Diesel Exhaust Particles Induce the Over expression of Tumor Necrosis Factor-α (TNF-α) Gene in Alveolar Macrophages and Failed to Induce Apoptosis through Activation of Nuclear Factor-κB (NF-κB)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Introduction
Materials and Methods
Materials
Alveolar Macrophages (AM)
Measurement of TNF-α Release
Western Blot Analysis of TNF-α Protein
Detection of Apoptosis
Statistical Analysis
Results
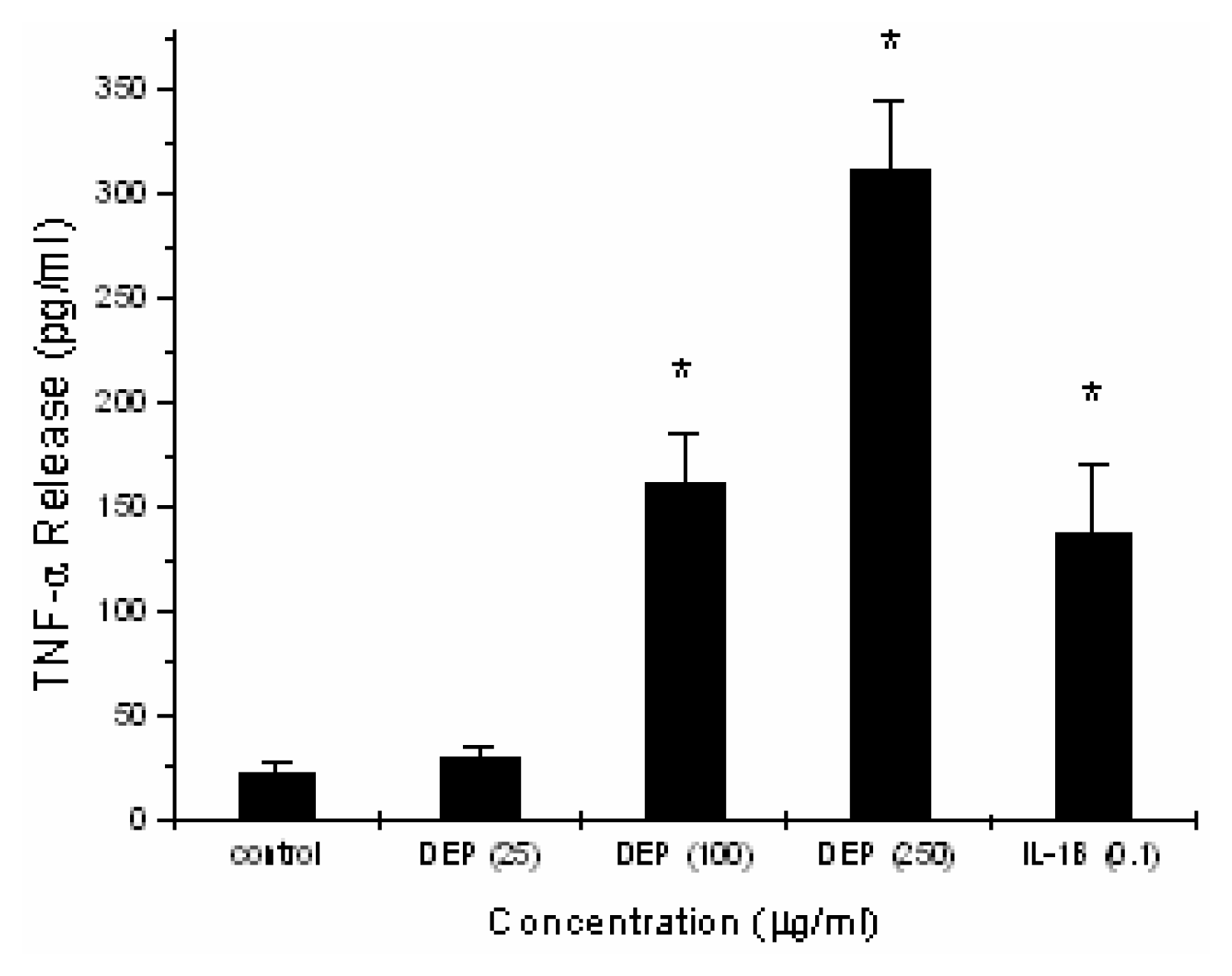
Induction of TNF-α by DEP in Alveolar Macrophages
Determination of TNF-α Protein by Western Blotting
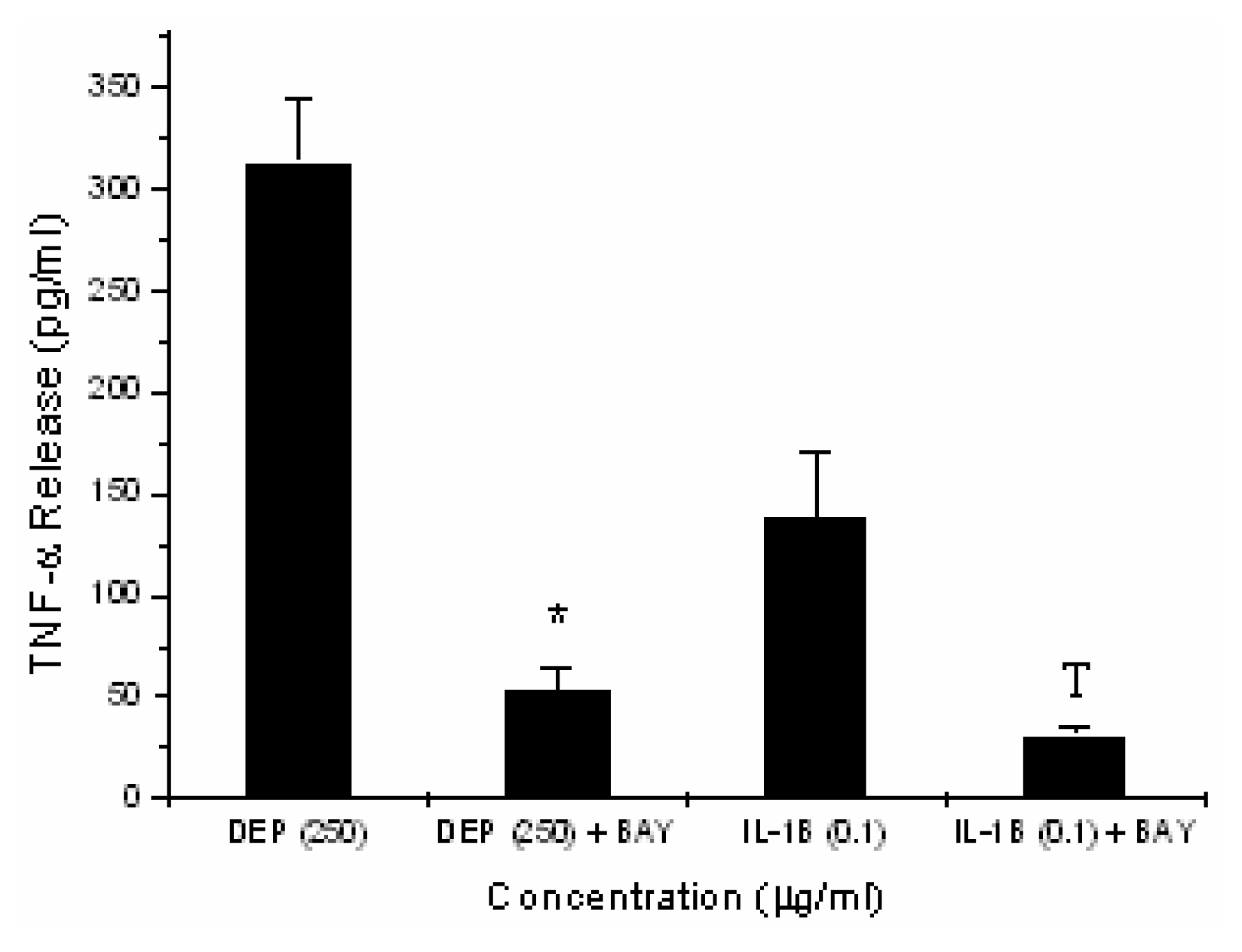
Inhibition of DEP-induced TNF-α Expression by Inhibiting NF-κB Binding Activity
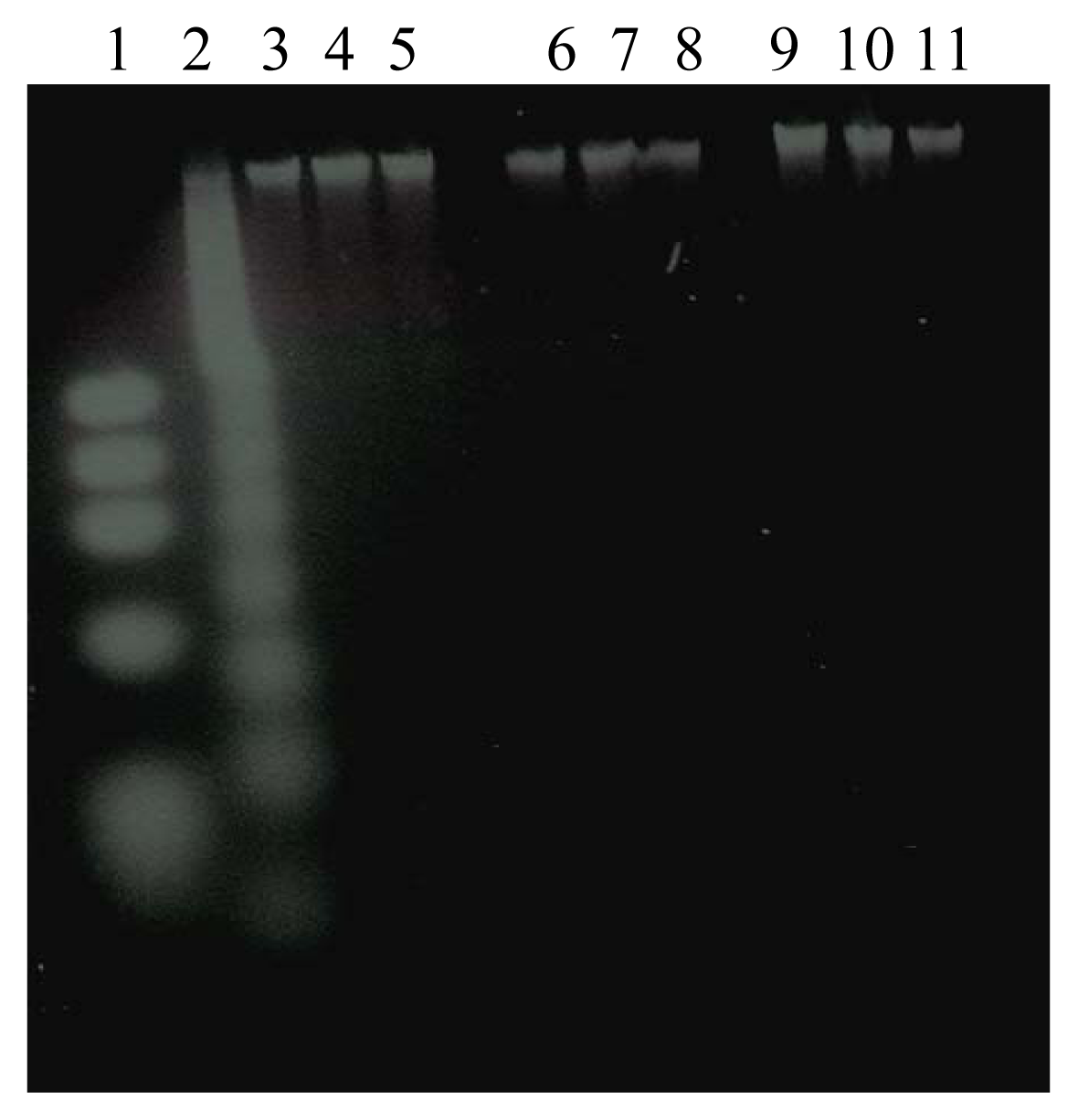
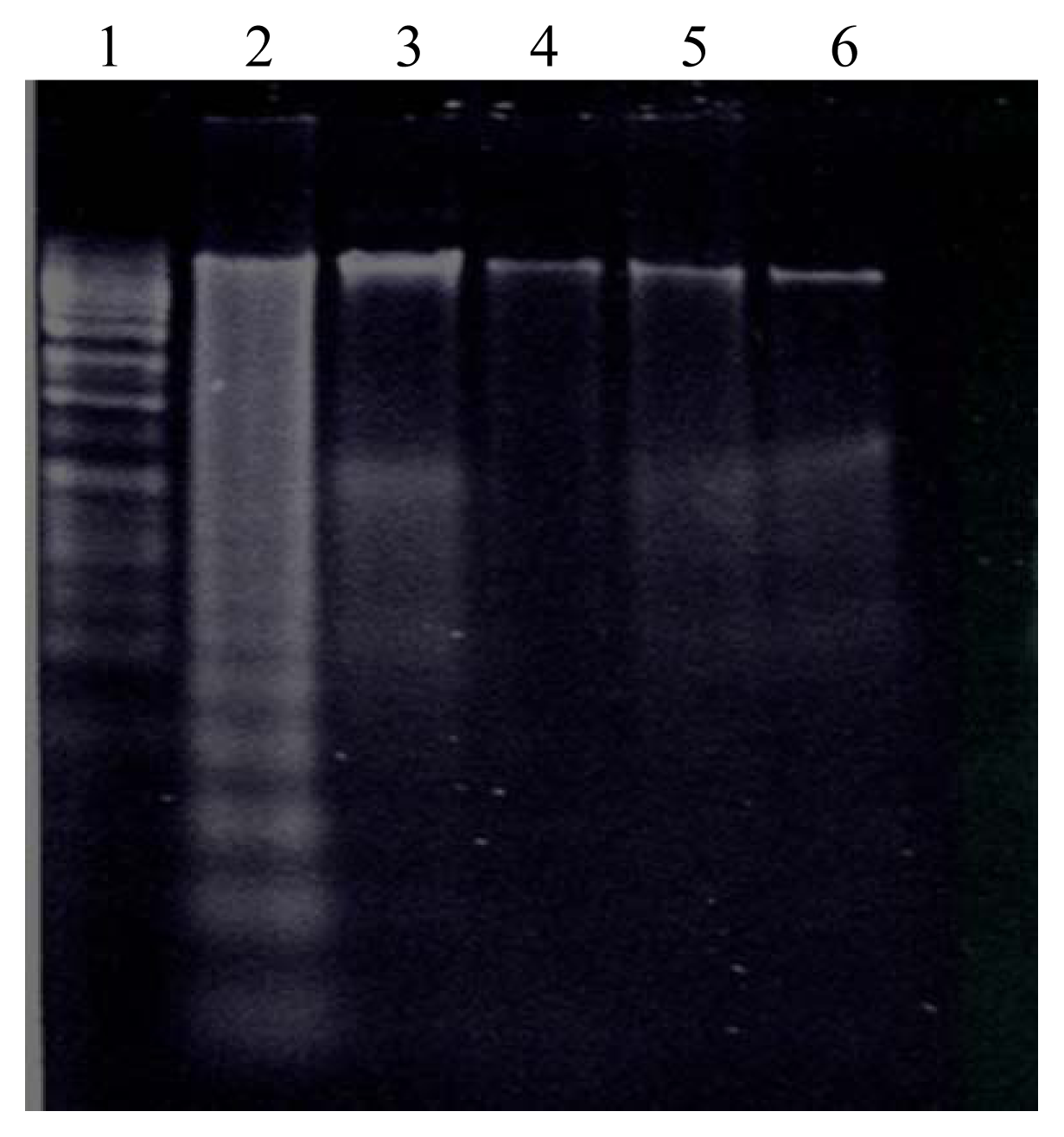
Effect of DEP Exposure on Alveolar Macrophage Apoptotic Response
Discussion





Acknowledgments
References
- Ohtoshi, T.; Tazikawa, H.; Okazaki, H.; Kawasaki, S.; Takeuchi, N.; Ohta, K.; Ito, K. Diesel exhaust particles (DEP) stimulate human airway epithelial cells to produce cytokines relevant to airway inflammation in vitro. J. Allergy Clin. Immunol 1998, 101, 778–785. [Google Scholar]
- Sagai, M.; Furuyama, A.; Ichinose, T. Biological effects of diesel exhaust particles (DEP) HI. Pathogenesis of asthma-like symptoms in mice. Free Rod. Biol. Med 1996, 21, 199. [Google Scholar]
- Nel, A. E.; Diaz-Sanchez, D.; Ng, D.; Hiura, J.; Saxon, A. Enhancement of allergic inflammation by the interaction between diesel exhaust particles and the immune system. J. Allergy Clin. Immunol 1998, 102, 539–554. [Google Scholar]
- Peterson, B. A.; Saxon, A. Global increases in allergic respiratory disease: The possible roles of diesel exhaust particles. Ann. Allergy Asthma Immunol 1996, 77(4), 263–268. [Google Scholar]
- Diaz-Sanchez, D. The roles of diesel exhaust particles and their associated polyaromatic hydrocarbons in the induction of allergic airway disease. Allergy 1997, 52(38s), 52–56. [Google Scholar]
- Jakab, G. J.; Risby, T. H.; Sehnert, S. S.; Hmieleski, R. R.; Farrington, J. E. Suppression of alveolar macrophage membrane receptor-mediated phagocytosis by model and actual particle-adsorbate complexes; initial contact with the alveolar macrophage membrane. Environ. Health Perspect 1990, 86, 33744. [Google Scholar]
- Monn, C.; Fendt, R.; Koller, T. Ambient pm (10) extracts inhibit phagocytosis of defined inert model particles by alveolar macrophages. Inhal. Toxicol 1990, 14(4), 369–85. [Google Scholar]
- Hiura, T. S.; Kaszubowski, M. P.; LI, N.; Nel, A. E. Chemicals in diesel exhaust particles generate reactive oxygen radicals and induce apoptosis in macrophages. J. Immunol 1999, 163, 5582–5591. [Google Scholar]
- Holian, A.; Hamilton, R. F., Jr.; Morandi, M. T.; Brown, S. D.; Li, L. Urban particule-induced apoptosis and phenotype shifts in human alveolar macrophages. Environ. Health Perspect 1998, 106, 127–132. [Google Scholar]
- Berry, J.; Henoc, P.; Galle, P. Phagocytosis by cells of the pulmonary alveoli. Am. J. Pathol 1978, 93, 2744. [Google Scholar]
- Takano, H.; Yanagisawa, R.; Ichinose, T.; Sadakane, K.; Yoshinos, S.; Yoshikawa, T.; Morita, M. Diesel exhaust particles enhance lung injury related to bacterial endotoxin through expression of proinflammatory cytokines, chemokines, and intercellular adhesion molecule-1. Am. J. Resipr. Crit. Care Med 2002, 165, 1329–1335. [Google Scholar]
- Hiura, T. S.; Li, N.; Kaplan, R.; Horwitz, M.; Seagraave, J. C.; Nel, A. E. The role of a mitochondrial pathway in the induction of apoptosis by chemicals extracted from diesel exhaust particles. J. Immunol 2000, 165(5), 2703–11. [Google Scholar]
- Koay, A. M.; Gao, X.; Washington, M. K.; et al. Macrophages are necessary for maximal nuclea factor-KP activation in response to endotoxin. Am. J. Respir. Cell. Mol. Biol 2002, 26, 572–578. [Google Scholar]
- Sen, R.; Baltimore, D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 1986, 46(5), 705–716. [Google Scholar]
- Baldwin, A. S., Jr. Series introduction: the transcription factor NF-KB and human disease. J. Clin. Invest 2001, 707(7), 3–6. [Google Scholar]
- Stein, B.; Baldwin, A. S. Distinct mechanisms for the regulation of the interleukin-8 gene involve synergism and cooperativity between CIEBP and NF-KB. Mol. Cell Biol 1993, 13, 7191–7198. [Google Scholar]
- Christman, J. W.; Sadikot, R. T.; Blackwell, T. S. The role of nuclear factor-KP in pulmonary disease. Chest 2000, 777, 1482–1487. [Google Scholar]
- Graziano, F. M.; Cook, E. B.; Stahl, J. L. Cytokines, chemokines, RANTES, and eotaxin. Allergy Asthma Proc 1999, 20, 141–146. [Google Scholar]
- Anderson, J. P. Resolution of chronic inflammation by therapeutic induction of apoptosis. Trends Pharmacol. Sci 1996, 77, 438–442. [Google Scholar]
- Daniel, W. W. Biostatistics: A Foundation for Analysis in Health Sciences, 5th ed; John Wiley: New York, 1991; pp. 274–366. [Google Scholar]
- Bousquet, J.; Jeffery, R. K.; Busse, W. W.; Johnson, M.; Vignola, A. M. Asthma: from bronchoconstriction to airways inflammation and remodeling. Am. J. Respir. Crit. Care Med 2000, 161(5), 1720–1745. [Google Scholar]
- Borish, L.; Mascali, J. J.; Dishuck, J.; Beam, W. R.; Martin, R. J.; Rosenwasser, L. J. Detection of alveolar macrophage-derived IL-lbeta in asthma inhibition with corticosteroids. J. Immunol 1992, 49, 3078–3082. [Google Scholar]
- Gosset, P.; Garcon, G.; Casset, A.; Fleurisse, L.; Hannothiaux, M. H.; Craesy, C.; Shirali, P. Benzo(a)pyrene-coated onto Fe2O3 particles-induced apoptotic events in the lungs of Sprague Dawley rats. Toxicol. Lett 2003, 143, 223–232. [Google Scholar]
- Driscoll, K. E.; Hassenbein, D. G.; Carter, J. M.; Kunkel, S. L.; Quinlan, T. R.; Mossman, B. T. TNF alpha and increased chemokine expression in rat lung after particle exposure. Toxicol. Lett 1995, 82–83, 483–489. [Google Scholar]
- Driscoll, K. E. TNF-alpha and MIP-2: role in particle-induced inflammation and regulation by oxidative stress. Toxicol. Lett 1995, 112–113, 177–183. [Google Scholar]
- Fiers, W. Tumor necrosis factor: characterization of the molecular, cellular, and in vivo level. FEBS Lett 1991, 285, 199–212. [Google Scholar]
- Adaamson, I. Y.; Prieditis, H.; Vincent, R. Soluble and insoluble air particle fractions induce differential production of tumor necrosis factor alpha in rat lung. Exp. Lung Res 2004, 30(5), 355–368. [Google Scholar]
- Baeuerle, P. A. IKB-NF-KB structures: at the interface of inflammation control. Cell 1998, 95(6), 729–731. [Google Scholar]
- Karin, M.; Ben-Neriah, Y. Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu. Rev. Immunol 2000, 18, 621–663. [Google Scholar]
- Phelps, C. B.; Sengchanthalangsy, L. L.; et al. Mechanism of IKBOC binding to NF-κB dimmers. J. Biol. Chem 2000, 275(38), 29840–29846. [Google Scholar]
- Grimm, S.; Bauer, M. K. A.; Bauerle, P. A.; Schulze-Osthoff, K. Bcl-2 down-regulates the activity of transcription factor NF-KB induced apoptosis. J. Cell Biol 1996, 134, 13–23. [Google Scholar]
- Van Antwerp, D. J.; Martin, S. J.; Venna, I. M.; Green, D. R. Inhibition of TNF-induced apoptosis by NF-KB. Trends Cell Biol 1998, 8, 107–111. [Google Scholar]
- Manna, S. K.; Aggarwal, B. B. Lipopolysaccharide inhibits TNF-induced apoptosis: role of nuclear factor-kappa B activation and reactive oxygen intermediates. J. Immunol 1999, 162, 1510–1518. [Google Scholar]
- Kaltschmidt, B.; Kaltschmidt, C.; Hofmann, T. G.; Hehner, S. P.; Droge, W.; Schmitz, L. M. The pro- or anti-aapoptotic function of NF-KB is determined by the nature of the apoptotic stimulus. Eur. J. Biochem 2000, 267, 3828–3835. [Google Scholar]
- Wang, C. Y.; Mayo, M. W.; Baldwin, A. S., Jr. TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-kappa B. Science 1996, 274, 784–787. [Google Scholar]
- van Antwerp, D. J.; Martin, S. J.; Kafri, T.; Green, D. R.; Verma, I. M. Suppression of TNF-alpha- induced apoptosis by NF-kappa B. Science 1996, 274, 787–789. [Google Scholar]
- Beg, A. A.; Baltimore, D. An essential role for NF- kappa B in preventing TNF-a-induced cell death. Science 1996, 274, 782–784. [Google Scholar]
- Liu, Z.; Hsu, H.; Goeddel, D. V.; Karin, M. Dissection of TNF receptor I effector functions: JNK activation is not linked to apoptosis while NF-κB activation prevents cell death. Cell 1996, 87, 565–576. [Google Scholar]
- Maiuri, M. C.; Tajana, G.; Iuvone, T.; De Stefano, D.; Mele, G.; Ribecca, M. T.; Cinelli, M. P.; Romano, M. F.; Turco, M. C.; Carnuccio, R. Nuclear factor-kappa B regulates inflammatory cell apoptosis and phagocytosis in rat carrageenin-sponge implant model. Am. J. PathoL 2004, 165(1), 115–1126. [Google Scholar]
- D’Acquisto, F.; De Cristofaro, F.; Maiuri, M. C.; Tajana, G.; Crnuccio, R. Protective role of nuclear factor kappa B against nitric oxide-induced apoptosis in J774 macrophages. Cell Death Differ 2001, 8(2), 144–151. [Google Scholar]
- Rahman, I. Oxidative stress and gene transcription in asthma and chronic obstructive pulmonary disease: antioxidant therapeutic targets. Curr. Drug Targets Inflamm. Allergy 2002, 1(3), 291–315. [Google Scholar]
- Barnes, P. J.; Karin, M. Nuclear factor-κB: a pivotal transcription factor in chronic inflammatory diseases. N. Engl. J. Med 1997, 336(15), 1066–1071. [Google Scholar]
- Baulig, A.; Garlatti, M.; Bonvalott, V.; Marchand, A.; Barouki, R.; Marano, F.; Baeza-Squiban, A. Involvement of reactive oxygen species in the metabolic pathways triggered by diesel exhaust particles in human airway epithelial cells. Am. J. Physiol 2003, 285, L671–L679. [Google Scholar]
- Schreck, R.; Reiber, P.; Baeurlef, P. A. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-KB transcription factor and HIV-1. EMBO J 1991, 10, 2247–2258. [Google Scholar]
- Staal, F. J.; Anderson, M. T.; Herzenberg, L. A. Redox regulation regulation of aactivaation of NF-κB transcription factor complex: effects of N- acetylcysteine. Methods Enzymol 1995, 252, 168–174. [Google Scholar]
- Kumagai, Y.; Arimoto, T.; Shinyashiki, M.; Shimojo, N.; Nakai, Y.; Yoshikawa, T.; Sagai, M. Generation of reactive oxygen species during interaction of diesel exhaust particle component with NADPH-cytochrome p450 reductase and involvement of the bioactivation in the DNA damage. Free Rad. Biol. Med 1997, 22(3), 479–487. [Google Scholar]
- Janssen-Heininger, Y. M. W.; Macara, I.; Mossman, B. T. Cooperatively between oxidants and tumor necrosis factor in the activation of nuclear factor (NF)-KB: requirement of Ras/mitoge-activated protein kinases in the activation of NF-κB by oxidants. Am. J. Respir. Cell Mol. Biol 1999, 20, 942–952. [Google Scholar]
- Malinin, N. L.; Boldin, M. P.; Kovalenka, A. V.; Wallach, D. MAP3K-related kinase involved in NF-κB induction by TNF, CD95, and IL-1. Nature 1997, 385, 540–544. [Google Scholar]
© 2005 MDPI. All rights reserved.
Share and Cite
Kafoury, R.M.; Madden, M.C. Diesel Exhaust Particles Induce the Over expression of Tumor Necrosis Factor-α (TNF-α) Gene in Alveolar Macrophages and Failed to Induce Apoptosis through Activation of Nuclear Factor-κB (NF-κB). Int. J. Environ. Res. Public Health 2005, 2, 107-113. https://doi.org/10.3390/ijerph2005010107
Kafoury RM, Madden MC. Diesel Exhaust Particles Induce the Over expression of Tumor Necrosis Factor-α (TNF-α) Gene in Alveolar Macrophages and Failed to Induce Apoptosis through Activation of Nuclear Factor-κB (NF-κB). International Journal of Environmental Research and Public Health. 2005; 2(1):107-113. https://doi.org/10.3390/ijerph2005010107
Chicago/Turabian StyleKafoury, Ramzi M., and Michael C. Madden. 2005. "Diesel Exhaust Particles Induce the Over expression of Tumor Necrosis Factor-α (TNF-α) Gene in Alveolar Macrophages and Failed to Induce Apoptosis through Activation of Nuclear Factor-κB (NF-κB)" International Journal of Environmental Research and Public Health 2, no. 1: 107-113. https://doi.org/10.3390/ijerph2005010107