Cytokines and Adhesion Molecules Expression in the Brain in Human Cerebral Malaria

Abstract
:Introduction
Materials and Methods
Case Selection
- (1)
- Cerebral malaria [CM].
- (2)
- Malaria complicated by severe anaemia/severe malarial anaemia [SMA].
- (3)
- Purulent bacterial meningitis [PBM] (i.e. central nervous system infection other than cerebral malaria)
- (4)
- Non-central nervous system infection [NCNSI] (i.e. infection in an anatomic organ-system other than the central nervous system), and (5) Non-infection deaths [NI] (i.e. no focus of infection found clinically or at autopsy).
Brain Tissue Preparation and Immunohistochemistry
Evaluation of Immunostaining
Data Analysis
Results
Clinical and Diagnostic Details
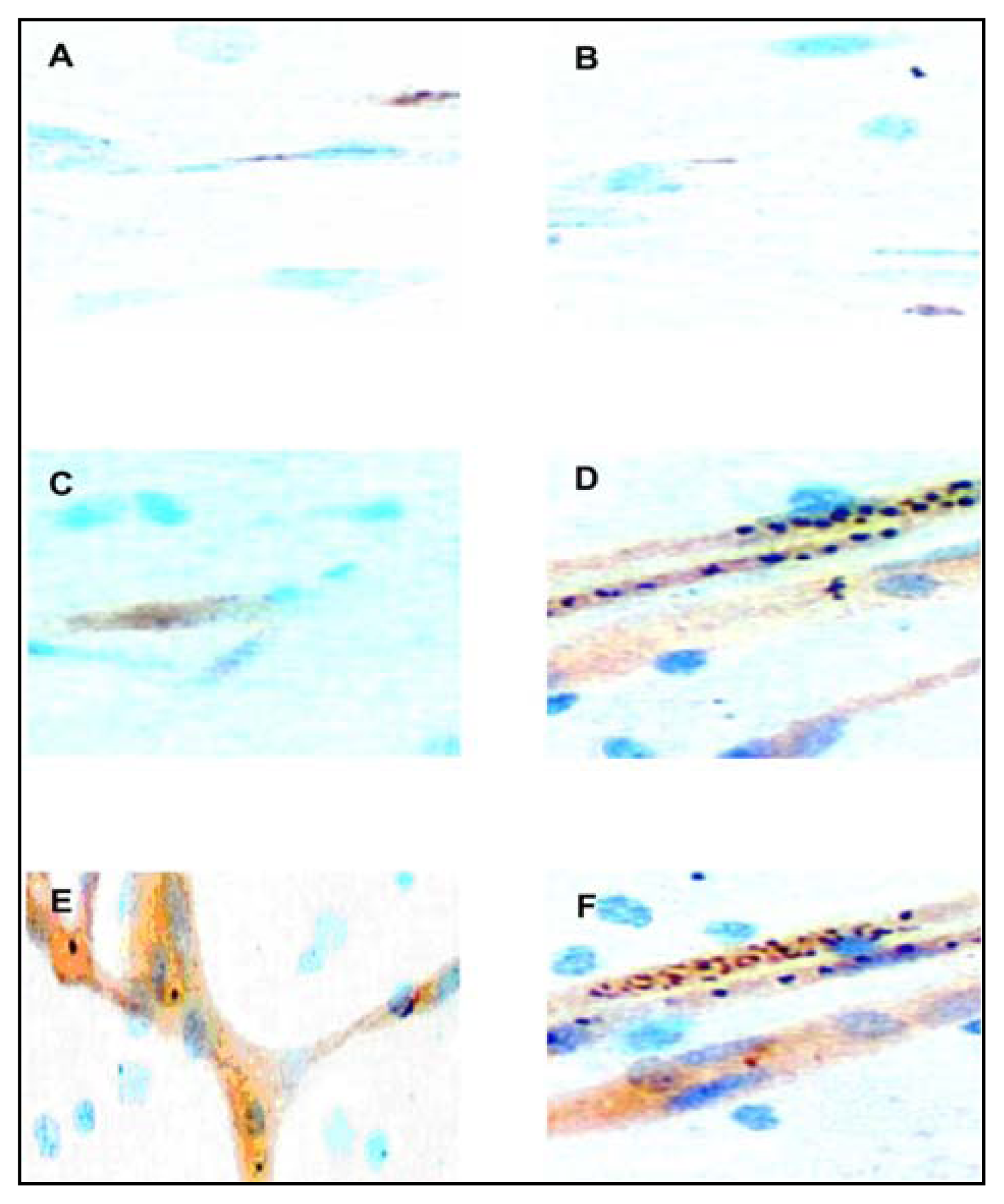
Adhesion Molecule Immunostaining
Cytokine Immunostaining
Discussion



{kind=link}
{kind=link}
| Case | Age (months) | Sex | Duration of Admission (hours) | Hb (g/dl) | Blantyre Coma Score | Peripheral Parasitaemia on Admission (μl) | Brain Smear# | Final Diagnosis |
|---|---|---|---|---|---|---|---|---|
| 1 | 56 | M | 28.5 | 8.1 | 2 | 14,654 | Pos | CM |
| 2 | 96 | M | 13 | 12.2 | 2 | 54,392 | Pos | CM |
| 3 | 50 | F | 4 | 6.0 | 0 | 21,413 | Pos | CM |
| 4 | 48 | F | 3.5 | 4.7 | 4 | 88,796 | Neg | SMA |
| 5 | 108 | F | 12 | 7.4 | 2 | 14,486 | Pos | CM |
| 6 | 132 | M | 31 | 9.2 | 1 | 78,624 | Pos | CM |
| 7 | 48 | F | 14.5 | 6.9 | 2 | 74,092 | Pos | CM |
| 8 | 18 | M | 8 | 8.1 | 5 | 0 | Neg | SBP* |
| 9 | 84 | M | 6 | 4.1 | 5 | 0 | Neg | HSC** |
| 10 | 39 | F | 3 | 6.4 | 2 | 54,867 | Pos | CM |
| 11 | 9 | F | 1 | 3.4 | 4 | 440,262 | Neg | SMA |
| 12 | 144 | M | 1.5 | 2.5 | 5 | 0 | Neg | BDU† |
| 13 | 72 | M | 5 | 8.5 | 3 | 0 | Neg | PBM‡ |
| 14 | 8 | F | 2.5 | 4.0 | 4 | 140,251 | Neg | SMA |
| 15 | 90 | M | 48 | 8.7 | 5 | 0 | Neg | TP&S¶ |
| 16 | 18 | M | 2 | 3.0 | 4 | 231,175 | Neg | SMA |
| 17 | 18 | F | 3.5 | 2.8 | 3 | 91,782 | Neg | SMA |
| 18 | 48 | F | 31 | 8.0 | 2 | 80,640 | Pos | CM |
| 19 | 72 | F | 2 | 7.2 | 2 | 17,789 | Pos | CM |
| 20 | 42 | F | 1 | 6.6 | 1 | 40,115 | Pos | CM |
| 21 | 36 | M | 37 | 10.4 | 5 | 0 | Neg | NPS§ |
| Antigen | Total Number of Vessels | R+S+ | R+S− | R−S+ | R−S− | X2Value | P Value (df = 1) | Relative Risk |
|---|---|---|---|---|---|---|---|---|
| ICAM-1 | 860 | 350 | 173 | 130 | 207 | 66.55 | 3.1 X 10−16 | 1.73 |
| VCAM-1 | 821 | 305 | 127 | 180 | 209 | 50.52 | 1.2 X 10−12 | 1.53 |
| E-Selectin | 843 | 317 | 140 | 160 | 226 | 65.43 | 6.1 X 10−16 | 1.67 |
References
- Tropical Disease Research (TDR), Malaria. In Tropical Research; 7th Program Report, UNDPA World Bank/WHO: Geneva, 1998; pp. 3–40.
- Kwiatkowski, D.; Hill, A. V.; Sambou, I.; Twumasi, P.; Castracane, J.; Manogue, K. R.; Cerami, A.; Brewster, D. R.; Greenwood, B. M. TNF concentration in fatal cerebral, non-fatal cerebral and uncomplicated Plasmodium falciparum malaria. Lancet 1990, 336, 1201–1204. [Google Scholar]
- Newton, C. R. J. C.; Krishna, S. Severe falciparum malaria in children: current understanding of pathophysiology and supportive treatment. Pharmacol Ther 1998, 79, 1–53. [Google Scholar]
- Artavanis-Tsakonas, K.; Tongren, J. E.; Riley, E. M. The war between the malaria parasite and the immune system: immunity, immunoregulation and immuno-pathology. Clin Exp Immunol 2003, 133, 145–52. [Google Scholar]
- Brown, H.; Turner, G.; Rogerson, S.; Tembo, M.; Mwenechanya, J.; Molyneux, M.; Taylor, T. Cytokine Expression in the Brain in Human Cerebral Malaria. J Infect Dis 1999, 180, 1742–1746. [Google Scholar]
- Mazier, D.; Idrissa-Boubou, M. Immunogenetics and cerebral malaria. Bull Soc. Pathol Exot 1999, 92, 249–255. [Google Scholar]
- Rogerson, S. J.; Tembenu, R.; Dobano, C.; Plitt, S.; Taylor, T. E.; Molyneux, M. E. Cytoadherence characteristics of Plasmodium falciparum-infected erythrocytes from Malawian children with severe and uncomplicated malaria. Am J. Trop. Med. Hyg 1999, 61, 461–412. [Google Scholar]
- Silamut, K.; Phu, N. R.; Whitty, C.; Turner, G. D.; Louwrier, K.; Mai, N. T.; Simpson, J. A.; Hien, T. T.; White, N. J. A quantitative analysis of the microvascular sequestration of malaria parasites in the Human Brain. Am J. Pathol 1999, 755, 395–410. [Google Scholar]
- Ockenhouse, C. F.; Ho, M.; Tandon, N. N.; van Seventer, G. A.; Shaw, S.; White, N. J.; Jamieson, G. A.; Chulay, J. D.; Webster, H. K. Molecular basis of sequestration in severe and uncomplicated Plasmodium falciparum malaria: differential adhesion of infected erythrocytes to CD36 and ICAM-1. J Infect Dis 1991, 164, 163–169. [Google Scholar]
- Turner, G. D. H.; Morrison, H.; Jones, M.; Davis, T. M. E.; Looareesuwan, S.; Buley, I. D.; Gatter, K. C.; Newbold, C. I.; Pukrittayakamee, S.; Nagachinta, B.; White, N. J.; Berendt, A. R. An immunohistochemical study of the pathology of fatal Malaria: evidence for systemic endothelial activation. Am J. Pathol 1994, 145, 1057–1069. [Google Scholar]
- Turner, G. D.; Ly, V. C.; Nguyen, T. H.; Tran, T. H.; Nguyen, H. P.; Bethell, D.; Wyllie, S.; Louwrier, K.; Fox, S. B.; Gatter, K. C.; Day, N. P.; Tran, T. H.; White, N. J.; Berendt, A. R. Systemic endothelial activation in both mild and severe malaria: correlating dermal microvascular endothelial cell phenotype and soluble cell adhesion molecules with disease severity. Am J. Pathol 1998, 152, 1477–1487. [Google Scholar]
- Ockenhouse, C.; Tegoshi, T.; Maeno, Y.; Benjamin, C.; Ho, M.; Kan, K.; Thway, Y.; Win, K.; Aikawa, M.; Lobb, R. Human vascular endothelial cell adhesion receptors for Plasmodium falciparum infected erythrocytes: roles for endothelial leukocytes adhesion molecule-1 and vascular cell adhesion molecule-1. J. Exp Med 1992, 176, 1183–1189. [Google Scholar]
- Aikawa, M.; Brown, A.; Smith, CD.; Tegoshi, T.; Howard, R. J.; Hasler, T. H.; Ito, Y.; Perry, G.; Collins, W. E.; Webster, K. A primate model for human cerebral malaria: Plasmodium coatneyi- infected rhesus monkeys. Am J. Trop. Med. Hyg 1992, 46, 391–397. [Google Scholar]
- Weber, C.; Negrescu, E.; Erl, W.; Pietsch, A.; Frankenberger, M.; Ziegler-Heitbrock, H. W.; Siess, W.; Weber, P. C. hihibitors of protein tyrosine kinase suppress TNF-stimulated induction of endothelial cell adhesion molecules. J. Immunol 1995, 755, 445–451. [Google Scholar]
- Udomsangpetch, R.; Chivapat, S.; Viriyavejakul, P.; Riganti, M.; Wilairatana, P.; Pongpontatn, E.; Looaresuwan, S. Involvement of cytokines in the histopathology of Malaria. Am J. Trop. Med. Hyg 1997, 57, 501–506. [Google Scholar]
- Porta, J.; Carota, A.; Pizzolato, G. P.; Wildi, E.; Widmer, M. C.; Margairaz, C.; Grau, G. E. Immunopathological changes in Human Cerebral Malaria. Clin Neuropathol 1993, 12, 142–146. [Google Scholar]
- Medana, I. M.; Hunt, N. H.; Chaudhri, G. TNF-αexpression in the brain during fatal murine cerebral malaria: evidence for production by microglia and astrocytes. Am J. Pathol 1997, 750, 1473–1483. [Google Scholar]
- Polder, T. W.; Eling, W. M.; Kubat, K.; Jerusalem, C. R. Histochemistry of cerebral lesions in mice infected with. Plasmodium berghei. Trop. Med. Parasitol 1988, 39, 277–283. [Google Scholar]
- Polder, T. W.; Eling, W. M.; Jerusalem, C. R.; Wijers-Rouw, M. J Neural Sci 1991a, 707, 24–34.
- Polder, T. W.; Jerusalem, C. R.; Eling, W. M. Morphological characteristics of intracerebral arterioles in clinical (Plasmodium falciparum) and experimental (Plasmodium berghei) cerebral malaria. J Neurol Sci 1991b, 707, 35–46. [Google Scholar]
- Flanders, K. C.; Ren, R. F.; Lippa, C. F. Transforming growth factor-betas in neurodegenerative disease. Prog Neurobiol 1998, 54, 71–85. [Google Scholar]
- Johnson, M. D.; Gold, L. L. Distribution of transforming growth factor-beta isoforms in HIV-1 encephalitis. Hum Pathol 1996, 27, 643–9. [Google Scholar]
- Omer, F. M.; Riley, E. M. Transforming growth factor beta production is inversely correlated with severity of murine malaria infection. J. Exp. Med 1998, 188, 39–48. [Google Scholar]
- Bitsch, A.; Trostdorf, F.; Brack, W.; Schmidt, H.; Fischer, F. R.; Nau, R. Central nervous system TNF-alpha mRNA expression during rabbit experimental pneumococcal meningitis. Neurosci. Lett 1997, 237, 105–108. [Google Scholar]
- Gahring, L. C.; Carlson, N. G.; Kulmar, R. A.; Rogers, S. W. Neuronal expression of tumour necrosis factor alpha in the murine brain. Neuroimmunomodulation 1996, 3, 289–303. [Google Scholar]
- Rothwell, N. J.; Strijbos, P. J. Cytokines in neurodegeneration and repair. Int J Dev Neurosci 1995, 13, 179–185. [Google Scholar]
- Tongren, J. E.; Yang, C.; Collins, W. E.; Sullivan, J. S.; Lai, A. A.; Xiao, L. Expression of proinflammatory cytokines in four regions of the brain in Macaque Mulatta ( rhesus) monkeys infected with Plasmodium coatneyi. Am J. Trop. Med. Hyg 2000, 62, 530–534. [Google Scholar]
- Smith, C. D.; Brown, A. E.; Nakazawa, S.; Fujioka, H.; Aikawa, M. Multi-organ erythrocyte sequestration and ligand expression in rhesus monkeys infected with Plasmodium coatneyi malaria. Am J. Trop. Med. Hyg 1996, 55, 379–383. [Google Scholar]
- Sein, K. K.; Maeno, Y.; Thuc, H. V.; Anh, T. K.; Aikawa, M. Differential sequestration pattern of parasitized erythrocytes in the cerebrum and cerebellum in Human Cerebral Malaria. Am J Trop Med Hyg 1993a, 48, 504–511. [Google Scholar]
- Sein, K. K.; Brown, A. E.; Maeno, Y.; Smith, C. D.; Corcoran, K. D.; Hansukjariya, P.; Webster, H. K.; Aikawa, M. Sequestration pattern of parasitized erythrocytes in cerebrum, mid-brain and cerebellum of Plasmodium coatneyi-iniected rhesus monkeys (Macaca mulatto). Am J Trop Med Hyg 1993b, 49, 513–519. [Google Scholar]
- Engwerda, C. R.; Mynott, T. L.; Sawhney, S.; de Souza, J. B.; Bickle, Q. D.; Kaye, P. M. Locally up-regulated lymphotoxin alpha, not systemic tumour necrosis factor alpha is the principal mediator of murine cerebral malaria. J. Exp Med 2002, 195, 1371–1377. [Google Scholar]
- Raja, R. N. Postmortem examination in cerebral malaria: a new simple method of demonstrating parasites in the capillaries of the brain. Indian Med. Gazette 1922, 57, 298–299. [Google Scholar]
- World Health Organization, Communicable Diseases Cluster: Severe falciparum malaria. In Trans Roy Soc Trop Med Hyg; 2000; Volume 94, pp. 81–90.
- Grau, G. E.; Mackenzie, C. D.; Carr, R. A.; Redard, M.; Pizzolato, G.; Allasia, C.; Cataldo, C.; Taylor, T. E.; Molyneux, M. E. Platelet accumulation in brain microvessels in fatal paediatric cerebral malaria. J. Infect Dis 2003, 187, 461–466. [Google Scholar]
- Wassmer, S. C.; Combes, V.; GRAU, G. E. Pathophysiology of cerebral malaria: role of host cells in the modulation of cytoadhesion. Ann NY Acad Scl 2003a, 992, 30–38. [Google Scholar]
- Wassmer, S. C.; Coltel, N.; Combes, V.; Grau, G. E. Pathogenesis of cerebral malaria: facts and hypotheses. Med Trop (Mars) 2003b, 63, 254–257. [Google Scholar]
- Wassmer, S. C.; Lepolard, C.; Traore, B.; Pouvelle, B.; Gysin, J.; Grau, G. E. Platelets reorient Plasmodium falciparum-infected erythrocyte cytoadhesion activated endothelial cells. J. Infect Dis 2004, 189, 180–189. [Google Scholar]
- Sarfo, B. Y.; Singh, S.; LiUard, J. W.; Quarshie, A.; Gyasi, R. K.; Armah, H.; Adjei, A. A.; Jolly, P.; Stiles, J. K. The cerebral-malaria-associated expression of RANTES, CCR3 and CCR5 in post mortem tissue samples. Ann Trop Med. Parasitol 2004, 98, 297–303. [Google Scholar]
- Belnoue, E.; Kayabanda, M.; Deschemin, J. C. CCR5 deficiency decreases susceptibility to experimental cerebral malaria. Blood 2003, 101, 4253–4259. [Google Scholar]
- Nitcheu, J.; Bonduelle, O.; Combadiere, C.; Tefit, M.; Seilhean, D.; Mazier, D.; Combadiere, B. Perforin-dependent brain-infiltrating cytotoxic CD8+ T lymphocytes mediate experimental cerebral malaria pathogenesis. J. Immunol 2003, 170, 2221–2228. [Google Scholar]
- Holding, P. A.; Stevenson, J.; Peshu, N.; Marsh, K. Cognitive sequelae of severe malaria with impaired consciousness. Trans Roy Soc Trop Med Hyg 1999, 93, 529–534. [Google Scholar]
- Schmahmann, J. D. An emerging concept: The cerebellar contribution to higher functions. Arch. Neurol 1991, 45, 1178–87. [Google Scholar]
© 2005 MDPI. All rights reserved.
Share and Cite
Armah, H.; Wiredu, E.K.; Dodoo, A.K.; Adjei, A.A.; Tettey, Y.; Gyasi, R. Cytokines and Adhesion Molecules Expression in the Brain in Human Cerebral Malaria. Int. J. Environ. Res. Public Health 2005, 2, 123-131. https://doi.org/10.3390/ijerph2005010123
Armah H, Wiredu EK, Dodoo AK, Adjei AA, Tettey Y, Gyasi R. Cytokines and Adhesion Molecules Expression in the Brain in Human Cerebral Malaria. International Journal of Environmental Research and Public Health. 2005; 2(1):123-131. https://doi.org/10.3390/ijerph2005010123
Chicago/Turabian StyleArmah, Henry, Edwin Kwame Wiredu, Alfred Kofi Dodoo, Andrew Anthony Adjei, Yao Tettey, and Richard Gyasi. 2005. "Cytokines and Adhesion Molecules Expression in the Brain in Human Cerebral Malaria" International Journal of Environmental Research and Public Health 2, no. 1: 123-131. https://doi.org/10.3390/ijerph2005010123