Aniquinazolines A–D, Four New Quinazolinone Alkaloids from Marine-Derived Endophytic Fungus Aspergillus nidulans


Abstract
:1. Introduction
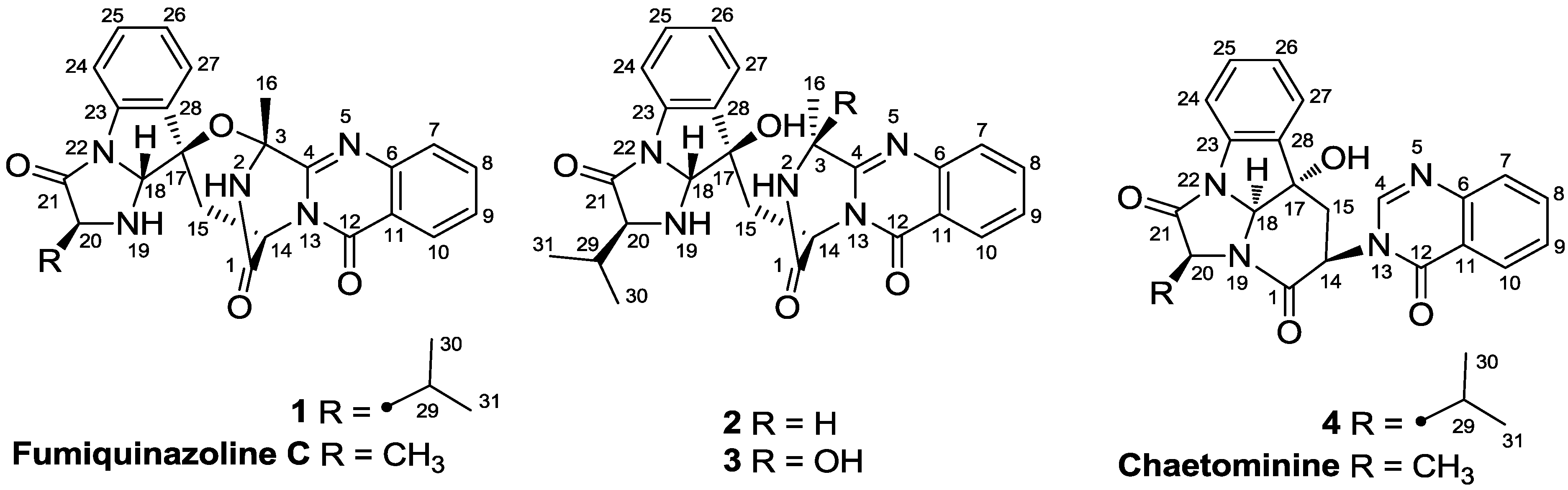
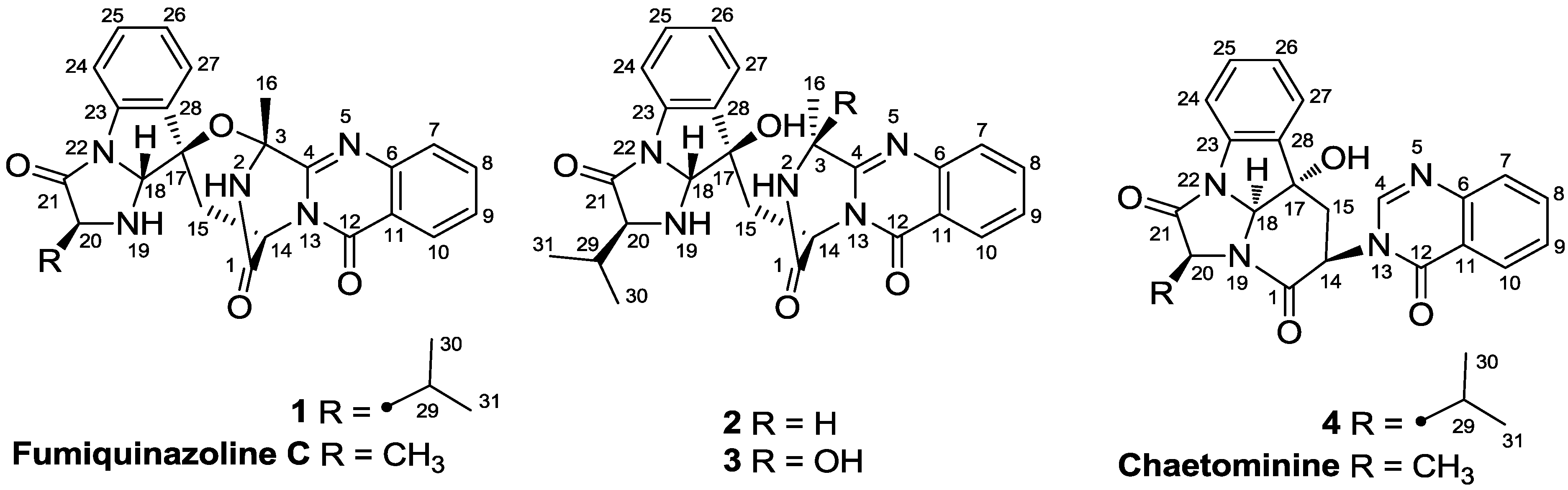
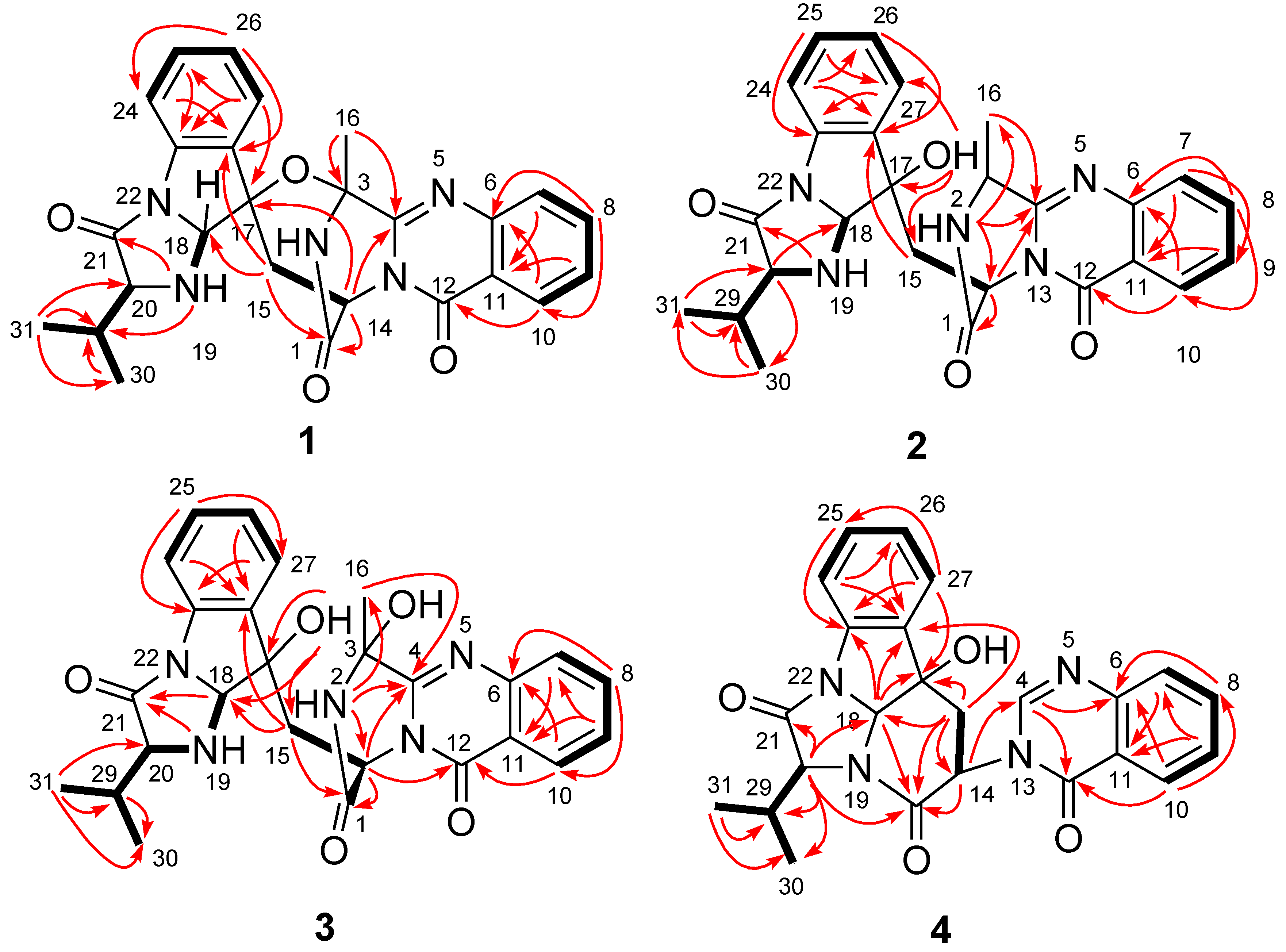
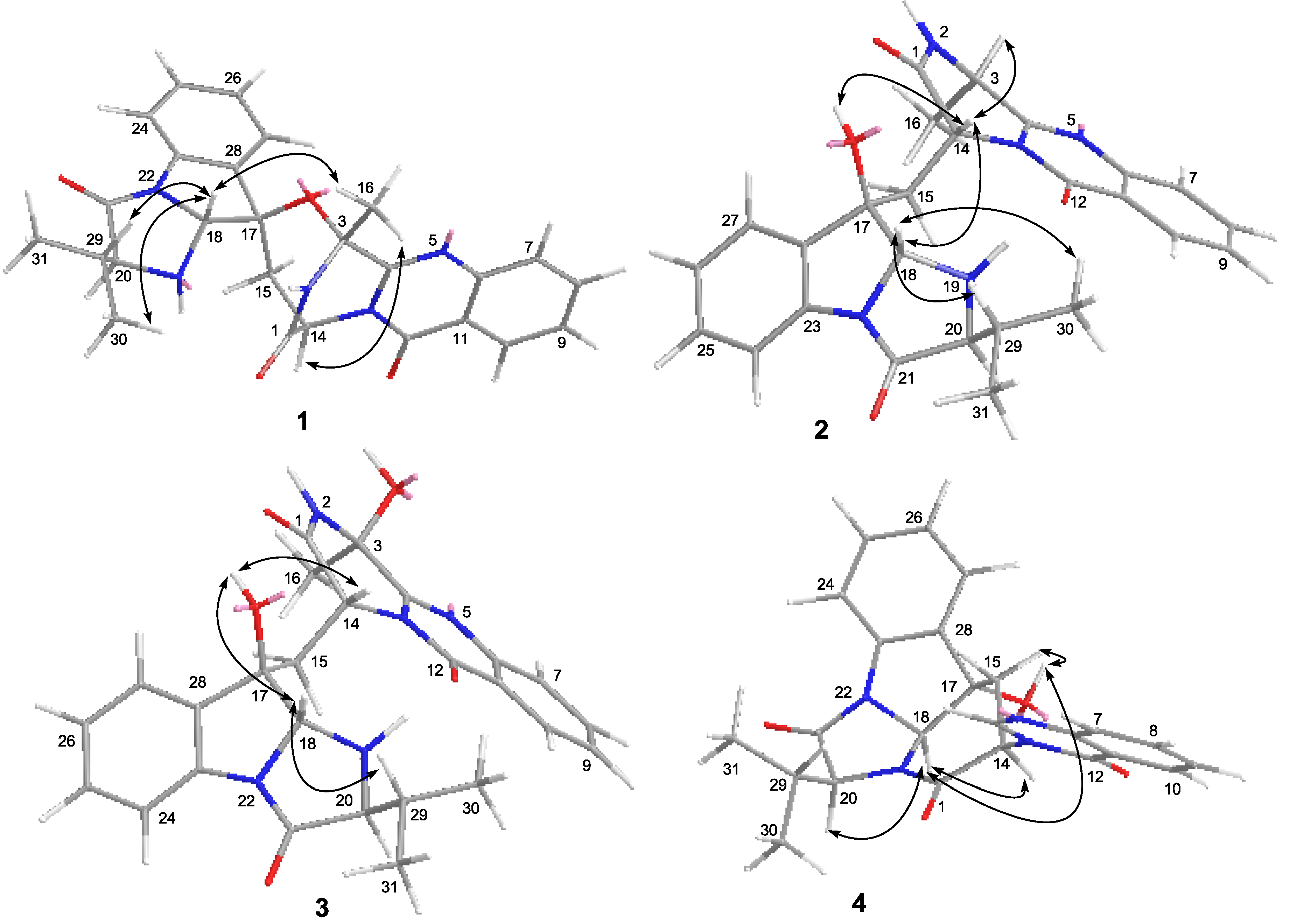
2. Results and Discussion
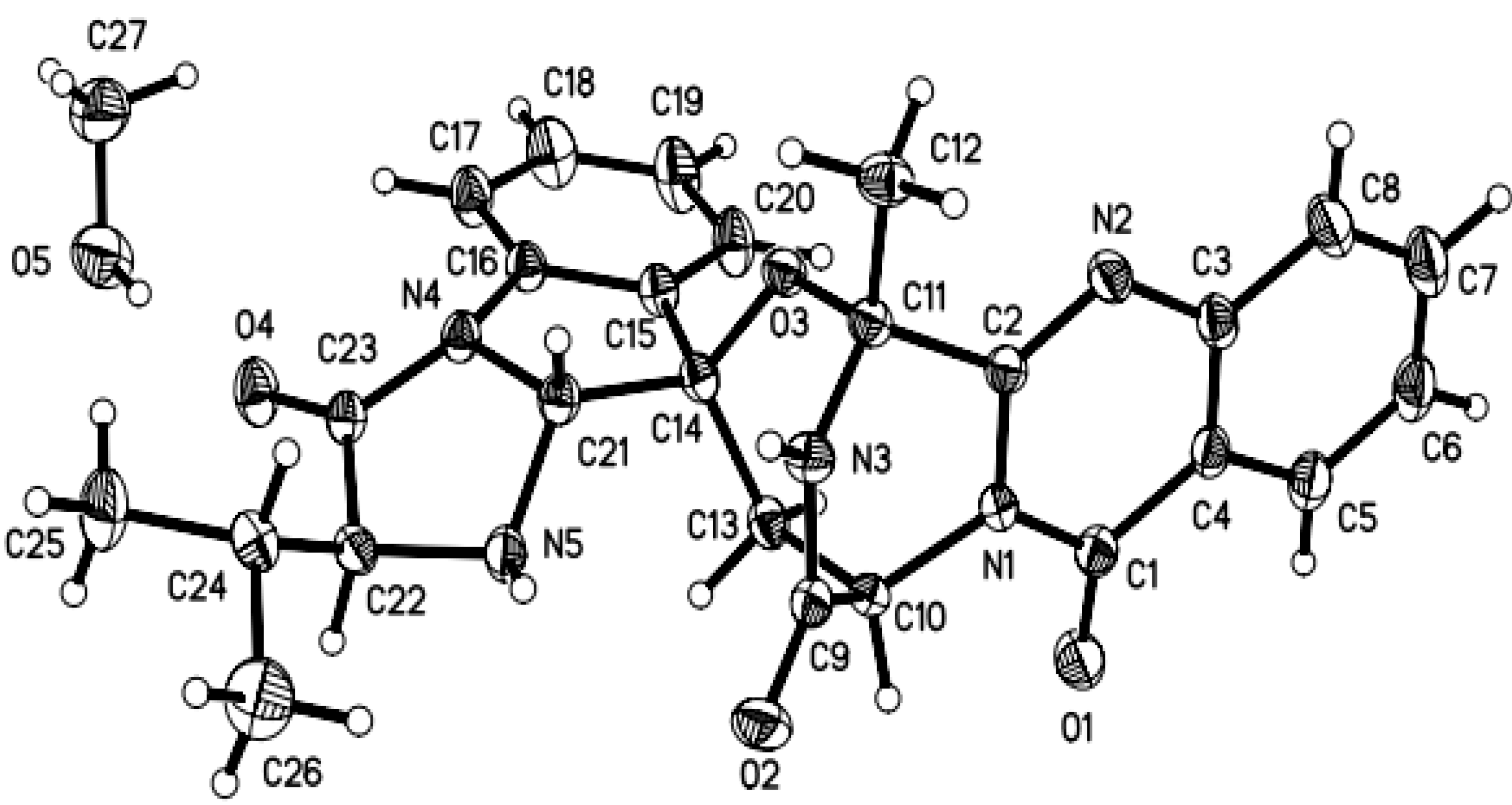
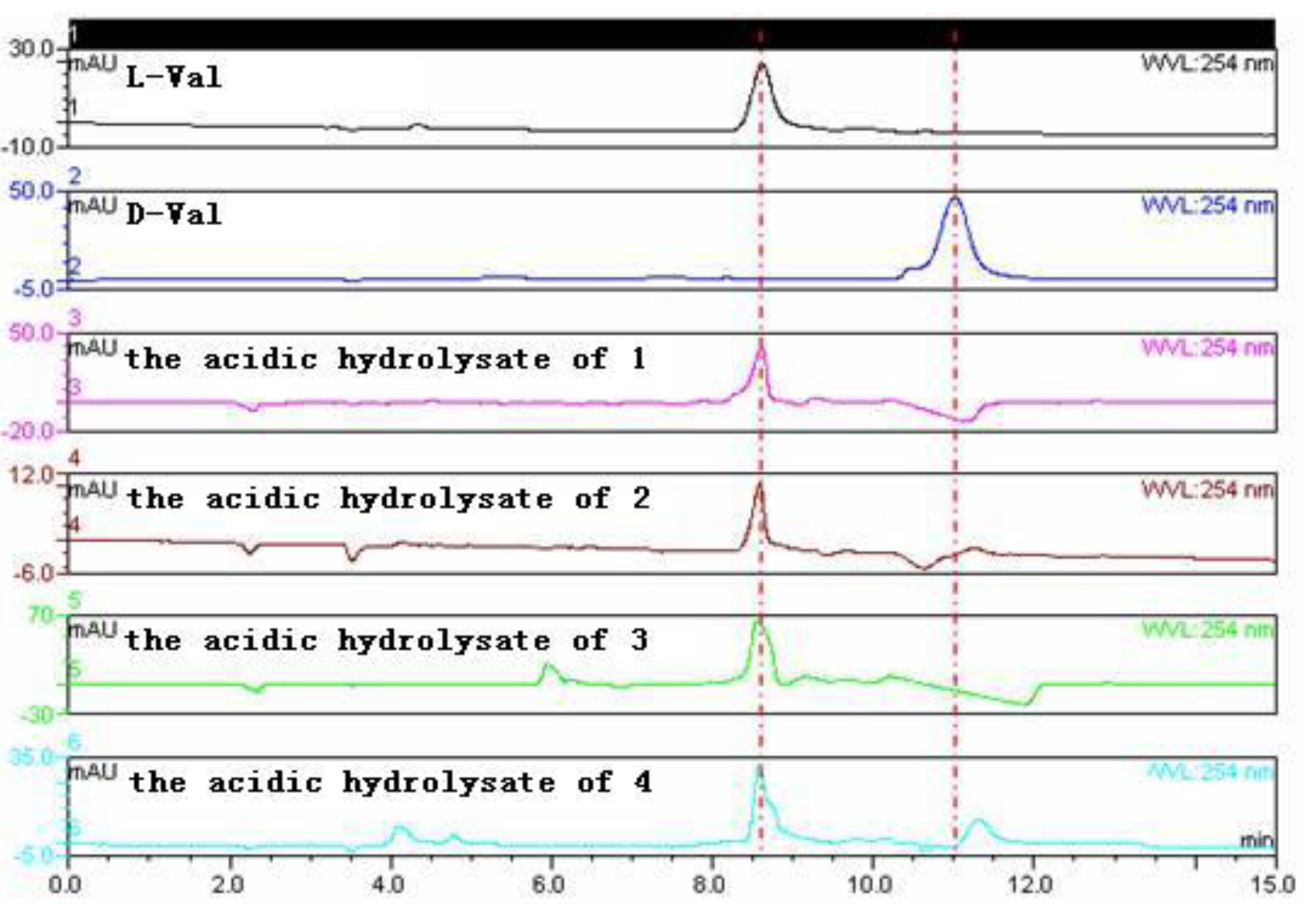
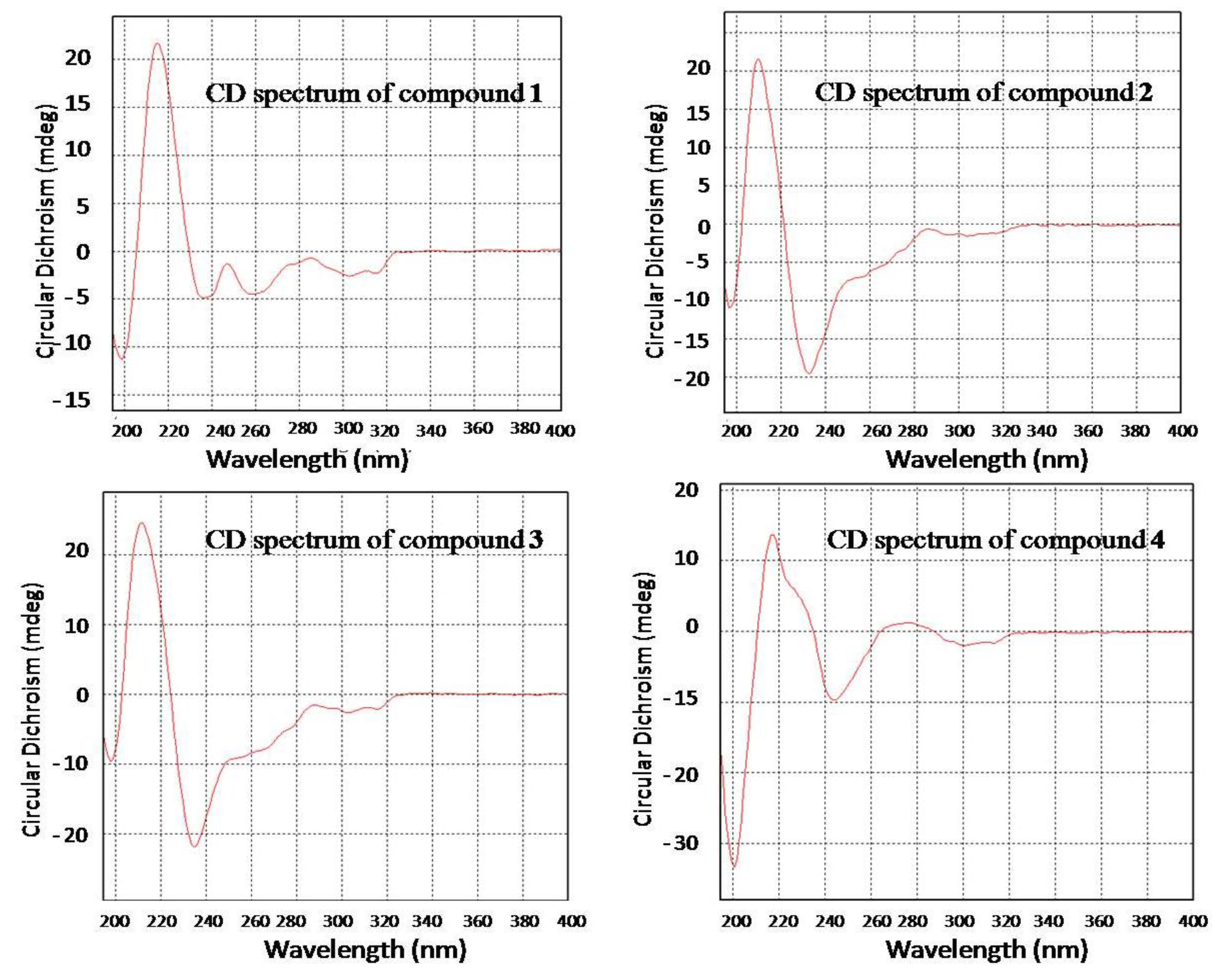
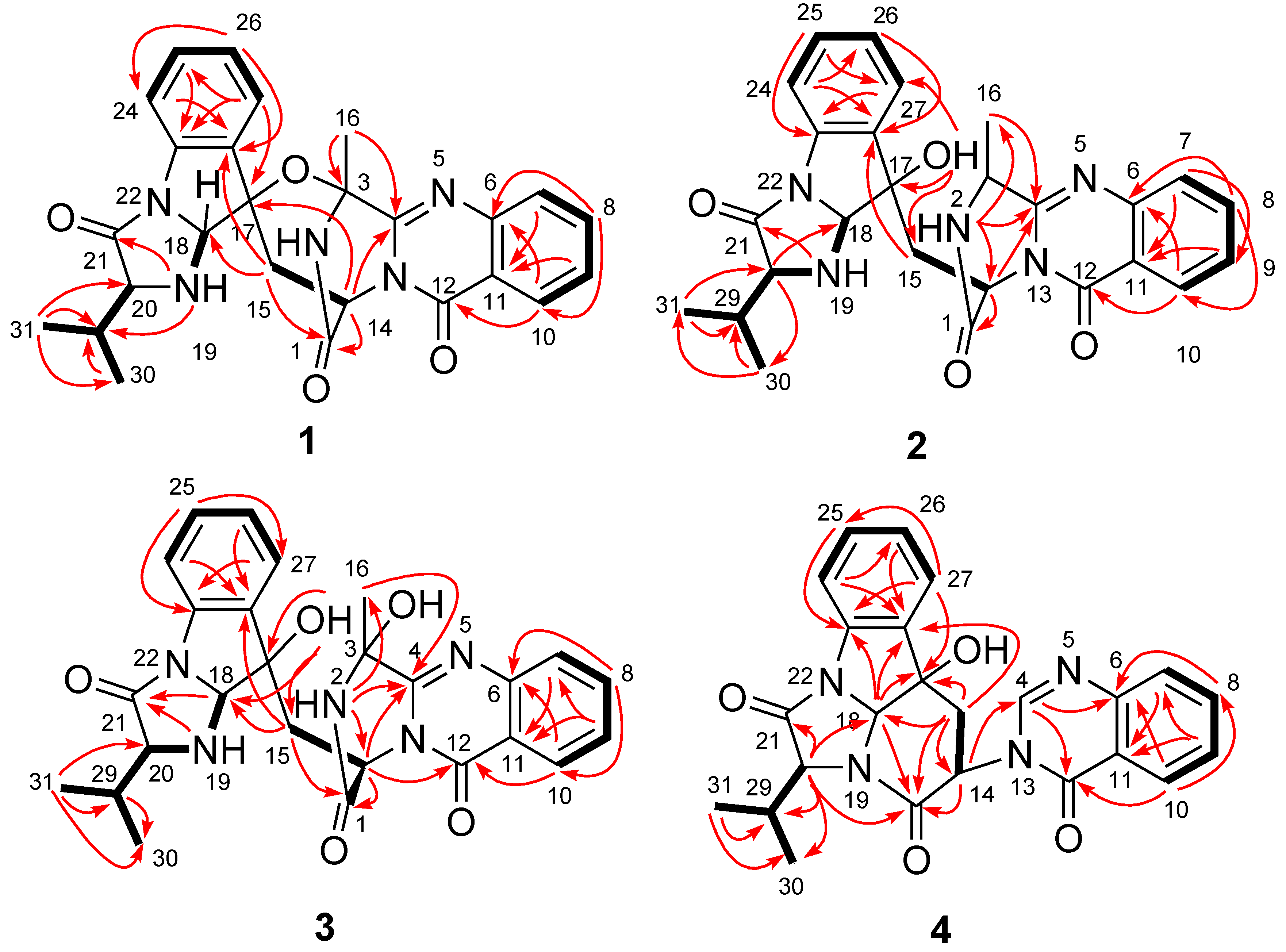
2.1. Structure Elucidation of the New Compounds 1–4

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 (J in Hz) | 2 (J in Hz) | 3 (J in Hz) | 4 (J in Hz) |
|---|---|---|---|---|
| 2-NH | 9.84, br s | 8.61, br s | 9.49, br s | |
| 3 | 4.94, q (6.5) | |||
| 4 | 8.30, s | |||
| 7 | 7.75, dd (7.9, 0.9) | 7.66, d (7.8) | 7.70, m | 7.71, dd (7.9, 0.8) |
| 8 | 7.88, td (7.9, 1.3) | 7.82, t (7.8) | 7.86, td (7.8, 1.2) | 7.87, td (7.9, 1.3) |
| 9 | 7.61, td (7.9, 0.9) | 7.53, t (7.8) | 7.58, td (7.8, 1.2) | 7.59, td (7.9, 0.8) |
| 10 | 8.18, dd (7.9, 1.3) | 8.12, d (7.8) | 8.16, dd (7.8, 1.2) | 8.18, dd (7.9, 1.3) |
| 14 | 5.36, dd (5.5, 1.5) | 5.58, dd (9.6, 4.3) | 5.44, dd (7.4, 6.5) | 4.86, dd (8.1, 5.3) |
| 15 | 2.98, dd (14.0, 5.5) 1.92,dd (14.0, 1.5) | 2.59, dd (14.8, 9.6) 1.83, dd (14.8, 4.3) | 2.61, dd (14.5, 7.4) 2.20, dd (14.5, 6.5) | 3.01, dd (14.0, 8.1) 2.77, dd (14.0, 5.3) |
| 16 | 1.91, s | 1.59, d (6.5) | 1.76, s | |
| 17-OH | 5.67, br s | 5.67, br s | 5.45, br s * | |
| 18 | 5.70, dd (6.0, 1.5) | 5.26, d (5.8) | 5.31, dd (7.3, 1.5) | 5.82, s |
| 19-NH | 2.67, d (5.5) | 3.51, m | 3.38, dd (7.3, 2.8) | |
| 20 | 3.59, br d (5.5) | 3.55, br s | 3.53, br s | 4.21, d (9.3) |
| 24 | 7.38, d (7.7) | 7.35, d (7.8) | 7.34, d (7.6) | 7.47, d (7.4) |
| 25 | 7.31, td (7.7, 0.9) | 7.26, t (7.8) | 7.27, td (7.6, 1.0) | 7.43, td (7.4, 1.2) |
| 26 | 7.04, td (7.7, 0.9) | 7.09, t (7.8) | 7.15, td (7.6, 1.0) | 7.26, td (7.4, 1.2) |
| 27 | 7.25, d (7.7) | 7.82, d (7.8) | 7.70, m | 7.54, d (7.4) |
| 29 | 2.09, dq (13.4, 6.8) | 1.99, m | 1.98, m | 2.45, m |
| 30 | 1.07, d (6.8) | 0.93, d (6.8) | 0.91, d (6.6) | 1.15, d (6.7) |
| 31 | 1.07, d (6.8) | 0.96, d (6.8) | 0.92, d (6.6) | 1.12, d (6.7) |
| Position | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| 1 | 168.6, C | 168.9, C | 169.7, C | 164.8, C |
| 3 | 84.2, C | 48.5, CH | 84.4, C | |
| 4 | 151.5, C | 153.3, C | 150.3, C | 146.7, CH |
| 6 | 146.6, C | 146.6, C | 146.0, C | 147.5, C |
| 7 | 127.8, CH | 126.9, CH | 127.3, CH | 127.1, CH |
| 8 | 134.6, CH | 134.4, CH | 134.6, CH | 134.6, CH |
| 9 | 127.5, CH | 126.7, CH | 127.4, CH | 127.3, CH |
| 10 | 126.4, CH | 126.4, CH | 126.4, CH | 126.1, CH |
| 11 | 120.5, C | 120.0, C | 120.1, C | 121.5, C |
| 12 | 158.8, C | 160.2, C | 160.5, C | 159.7, C |
| 14 | 52.7, CH | 51.8, CH | 52.6, CH | 55.6, CH |
| 15 | 33.7, CH2 | 35.5, CH2 | 39.1, CH2 | 34.1, CH2 |
| 16 | 23.7, CH3 | 16.5, CH3 | 20.3, CH3 | |
| 17 | 86.3, C | 80.1, C | 80.3, C | 73.8, C |
| 18 | 88.4, CH | 88.1, CH | 88.1, CH | 83.7, CH |
| 20 | 69.0, CH | 69.1, CH | 69.0, CH | 69.3, CH |
| 21 | 171.5, C | 172.1, C | 172.6, C | 173.8, C |
| 23 | 136.6, C | 137.2, C | 137.7, C | 140.2, C |
| 24 | 114.3, CH | 115.1, CH | 115.2, CH | 114.2, CH |
| 25 | 129.7, CH | 128.8, CH | 128.6, CH | 130.0, CH |
| 26 | 125.1, CH | 124.6, CH | 124.6, CH | 125.1, CH |
| 27 | 126.2, CH | 125.4, CH | 125.2, CH | 124.5, CH |
| 28 | 137.4, C | 138.7, C | 138.8, C | 134.9, C |
| 29 | 31.4, CH | 31.0, CH | 31.0, CH | 28.2, CH |
| 30 | 17.9, CH3 | 17.6, CH3 | 17.4, CH3 | 18.6, CH3 |
| 31 | 18.5, CH3 | 18.6, CH3 | 18.5, CH3 | 20.1, CH3 |
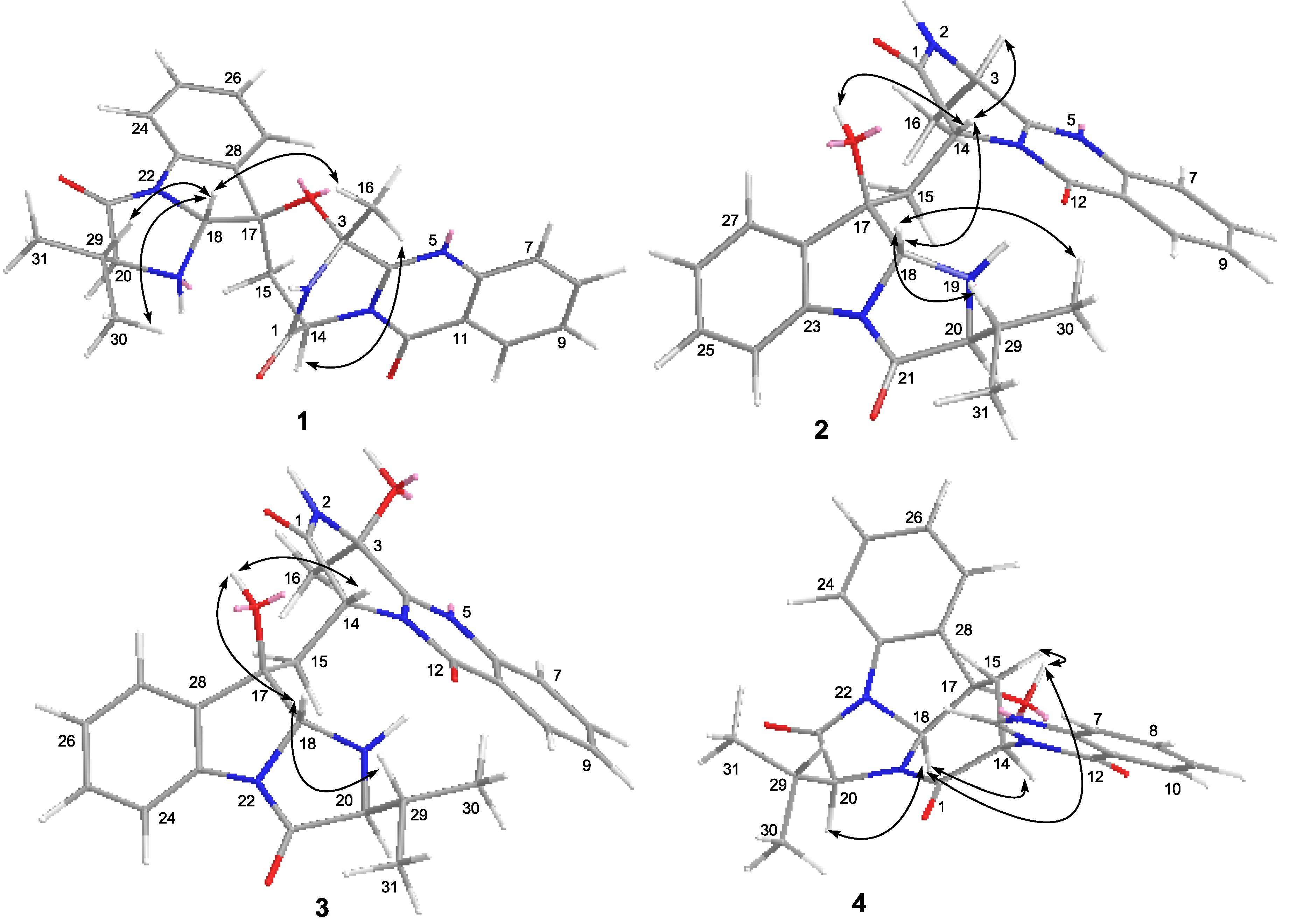
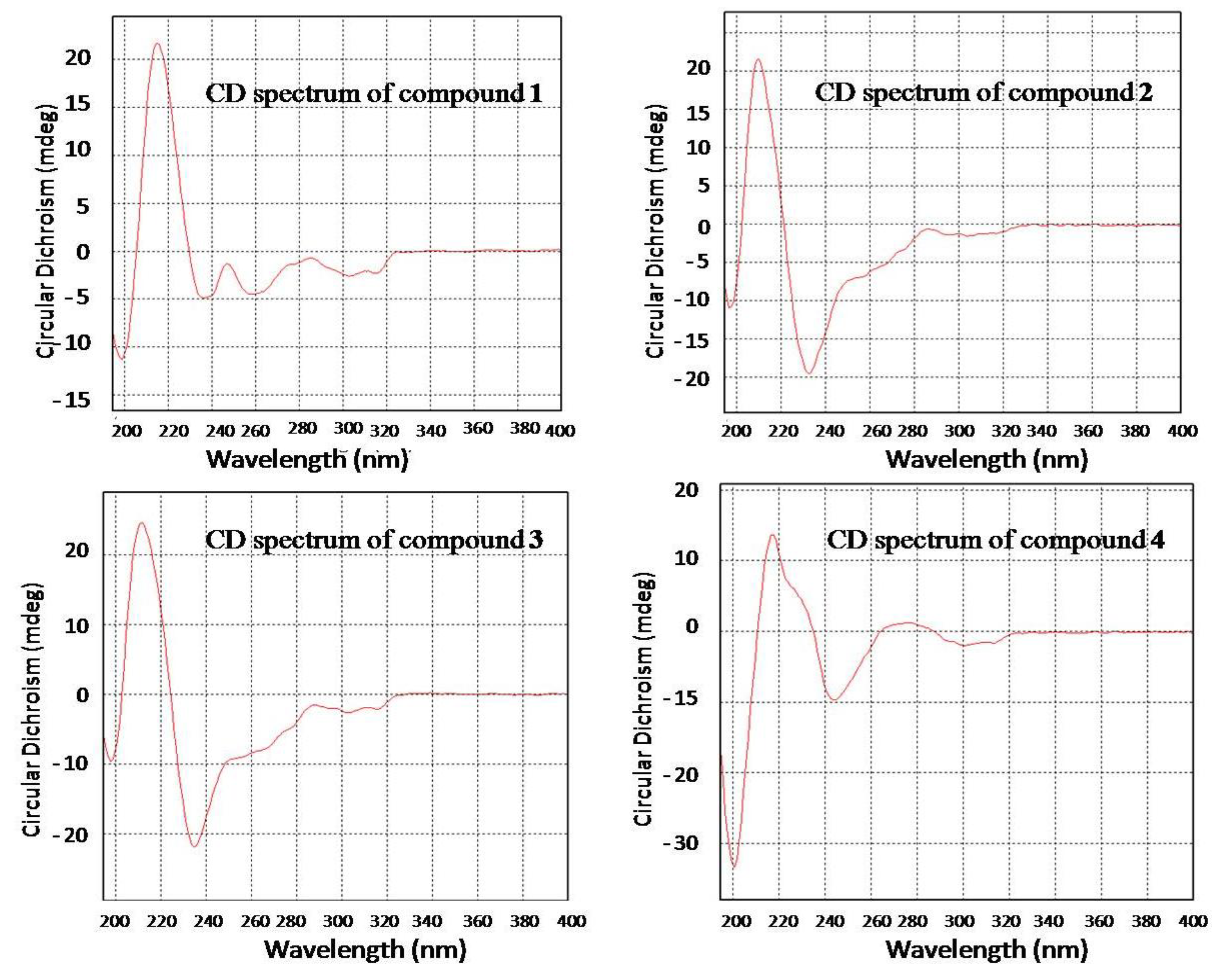
2.2. Biological Activities of the Isolated Compounds
3. Experimental Section
3.1. General
3.2. Fungal Material
3.3. Fermentation
3.4. Extraction and Isolation
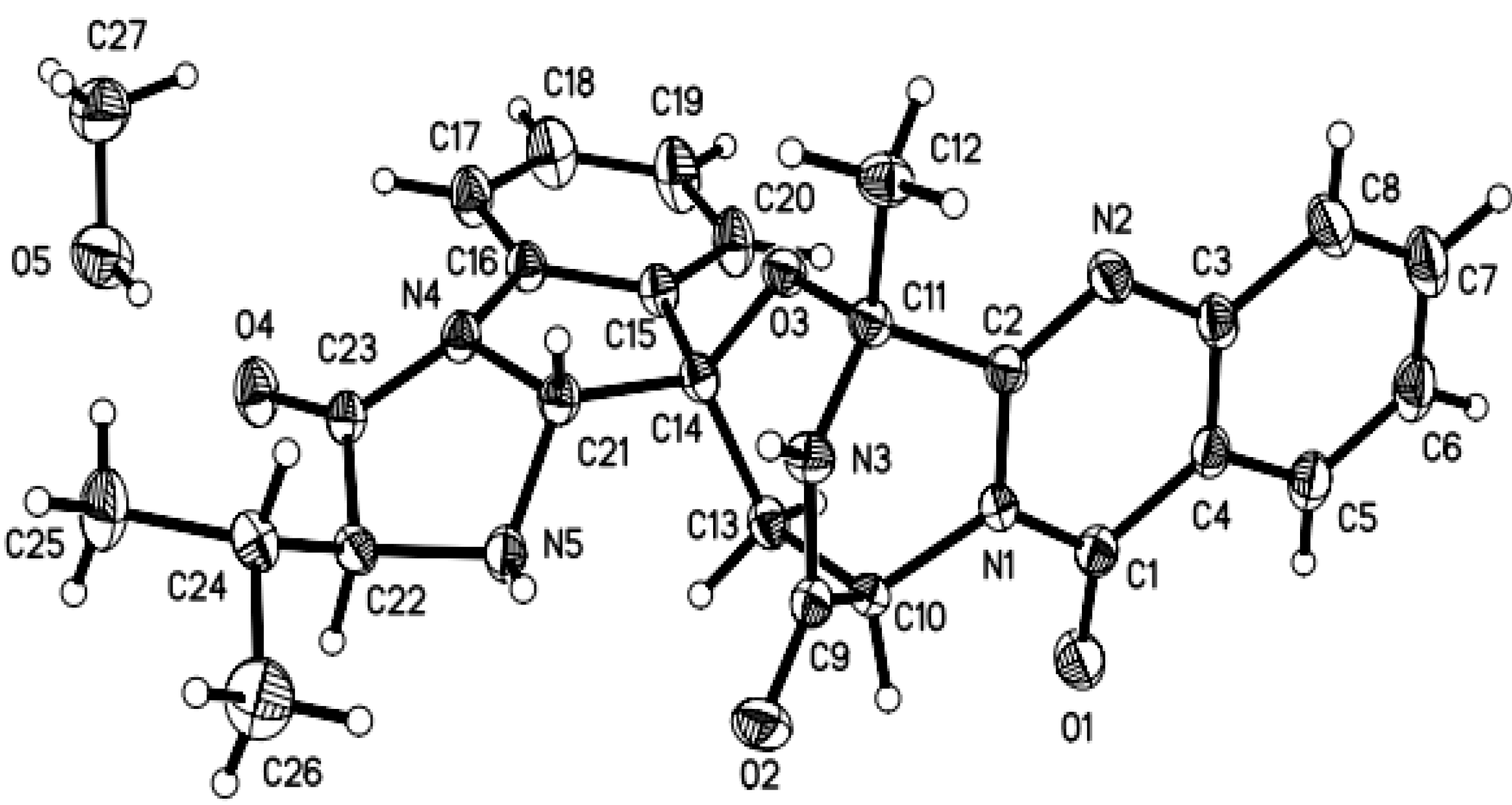
3.5. X-ray Crystallographic Analysis of Compounds 1
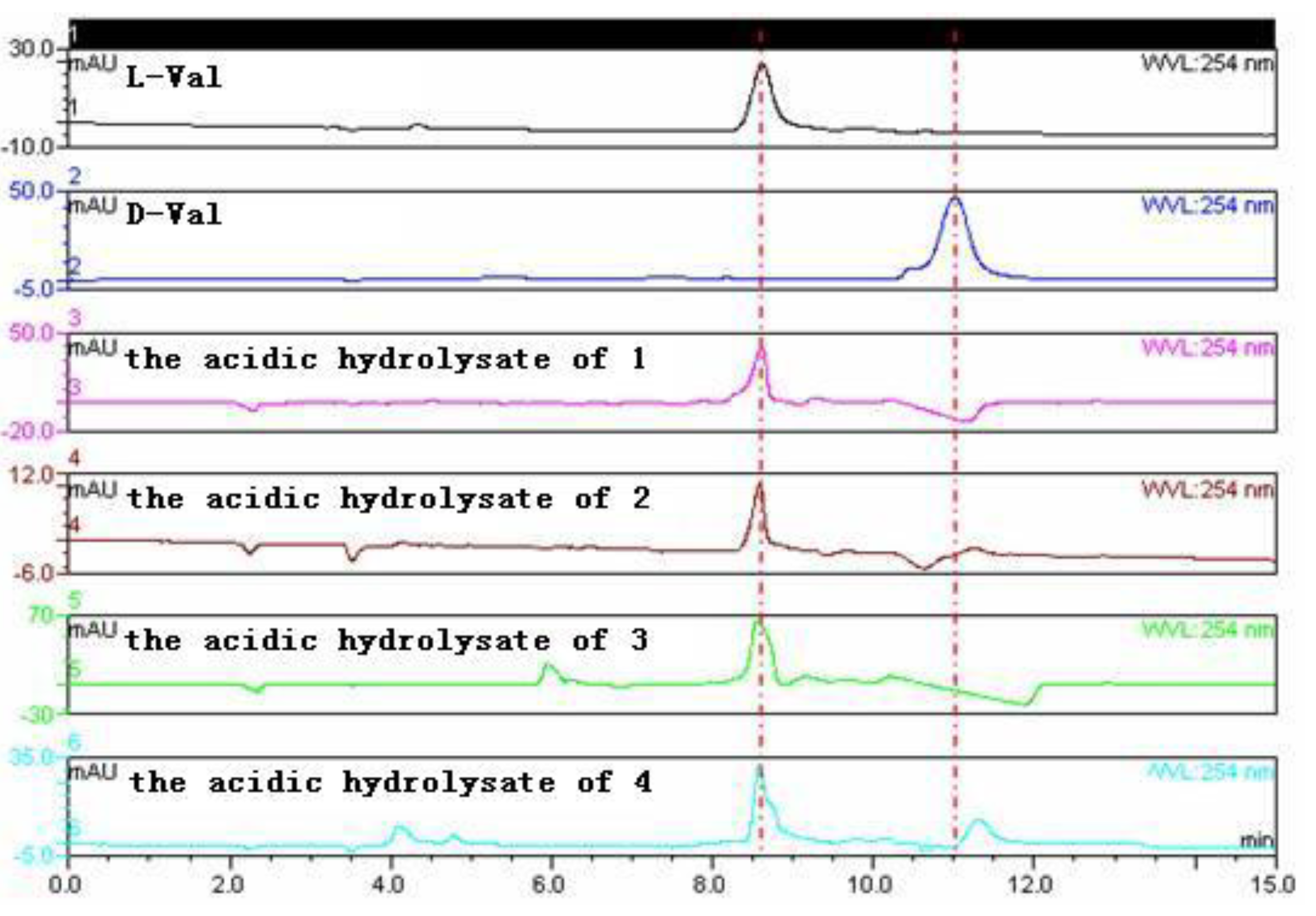
3.6. Amino Acid Analysis
3.7. Reduction of Compound 1
3.8. Brine Shrimp Toxicity
3.9. Cytotoxicity Assay
3.10. Antibacterial Assay
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Mhaske, S.B.; Argade, N.P. The chemistry of recently isolated naturally occurring quinazolinone alkaloids. Tetrahedron 2006, 62, 9787–9826. [Google Scholar] [CrossRef]
- Takahashi, C.; Matsushita, T.; Doi, M.; Minoura, K.; Shingu, T.; Kumeda, Y.; Numata, A. Fumiquinazolines A–G, novel metabolites of a fungus separated from a Pseudolabrus marine fish. J. Chem. Soc. Perkin. Trans. I 1995, 2345–2353. [Google Scholar] [CrossRef]
- Larsen, T.O.; Frydenvang, K.; Frisvad, J.C.; Christophersen, C. UV-guided isolation of alantrypinone, a novel Penicillium alkaloid. J. Nat. Prod. 1998, 61, 1154–1157. [Google Scholar] [CrossRef]
- Numata, A.; Takahashi, C.; Matsushita, T.; Miyamoto, T.; Kawai, K.; Usami, Y.; Matsumura, E.; Inoue, M.; Ohishi, H.; Shingu, T. Fumiquinazolines, novel metabolites of a fungus isolated from a saltfish. Tetrahedron Lett. 1992, 33, 1621–1624. [Google Scholar] [CrossRef]
- Jiao, R.H.; Xu, S.; Liu, J.Y.; Ge, H.M.; Ding, H.; Xu, C.; Zhu, H.L.; Tan, R.X. Chaetominine, a cytotoxic alkaloid produced by endophytic Chaetomium sp. IFB-E015. Org. Lett. 2006, 8, 5709–5712. [Google Scholar] [CrossRef]
- Zhuang, Y.B.; Teng, X.C.; Wang, Y.; Liu, P.P.; Li, G.Q.; Zhu, W.M. New quinazolinone alkaloids within rare amino acid residue from coral-associated fungus, Aspergillus versicolor LCJ-5-4. Org. Lett. 2011, 13, 1130–1133. [Google Scholar] [CrossRef]
- Fremlin, L.J.; Piggott, A.M.; Lacey, E.; Capon, R.J. Cottoquinazoline A and cotteslosins A and B, metabolites from an Australian marine-derived strain of Aspergillus versicolor. J. Nat. Prod. 2009, 72, 666–670. [Google Scholar] [CrossRef]
- Barrow, C.J.; Sun, H.H. Spiroquinazoline, a novel substance P inhibitor with a new carbon skeleton, isolated from Aspergillus flavipes. J. Nat. Prod. 1994, 57, 471–476. [Google Scholar] [CrossRef]
- Li, X.J.; Zhang, Q.; Zhang, A.L.; Gao, J.M. Metabolites from Aspergillus fumigatus, an endophytic fungus associated with Melia azedarach, and their antifungal, antifeedant, and toxic Activities. J. Agric. Food Chem. 2012, 60, 3424–3431. [Google Scholar] [CrossRef]
- Ames, B.D.; Haynes, S.W.; Gao, X.; Evans, B.S.; Kelleher, N.L.; Tang, Y.; Walsh, C.T. Complexity generation in fungal peptidyl alkaloid biosynthesis: Oxidation of fumiquinazoline A to the heptacyclic hemiaminal fumiquinazoline C by the flavoenzyme Af12070 from Aspergillus fumigatus. Biochemistry 2011, 50, 8756–8769. [Google Scholar] [CrossRef]
- Sun, H.F.; Li, X.M.; Meng, L.; Cui, C.M.; Gao, S.S.; Li, C.S.; Huang, C.G.; Wang, B.G. Asperolides A–C, tetranorlabdane diterpenoids from the marine alga-derived endophytic fungus Aspergillus wentii EN-48. J. Nat. Prod. 2012, 75, 148–152. [Google Scholar] [CrossRef]
- Du, F.Y.; Li, X.M.; Li, C.S.; Shang, Z.; Wang, B.G. Cristatumins A–D, new indole alkaloids from the marine-derived endophytic fungus Eurotium cristatum EN-220. Bioorg. Med. Chem. Lett. 2012, 22, 4650–4653. [Google Scholar] [CrossRef]
- Li, C.S.; An, C.Y.; Li, X.M.; Gao, S.S.; Cui, C.M.; Sun, H.F.; Wang, B.G. Triazole and dihydroimidazole alkaloids from the marine sediment-derived fungus Penicillium paneum SD-44. J. Nat. Prod. 2011, 74, 1331–1334. [Google Scholar] [CrossRef]
- Gao, S.S.; Li, X.M.; Du, F.Y.; Li, C.S.; Proksch, P.; Wang, B.G. Secondary metabolites from a marine-derived endophytic fungus Penicillium chrysogenum QEN-24S. Mar. Drugs 2011, 9, 59–70. [Google Scholar]
- Liu, D.; Li, X.M.; Meng, L.; Li, C.S.; Gao, S.S.; Shang, Z.; Proksch, P.; Huang, C.G.; Wang, B.G. Nigerapyrones A–H, α-pyrone derivatives from the marine mangrove-derived endophytic fungus Aspergillus niger MA-132. J. Nat. Prod. 2011, 74, 1787–1791. [Google Scholar] [CrossRef]
- Wang, S.; Li, X.M.; Teuscher, F.; Li, D.L.; Diesel, A.; Ebel, R.; Proksch, P.; Wang, B.G. Chaetopyranin, a benzaldehyde derivative, and other related metabolites from Chaetomium globosum, an endophytic fungus derived from the marine red alga Polysiphonia urceolata. J. Nat. Prod. 2006, 69, 1622–1625. [Google Scholar] [CrossRef]
- Cambridge Crystallographic Data Centre (CCDC). Available online: http://www.ccdc.cam.ac.uk/data_request/cif (accessed on 5 March 2013).
- Sheldrick, G.M. SADABS, Software for Empirical Absorption Correction; University of Göttingen: Göttingen, Germany, 1996. [Google Scholar]
- Sheldrick, G.M. SHELXTL, Structure Determination Software Programs; Bruker Analytical X-ray System Inc.: Madison, WI, USA, 1997. [Google Scholar]
- Sheldrick, G.M. SHELXL-97 and SHELXS-97, Program for X-ray Crystal Structure Solution and Refinement; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Gerwick, W.H.; Proteau, P.J.; Nagle, D.G.; Hamel, E.; Blokhin, A.; Slate, D.L. Structure of curacin A, a novel antimitotic, antiproliferative, and brine shrimp toxic natural product from the marine Cyanobacterium Lyngbya majuscula. J. Org. Chem. 1994, 59, 1243–1245. [Google Scholar]
- Bergeron, R.J.; Cavanaugh, P.F., Jr.; Kline, S.J.; Hughes, R.G., Jr.; Elliott, G.T.; Porter, C.W. Antineoplastic and antiherpetic activity of spermidine catecholamide iron chelators. Biochem. Biophys. Res. Commun. 1984, 121, 848–854. [Google Scholar] [CrossRef]
- Al-Burtamani, S.K.S.; Fatope, M.O.; Marwah, R.G.; Onifade, A.K.; Al-Saidi, S.H. Chemical composition, antibacterial and antifungal activities of the essential oil of Haplophyllum tuberculatum from Oman. J. Ethnopharmacol. 2005, 96, 107–112. [Google Scholar] [CrossRef]
Supplementary Files
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
An, C.-Y.; Li, X.-M.; Li, C.-S.; Wang, M.-H.; Xu, G.-M.; Wang, B.-G. Aniquinazolines A–D, Four New Quinazolinone Alkaloids from Marine-Derived Endophytic Fungus Aspergillus nidulans. Mar. Drugs 2013, 11, 2682-2694. https://doi.org/10.3390/md11072682
An C-Y, Li X-M, Li C-S, Wang M-H, Xu G-M, Wang B-G. Aniquinazolines A–D, Four New Quinazolinone Alkaloids from Marine-Derived Endophytic Fungus Aspergillus nidulans. Marine Drugs. 2013; 11(7):2682-2694. https://doi.org/10.3390/md11072682
Chicago/Turabian StyleAn, Chun-Yan, Xiao-Ming Li, Chun-Shun Li, Ming-Hui Wang, Gang-Ming Xu, and Bin-Gui Wang. 2013. "Aniquinazolines A–D, Four New Quinazolinone Alkaloids from Marine-Derived Endophytic Fungus Aspergillus nidulans" Marine Drugs 11, no. 7: 2682-2694. https://doi.org/10.3390/md11072682