Antiplasmodial Activities of Homogentisic Acid Derivative Protein Kinase Inhibitors Isolated from a Vanuatu Marine Sponge Pseudoceratina sp.

 and
and
Abstract
:1. Introduction
2. Results and Discussion
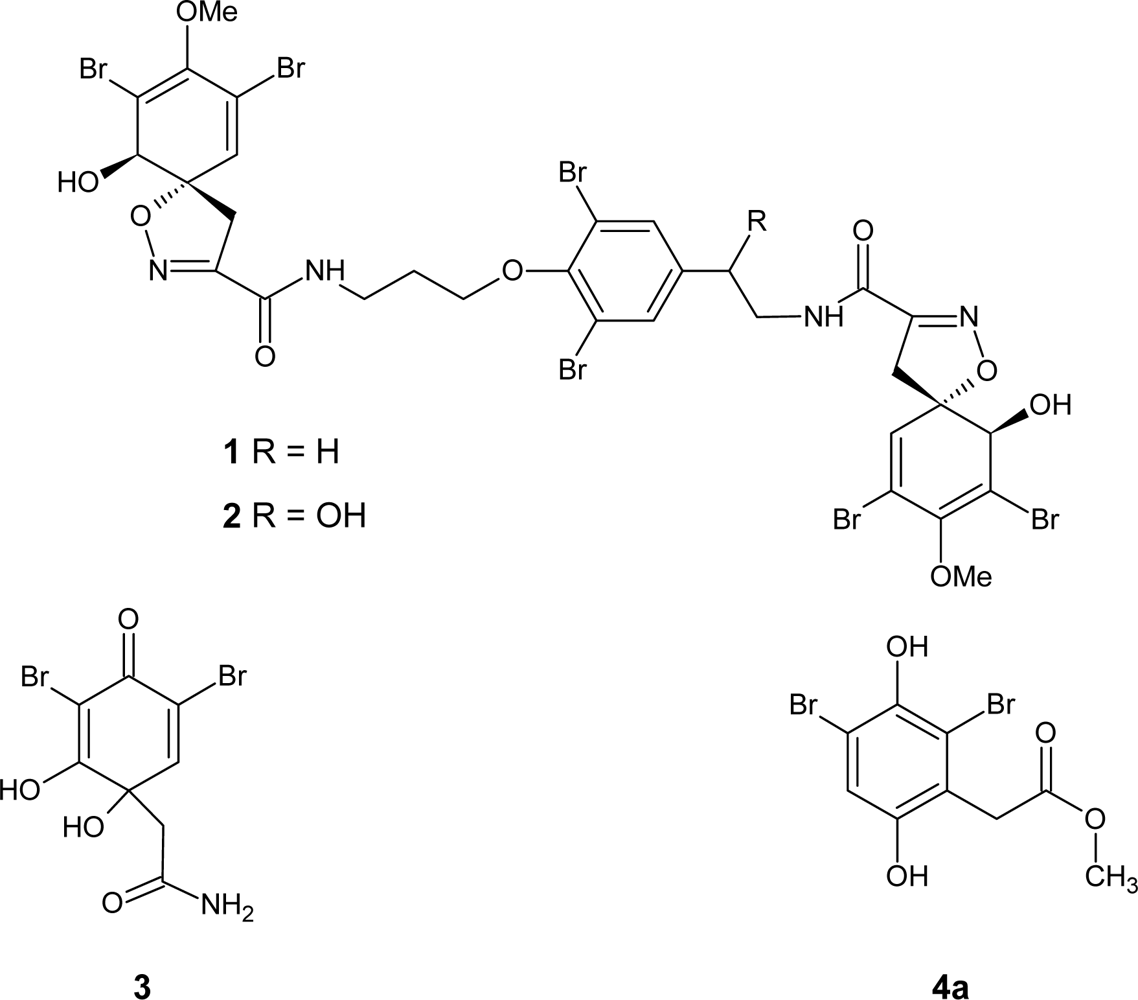
2.1. Chemistry
2.2. Biological Properties
3. Experimental Section
3.1. Materials
3.2. Extraction and Isolation
3.3. Protein Kinase Assay
3.4. Activity against Erythrocytic Stages of Cultured P. falciparum
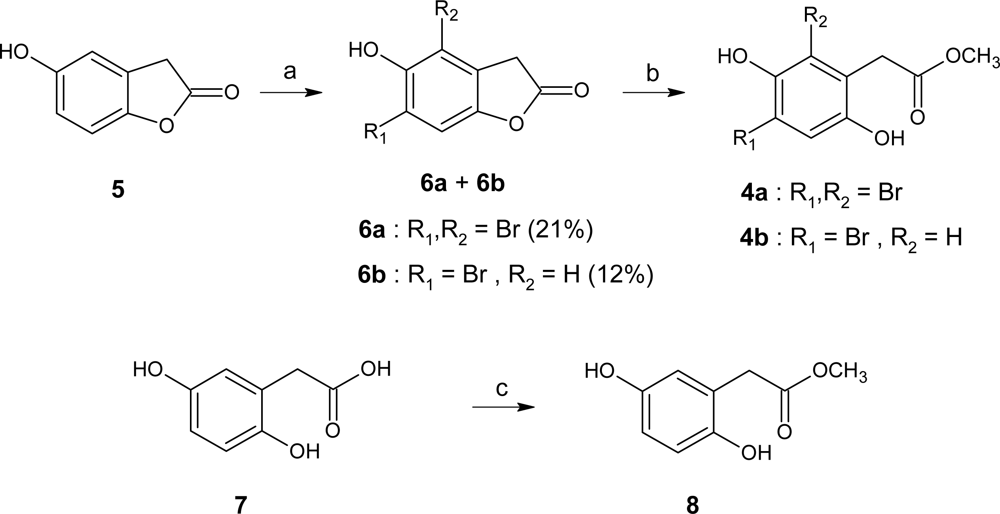
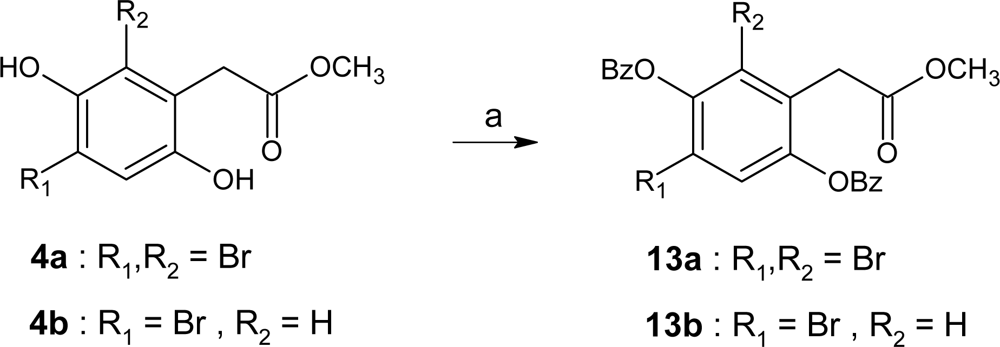
3.5. Synthesis
4,6-Dibromo-5-hydroxy-3H-benzofuran-2-one (6a) and 6-bromo-5-hydroxy-3H-benzofuran-2-one (6b)
Methyl (2,4-dibromo-3,6-dihydroxyphenyl)acetate (4a)
Methyl (4-bromo-3,6-dihydroxyphenyl)acetate (4b)
Methyl (2,5-dihydroxyphenyl)acetate (8)
2′,4′-Dibenzyloxyacetophenone (10)
Methyl (2,4-dihydroxyphenyl)acetate (11)
Methyl (3,5-dibromo-2,4-dihydroxyphenyl)acetate (12)
Methyl (3,6-dibenzoyl-2,4-dibromophenyl)acetate (13a)
Methyl (3,6-dibenzoyl-4-bromophenyl)acetate 13b
Acknowledgments
- Samples Availability: Available from the authors.
References and Notes
- Laurent, D; Pietra, F. Antiplasmodial marine natural products in the perspective of current chemotherapy and prevention of malaria. A Review. Mar Biotechnol 2006, 8, 433–447. [Google Scholar]
- Fattorusso, E; Taglialatela-Scafati, O. Marine Antimalarials. Mar Drugs 2009, 7, 130–152. [Google Scholar]
- Gademann, K; Kobylinska, J. Antimalarial natural products of marine and freshwater origin. Chem Rec 2009, 9, 187–198. [Google Scholar]
- Kaur, K; Jain, M; Kaur, T; Jain, R. Antimalarials from nature. Bioorg Med Chem 2009, 17, 3229–3256. [Google Scholar]
- Dorin, D; Le Roch, K; Sallicandro, P; Alano, P; Parzy, D; Poullet, P; Meijer, L; Doerig, C. Pfnek-1, a NIMA-related kinase from the human malaria parasite Plasmodium falciparum. Eur J Biochem 2001, 268, 2600–2608. [Google Scholar]
- Laurent, D; Jullian, V; Parenty, A; Knibiehler, M; Dorin, D; Schmitt, S; Lozach, O; Lebouvier, N; Frostin, M; Alby, F; Maurel, S; Doerig, C; Meijer, L; Sauvain, M. Antimalarial potential of xestoquinone, a protein kinase inhibitor isolated from a Vanuatu marine sponge Xestospongia sp. Bioorg Med Chem 2006, 14, 4477–4482. [Google Scholar]
- Takada, N; Watanabe, R; Suenaga, K; Yamada, K; Ueda, K; Kita, M; Uemura, D. Zamamistatin, a significant antibacterial bromotyrosine derivative, from the Okinawan sponge Pseudoceratina purpurea. Tetrahedron Lett 2001, 42, 5265–5267. [Google Scholar]
- Benharref, A; Païs, M; Debitus, C. Bromotyrosine Alkaloids from the Sponge Pseudoceratina verrucosa. J Nat Prod 1996, 59, 177–180. [Google Scholar]
- Tsukamoto, S; Kato, H; Hirota, H; Fusetani, N. Ceratinamides A and B: New antifouling dibromotyrosine derivatives from the marine sponge Pseudoceratina purpurea. Tetrahedron 1996, 52, 181–186. [Google Scholar]
- Kernan, MR; Cambie, RC; Bergquist, PR. Chemistry of Sponges, VII. 11,19-Dideoxyfistularin 3 and 11-hydroxyaerothionin, bromotyrosine derivatives from Pseudoceratina durissima. J Nat Prod 1990, 53, 615–622. [Google Scholar]
- Piña, IC; Gautschi, JT; Wang, GY; Sanders, ML; Schmitz, FJ; France, D; Cornell-Kennon, S; Sambucetti, LC; Remiszewski, SW; Perez, LB; Bair, KW; Crews, P. Psammaplins from the sponge Pseudoceratina purpurea: inhibition of both histone deacetylase and DNA methyltransferase. J Org Chem 2003, 68, 3866–3873. [Google Scholar]
- Aiello, A; Fattorusso, E; Menna, M; Pansini, M. Chemistry of Verongida Sponges-V. Brominated metabolites from the Caribbean Sponge Pseudoceratina sp. Biochem Syst Ecol 1995, 23, 377–381. [Google Scholar]
- Fusetani, N; Masuda, Y; Nakao, Y; Matsunaga, S; van Soest, RWM. Three new bromotyrosine derivatives lethal to crab from the marine sponge, Pseudoceratina purpurea. Tetrahedron 2001, 57, 7507–7511. [Google Scholar]
- Albrizio, S; Ciminiello, P; Fattorusso, E; Magno, S; Pansini, M. Chemistry of verongida sponges. I. Constituents of the Caribbean sponge Pseudoceratina crassa. Tetrahedron 1994, 50, 783–788. [Google Scholar]
- Jang, J-H; Van Soest, RWM; Fusetani, N; Matsunaga, S. Pseudoceratins A and B, Antifungal Bicyclic Bromotyrosine-Derived Metabolites from the Marine Sponge Pseudoceratina purpurea. J Org Chem 2007, 72, 1211–1217. [Google Scholar]
- Schoenfeld, RC; Ganem, B. Synthesis of ceratinamine and moloka’iamine: Antifouling agents from the marine sponge Pseudoceratina purpurea. Tetrahedron Lett 1998, 39, 4171–4150. [Google Scholar]
- Buchanan, MS; Carroll, AR; Fechner, GA; Boyle, A; Simpson, M; Addepalli, R; Avery, VM; Hooper, JNA; Cheung, THC; Quinn, RJ. Aplysamine 6, an Alkaloidal Inhibitor of Isoprenylcysteine Carboxyl Methyltransferase from the Sponge Pseudoceratina sp. J Nat Prod 2008, 71, 1066–1067. [Google Scholar]
- Thirionet, I; Daloze, D; Braekman, JC; Willemsen, P. 5-Bromoverongamine, a Novel Antifouling Tyrosine Alkaloid from the Sponge Pseudoceratina sp. Nat Prod Res 1998, 12, 209–214. [Google Scholar]
- Mancini, I; Guella, G; Laboute, P. C., D.; Pietra, F. Hemifistularin 3: A degraded peptide or biogenetic precursor? Isolation from a sponge of the order verongida from the coral sea or generation from base treatment of 11-oxofistularin 3. J Chem Soc, Perkin Trans 1 1993, 3121–3125. [Google Scholar]
- Krejcarek, GE; White, RH; Hager, LP; McClure, WO; Johnson, RD; Rinehart, KLJ; McMillan, JA; Paul, JC; Shaw, PD; Brusca, RC. A rearranged dibromotyrosine metabolite from Verongia aurea. Tetrahedron Lett 1975, 16, 507–510. [Google Scholar]
- Patil, AD; Westley, JW; Mattern, MR; Hofmann, GA. Homogentisic acid derivatives, methods of treatment of disease states mediated by protein kinase C using homogentisic acid derivatives, and pharmaceutical compositions there of. 28 september 1995, 1995.
- Krohn, K. Synthese des bactereostatischen 2.4-Dibrom-homogentisinsäureamidsund verwandter verbindungen. Tetrahedron Lett 1975, 16, 4667–4668. [Google Scholar]
- Abou-Shoer, MI; Shaala, LA; Youssef, DTA; Badr, JM; Habib, A-AM. Bioactive Brominated Metabolites from the Red Sea Sponge Suberea mollis. J Nat Prod 2008, 71, 1464–1467. [Google Scholar]
- Desoubzdanne, D; Marcourt, L; Raux, R; Chevalley, S; Dorin, D; Doerig, C; Valentin, A; Ausseil, F; Debitus, C. Alisiaquinones and alisiaquinol, dual inhibitors of Plasmodium falciparum enzyme targets from a new caledonian deep water sponge. J Nat Prod 2008, 71, 1189–1192. [Google Scholar]
- Mbah, JA; Tane, P; Ngadjui, BT; Connolly, JD; Okunji, CC; Iwu, MM; Schuster, BM. Antiplasmodial agents from the leaves of Glossocalyx brevipes. Planta Med 2004, 70, 437–440. [Google Scholar]
- Kerr, TJ; McHale, BB. Applications in General Microbiology: A Laboratory Manual, 6th ed; Hunter Textbooks: Winston-Salem, NC, USA, 2001; pp. 202–203. [Google Scholar]
- Tilvi, S; Rodrigues, C; Naik, CG; Parameswaran, PS; Wahidhulla, S. New bromotyrosine alkaloids from the marine sponge Psammaplysilla purpurea. Tetrahedron 2004, 60, 10207–10215. [Google Scholar]
- Yagi, H; Matsunaga, S; Fusetani, N. Purpuramines A-I, new bromotyrosine-derived metabolites from the marine sponge Psammaplysilla purpurea. Tetrahedron 1993, 49, 3749–3754. [Google Scholar]
- Kobayashi, J; Tsuda, M; Agemi, K; Shigemori, H; Ishibashi, M; Sasakia, T; Mikamib, Y. Purealidins B and C, new bromotyrosine alkaloids from the okinawan marine sponge Psammaplysilla purea. Tetrahedron 1991, 47, 6617–6622. [Google Scholar]
- Kim, D; Lee, IS; Jung, JH; Yang, S-I. Psammaplin A, a natural bromotyrosine derivative from a sponge, possesses the antibacterial activity against methicillin-resistant Staphylococcus aureus and the DNA gyrase-inhibitory activity. Arch Pharm Res 1999, 22, 25–29. [Google Scholar]
- Park, Y; Liu, Y; Hong, J; Lee, C-O; Cho, H; Kim, D-K; Im, KS; Jung, JH. New bromotyrosine derivatives from an association of two sponges, Jaspis wondoensis and Poecillastra wondoensis. J Nat Prod 2003, 66, 1495–1498. [Google Scholar]
- Pick, N; Rawat, M; Arad, D; Lan, J; Fan, J; Kende, AS; Av-Gay, Y. In vitro properties of antimicrobial bromotyrosine alkaloids. J Med Microbiol 2006, 55, 407–415. [Google Scholar]
- Matsunaga, S; Kobayashi, H; Van Soest, RBW; Fusetani, N. Novel Bromotyrosine Derivatives That Inhibit Growth of the Fish Pathogenic Bacterium Aeromonas hydrophila, from a Marine Sponge Hexadella sp. J Org Chem 2005, 70, 1893–1896. [Google Scholar]
- Makler, MT; Hinrichs, D. Measurement of the lactate dehydrogenase activity of Plasmodium falciparum as an assessment of parasitemia. Am J Trop Med Hyg 1993, 48, 205–210. [Google Scholar]
- Trager, W; Jensen, JB. Human malaria parasites in continuous culture. Science 1976, 193, 673–675. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1H, CD3OD, 300 MHz | 13C, CD3OD1 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 4a synthetic | 7.01 | 3.86 | 3.70 | 171.6 | 149.8 | 143.8 | 122.2 | 117.3 | 115.7 | 109.7 | 51.1 | 35.0 |
| 4a natural | 6.88 | 3.73 | 3.57 | 171.6 | 149.8 | 143.8 | 122.2 | 117.3 | 115.6 | 109.7 | 51.1 | 35.0 |
| Compound number | Pfnek-1 | FCB1 of P. falciparum |
|---|---|---|
| 1 | >50 | >100 |
| 2 | >50 | >100 |
| 3 | >50 | >100 |
| 4a | 1,8 | 12 |
| 4b | 10 | 26 |
| 5 | >50 | 29 |
| 6a | >50 | 35 |
| 6b | >50 | 17 |
| 8 | >50 | 36 |
| 12 | >50 | >100 |
| 13a | >50 | 22.5 |
| 13b | >50 | 13 |
| Compound number1 | Growth inhibition diameter (mm) | |
|---|---|---|
| S. aureus | E. coli | |
| 1 | 14 | 16 |
| 2 | 0 | 0 |
| 3 | 20 | 15 |
| 4a | 0 | 0 |
| gentamycin | 16 | - |
| chloramphenicol | - | 17 |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lebouvier, N.; Jullian, V.; Desvignes, I.; Maurel, S.; Parenty, A.; Dorin-Semblat, D.; Doerig, C.; Sauvain, M.; Laurent, D. Antiplasmodial Activities of Homogentisic Acid Derivative Protein Kinase Inhibitors Isolated from a Vanuatu Marine Sponge Pseudoceratina sp. Mar. Drugs 2009, 7, 640-653. https://doi.org/10.3390/md7040640
Lebouvier N, Jullian V, Desvignes I, Maurel S, Parenty A, Dorin-Semblat D, Doerig C, Sauvain M, Laurent D. Antiplasmodial Activities of Homogentisic Acid Derivative Protein Kinase Inhibitors Isolated from a Vanuatu Marine Sponge Pseudoceratina sp. Marine Drugs. 2009; 7(4):640-653. https://doi.org/10.3390/md7040640
Chicago/Turabian StyleLebouvier, Nicolas, Valérie Jullian, Isabelle Desvignes, Séverine Maurel, Arnaud Parenty, Dominique Dorin-Semblat, Christian Doerig, Michel Sauvain, and Dominique Laurent. 2009. "Antiplasmodial Activities of Homogentisic Acid Derivative Protein Kinase Inhibitors Isolated from a Vanuatu Marine Sponge Pseudoceratina sp." Marine Drugs 7, no. 4: 640-653. https://doi.org/10.3390/md7040640