: 2.23, m | a: 2.23, m | a: 2.08, m |
| | b: 1.17, m | b: 1.16, m | b: 1.17, m | b: 1.16, m | b: 1.87, m |
| 16 | 4.40, m | 4.40, m | 4.40, m | 4.40, m | 5.50, br s |
| 17 | 1.07, m | 1.07, m | 1.07, m | 1.07, m | |
| 18 | 0.92, s | 0.92, s | 0.92, s | 0.92, s | 1.00, s |
| 19 | 1.18, s | 1.18, s | 1.17, s | 1.18, s | 1.05, s |
| 20 | 1.86, m | 1.86, m | 1.86, m | 1.87, m | |
| 21 | 1.04, d (6.8) | 1.04, d (6.8) | 1.04, d (6.5) | 1.04, d (6.4) | 1.38, s |
| 22 | a: 1.68, m | a: 1.68, m | a: 1.68, m | a: 1.67, m | a: 1.59, m |
| | b: 1.22, m | b: 1.22, m | b: 1.22, m | b: 1.22, m | b: 1.48, m |
| 23 | a: 2.18, m | a: 2.18, m | a: 2.18, m | a: 2.18, m | 1.82, m |
| | b: 1.95, m | b: 1.95, m | b: 1.95, m | b: 1.95, m | |
| 24 | | | | | 1.06, m |
| 25 | 2.24, m | 2.25, m | 2.25, m | 2.25, m | 1.42, m |
| 26 | 1.03, d (7.2) | 1.03, d (7.0) | 1.03, d (7.0) | 1.03, d (6.8) | 0.86, d (6.4) |
| 27 | 1.03, d (7.2) | 1.03, d (7.0) | 1.03, d (7.0) | 1.03, d (6.8) | 0.89, d (6.4) |
| 28 | a: 4.76, s | a: 4.76, s | a: 4.76, s | a: 4.76, s | 0.76, d (6.8) |
| | b: 4.70, s | b: 4.70, s | b: 4.70, s | b: 4.70, s | |
| 29 | | | | | 0.78, d (7.2) |
| 1’ | | 5.02, d (4.0) | 5.12, d (4.0) | 5.04, d (4.0) | 5.04, d (4.0) |
| 2’ | | 5.07, dd (10.0, 4.0) | 4.02, m | 3.93, dd (10.0, 4.0) | 3.93, dd (10.0, 4.0) |
| 3’ | | 3.91, ddd (11.5,10.0, 3.5) | 4.87, dd (10.5, 4.0) | 3.74, br d (10.0) | 3.74, dd (10.0, 2.4) |
| 4’ | | 3.84, br s | 3.82, br s | 5.20, d (2.8) | 5.21, d (2.4) |
| 5’ | | 4.11, q (7.0) | 4.12, q (6.5) | 4.12, q (6.4) | 4.12, q (6.4) |
| 6’ | | 1.25, d (7.0) | 1.27, d (6.5) | 1.13, d (6.4) | 1.14, d (6.4) |
| OAc | | 2.18, s | 2.15, s | 2.17, s | 2.17, s |
| 3’-OH | | 1.90, d (11.5) | | | |
| 4’-OH | | 1.89, br s | 2.18, br s | | |
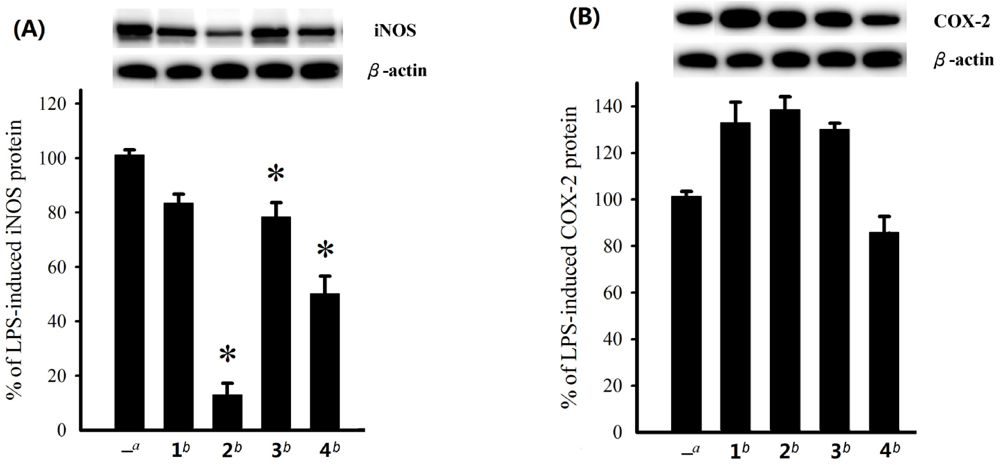
Figure 4.
Effect of compounds 1–4 at 10 μM on the LPS-induced pro-inflammatory iNOS and on COX-2 protein expression of RAW264.7 macrophage cells by immunoblot analysis. (A) Immunoblots for iNOS and β-actin, and relative density of iNOS. (B) Immunoblots for COX-2 and β-actin, and relative density of COX-2. The values are means ± SEM (n = 6). The relative intensity of the LPS alone stimulated group was taken as 100%. Under the same experimental conditions, 10 μM CAPE (caffeic acid phenethyl ester; Sigma Chemical Company, St. Louis, MO) reduced the levels of the iNOS and COX-2 protein to 0.8 ± 4.5% and 75.6 ± 12.2%, respectively, relative to the control cells stimulated with LPS. *Significantly different from LPS alone stimulated group (*P < 0.05).
Figure 4.
Effect of compounds 1–4 at 10 μM on the LPS-induced pro-inflammatory iNOS and on COX-2 protein expression of RAW264.7 macrophage cells by immunoblot analysis. (A) Immunoblots for iNOS and β-actin, and relative density of iNOS. (B) Immunoblots for COX-2 and β-actin, and relative density of COX-2. The values are means ± SEM (n = 6). The relative intensity of the LPS alone stimulated group was taken as 100%. Under the same experimental conditions, 10 μM CAPE (caffeic acid phenethyl ester; Sigma Chemical Company, St. Louis, MO) reduced the levels of the iNOS and COX-2 protein to 0.8 ± 4.5% and 75.6 ± 12.2%, respectively, relative to the control cells stimulated with LPS. *Significantly different from LPS alone stimulated group (*P < 0.05).
The absolute configuration of sterol
1 has been established by Mosher’s method in the present work. On the basis of biogenesis, the steroidal moieties of the glycosides
2–
5 should possess the same absolute configurations as shown in the formulae. Cytotoxicity of steroids
1–
5 against HepG2, HepG3, MCF-7, MDA-MB-231, and A-549 cancer cell lines was evaluated. The results showed that
1 exhibited cytotoxicity toward HepG2 cancer cell line with an IC
50 value of 14.9 µM, while
4 also showed cytotoxicity toward HepG2 and HepG3 cell lines with IC
50 values of 17.6 and 18.9 µM, respectively. The other compounds were found to be inactive (IC
50 > 20 μM) toward the above cancer cell lines after 72 h exposure. The effect of steroids
1–
4 on the expression of pro-inflammatory iNOS and COX-2 proteins in RAW264.7 macrophage cells stimulated with LPS was also evaluated using immunoblot analysis. At a concentration of 10 µM (
Figure 4), steroid
2 was found to significantly reduce the level of iNOS protein to 12.9 ± 4.3% and
4 could reduce the iNOS espression to 50.1 ± 6.3%. However, these compounds could not effectively reduce the level of COX-2. On the contrary,
1–
3 were found to stimulate the expression of COX-2.
3. Experimental Section
3.1. General Experimental Procedures
The melting point was determined using a Fisher-Johns melting point apparatus. Optical rotations were determined with a JASCO P1020 digital polarimeter. IR spectrum was obtained on a JASCO FT/IR-4100 spectrophotometer. The NMR spectra were recorded on a Varian 400 MR NMR or Varian Unity INOVA 500 FT-NMR instrument at 400 or 500 MHz for 1H (referenced to TMS, δH 0.00 ppm for CDCl3) and 100 or 125 MHz for 13C (referenced to δC 77.0 for CDCl3). ESIMS and HRESIMS were recorded by ESI FT-MS on a Bruker APEX II mass spectrometer. Silica gel 60 (Merck, 230−400 mesh) and LiChroprep RP-18 (Merck, 40–63 μm) were used for column chromatography. Precoated silica gel plates (Merck, Kieselgel 60 F254, 0.25 mm) and precoated RP-18 F254S plates (Merck, 1.05560) were used for TLC analyses. High-performance liquid chromatography (HPLC) was performed on a Hitachi L-7100 pump equipped with a Hitachi L-7400 UV detector at 210 nm and a semi-preparative reversed-phase column (Merck, Hibar Purospher RP-18e, 5 μm, 250 × 10 mm).
3.2. Animal Material
The soft coral Sinularia crassa was collected by hand using scuba off the coast of Sansiantai, Taitung county, Taiwan, in July 2008, at a depth of 10 m, and was stored in a freezer. This soft coral was identified by one of the authors (C.-F.D.). A voucher specimen (specimen no. SST-03) was deposited in the Department of Marine Biotechnology and Resources, National Sun Yat-sen University.
3.3. Extraction and Isolation
The frozen bodies of S. crassa (1.1 kg fresh wt) were minced and extracted with EtOH (3 × 2 L). The organic extract was concentrated to an aqueous suspension and was further partitioned between EtOAc and H2O. The EtOAc extract (17.0 g) was fractionated by open column chromatography on silica gel using n-hexane–EtOAc and EtOAc–MeOH mixtures of increasing polarity to yield 32 fractions. Fractions 25, eluting with EtOAc–MeOH (8:1), was further separated by silica gel column chromatography with gradient elution (n-hexane–acetone, 8:1 to 2:1) to yield four subfractions (25A–25D). Subfraction 25B was subjected to RP-18 column chromatography (MeOH–H2O, gradient, 50–90%), and subsequently purified by RP-18 HPLC (CH3CN–H2O, 65%) to obtain compounds 1 (6.6 mg) and 5 (1.2 mg). Compound 4 (1.8 mg) was obtained from subfraction 25C using RP-18 HPLC (CH3CN–H2O, 65%). In the same manner, Subfraction 25D was fractionated over RP-18 gel using gradient elution (MeOH–H2O, gradient, 50–90%) to yield two subfractions (25D-1 and 25D-2). Subfraction 25D-2 was separated by RP-18 HPLC (CH3CN–H2O, 85%) to yield compounds 2 (1.8 mg) and 3 (1.6 mg).
Crassarosterol A (
1): white powder; [α]
24D −45 (
c 0.66, CHCl
3); IR (KBr)
vmax 3389, 2962, 2925, 2854, 1462, 1048, 1024 cm
−1;
13C NMR and
1H NMR data, see
Table 1 and
Table 2; ESIMS
m/z 453 [M + Na]
+; HRESIMS
m/z 453.3342 [M + Na]
+ (calcd for C
28H
46O
3Na, 453.3344).
Crassarosteroside A (
2): white powder; [α]
24D−34 (
c 0.18, CHCl
3); IR (KBr)
vmax 3461, 2960, 2928, 2868, 1741, 1467, 1377, 1244, 1077, 1030 cm
−1;
13C NMR and
1H NMR data, see
Table 1 and
Table 2; ESIMS
m/z 641 [M + Na]
+; HRESIMS
m/z 641.4027 [M + Na]
+ (calcd for C
36H
58O
8Na, 641.4029).
Crassarosteroside B (
3): white powder; [α]
24D−17 (
c 0.16, CHCl
3); IR (KBr)
vmax 3388, 2963, 2930, 2857, 1732, 1458, 1375, 1258, 1041 cm
−1;
13C NMR and
1H NMR data, see
Table 1 and
Table 2; ESIMS
m/z 641 [M + Na]
+; HRESIMS
m/z 641.4026 [M + Na]
+ (calcd for C
36H
58O
8Na, 641.4029).
Crassarosteroside C (
4): white powder; [α]
24D−52 (
c 0.18, CHCl
3); IR (KBr)
vmax 3426, 2960, 2930, 2859, 1735, 1461, 1375, 1247, 1074, 1033 cm
−1;
13C NMR and
1H NMR data, see
Table 1 and
Table 2; ESIMS
m/z 641 [M + Na]
+; HRESIMS
m/z 641.4026 [M + Na]
+ (calcd for C
36H
58O
8Na, 641.4029).
Crassarosteroside D (
5): white powder; [α]
24D−45 (
c 0.12, CHCl
3); IR (KBr)
vmax 3440, 2960, 2925, 2855, 1737, 1461, 1377, 1244, 1074, 1036 cm
−1;
13C NMR and
1H NMR data, see
Table 1 and
Table 2; ESIMS
m/z 639 [M + Na]
+; HRESIMS
m/z 639.4234 [M + Na]
+ (calcd for C
37H
60O
7Na, 639.4237).
3.4. Preparation of (S)-and (R)-MTPA Esters of 1
To a solution of 1 (1.0 mg) in pyridine (0.4 mL) was added (R)-MTPA chloride (25 μL), and the mixture was allowed to stand for 3 h at room temperature. The reaction was quenched by the addition of 1.0 mL of H2O, and the mixture was subsequently extracted with EtOAc (3 × 1.0 mL). The EtOAc layers were combined, dried over anhydrous MgSO4, and evaporated. The residue was subjected to short silica gel column chromatography using n-hexane−EtOAc (3:1) to yield the (S)-MTPA ester, 1a (0.7 mg). The same procedure was used to prepare the (R)-MTPA ester, 1b (1.0 mg from 1.0 mg of 1), with (S)-MTPA chloride. Selected 1H NMR (CDCl3, 400 MHz) of 1a: δ 7.41−7.52 (5H, m, Ph), 5.48 (1H, br d, J = 6.0 Hz, H-6), 4.89 (1H, m, H-3), 4.76 (1H, s, H-28a), 4.70 (1H, s, H-28b), 4.41 (1H, m, H-16), 4.05 (1H, m, H-11), 3.57 (3H, s, OMe), 2.62 (1H, br d, J = 14.0 Hz, H-1a), 2.48 (1H, m, H-4a), 1.85 (1H, m, H-2a), 1.17 (3H, s, H3-19), 1.03 (6H, d, J = 7.2 Hz, H3-26 and 27), 0.92 (3H, s, H3-18); selected 1H NMR (CDCl3, 400 MHz) of 1b: δ 7.41−7.53 (5H, m, Ph), 5.47 (1H, br d, J = 5.2 Hz, H-6), 4.89 (1H, m, H-3), 4.76 (1H, s, H-28a), 4.70 (1H, s, H-28b), 4.41 (1H, m, H-16), 4.06 (1H, m, H-11), 3.57 (3H, s, OMe), 2.65 (1H, br d, J = 13.6 Hz, H-1a), 2.37 (1H, m, H-4a), 1.77 (1H, m, H-2a), 1.17 (3H, s, H3-19), 1.03 (6H, d, J = 7.2 Hz, H3-26 and 27), 0.92 (3H, s, H3-18).
3.5. Determination of Sugar Configuration
Authentic samples of D-fucose and L-cysteine methyl ester hydrochloride (each 0.5 mg) were dissolved in pyridine (0.1 mL) and heated at 60 °C for 1 h. To this mixture was added o-tolylisothiocyanate (0.5 mg in 0.1 mL pyridine) and heated at 60 °C for additional 1 h. The reaction mixture was directly analyzed by reversed-phase HPLC (Mightysil RP-18 GP column; 4.6 × 250 nm; 25% CH3CN in 50 mM H3PO4; 0.8 mL/min; 35 °C) and detected at 250 nm to give the retention time of the o-tolylthiocarbamate of sugar. The retention of the o-tolylthiocarbamate derived from L-fucose, L-cysteine methyl ester, and o-tolylisothiocyanate was obtained by the same manner.
A solution of the glycoside (0.4 mg for each) in 0.6 M HCl/dioxane (1:1 v/v, 0.2 mL) was heated at 80 °C for 24 h. After cooling, the solution was neutralized with Amberlite IRA-400, and the resin was removed by filtration. The filtrate was extracted with EtOAc. The aqueous layer was dried in vacuo and the afforded residue was dissolved in pyridine (0.1 mL) containing L-cysteine methyl ester (0.5 mg), followed by heating at 60 °C for 1 h. A 0.1 mL solution of o-tolylisothiocyanate (0.5 mg) in pyridine was added to the mixture, which was heated at 60 °C for additional 1 h, to yield the corresponding o-tolylthiocarbamate derivative. Reverse phase HPLC analysis of the o-tolylthiocarbamate derivatives derived from the hydrolytes of the glycosides 2–5 showed peaks at 28.2, 28.0, 28.1, and 27.9 min, respectively, while the tR values for standard L-fucose and D-fucose derivatives were observed at 28.0 and 25.4 min, respectively, suggesting the presence of a L-fucose residue in 2–5.
3.6. Cytotoxicity Testing
Cell lines were purchased from the American Type Culture Collection (ATCC). Compounds were assayed for cytotoxicity against human liver carcinoma (HepG2 and HepG3), human breast carcinoma (MCF-7 and MDA-MB-231), and human lung carcinoma (A-549) cells using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method [
19]. Freshly trypsinized cell suspensions were seeded in 96-well microtiter plates at densities of 5000–10000 cells per well with tested compounds added from DMSO-diluted stock. After 3 days in culture, attached cells were incubated with MTT (0.5 mg/mL, 1 h) and subsequently dissolved in DMSO. The absorbency at 550 nm was then measured using a microplate reader. The IC
50 is the concentration of agent that reduced cell growth by 50% under the experimental conditions.
3.7. In Vitro Anti-Inflammatory Assay
Macrophage (RAW264.7) cell line was purchased from ATCC. In vitro anti-inflammatory activities of tested compounds were measured by examining the inhibition of lipopolysaccharide (LPS) induced upregulation of iNOS and COX-2 proteins in macrophage cells using western blotting analysis [
20,
21].

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}


