Molecular Dynamics Simulations of Clathrate Hydrates on Specialised Hardware Platforms

Abstract
:1. Introduction
2. Simulation Methodology
3. Results and Discussion
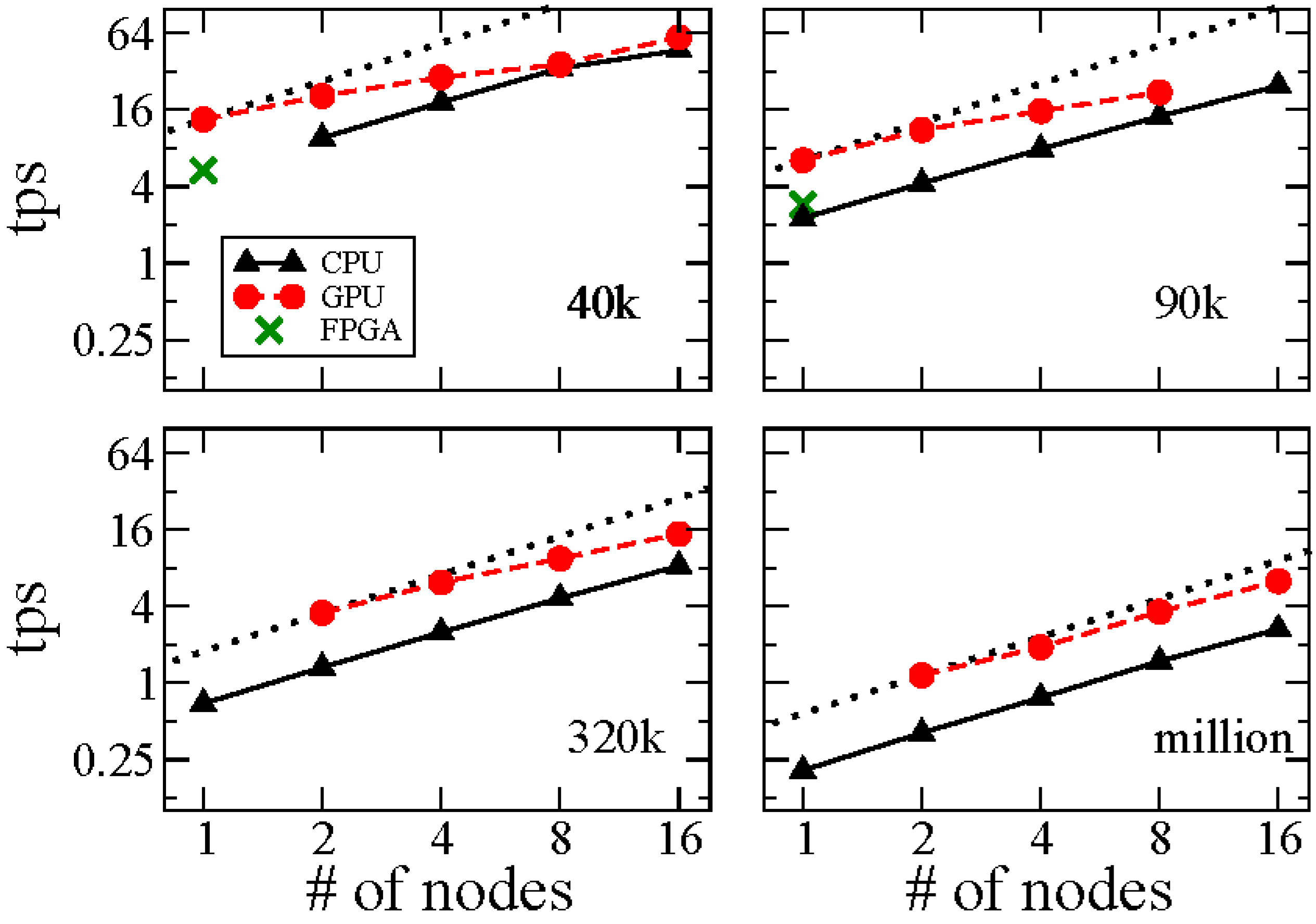

{kind=link}
| No. of Molecules | No. of Nodes | CPU | CPU-GPU |
|---|---|---|---|
| 39,788 | 1 | 0.0744 | |
| 39,788 | 2 | 0.104 | 0.0485 |
| 39,788 | 4 | 0.0545 | 0.0346 |
| 39,788 | 8 | 0.0294 | 0.0273 |
| 39,788 | 16 | 0.0212 | 0.0167 |
| 91,430 | 1 | 0.443 | 0.156 |
| 91,430 | 2 | 0.235 | 0.0898 |
| 91,430 | 4 | 0.127 | 0.0637 |
| 91,430 | 8 | 0.0699 | 0.0453 |
| 91,430 | 16 | 0.0405 | |
| 318,304 | 1 | 1.449 | |
| 318,304 | 2 | 0.755 | 0.283 |
| 318,304 | 4 | 0.400 | 0.162 |
| 318,304 | 8 | 0.217 | 0.105 |
| 318,304 | 16 | 0.120 | 0.0673 |
| 1,074,276 | 1 | 4.863 | |
| 1,074,276 | 2 | 2.471 | 0.869 |
| 1,074,276 | 4 | 1.305 | 0.524 |
| 1,074,276 | 8 | 0.675 | 0.275 |
| 1,074,276 | 16 | 0.379 | 0.158 |
4. Conclusions
Acknowledgements
References
- Makogon, Y.F. Hydrates of Hydrocarbons; PennWell Books: Tulsa, OK, USA, 1997. [Google Scholar]
- Sloan, E.D.; Koh, C.A. Clathrate Hydrates of Natural Gases, 3rd ed.; CRC Press, Taylor & Francis: Boca Raton, FL, USA, 2007. [Google Scholar]
- Waite, W.F.; Stern, L.A.; Kirby, S.H.; Winters, W.J.; Mason, D.H. Simultaneous determination of thermal conductivity, thermal diffusivity and specific heat in Si methane hydrate. Geophys. J. Int. 2007, 169, 767–774. [Google Scholar] [CrossRef]
- Boswell, R. Resource potential of methane hydrate coming into focus. J. Petrol. Sci. Eng. 2007, 56, 9–13. [Google Scholar] [CrossRef]
- Narumi, T.; Ohno, Y.; Okimoto, N.; Suenaga, A.; Yanai, R.; Taiji, M. A high-speed special-purpose computer for molecular dynamics simulations: MDGRAPE-3. John Von Neumann Inst. Comput. 2006, 34, 29–36. [Google Scholar]
- Taiji, M. MDGRAPE-3 Chip: A 165-Gflops Application-Specific LSI for Molecular Dynamics Simulations. In Proceedings of the 16th IEEE Hot Chips Symposium, Stanford, CA, USA, 22–24 August 2004.
- Kikugawa, G.; Apostolov, R.; Kamiya, N.; Taiji, M.; Himeno, R.; Nakamura, H.; Yonezawa, Y. Application of MDGRAPE-3, a special purpose board for molecular dynamics simulations, to periodic biomolecular systems. J. Comput. Chem. 2009, 30, 110. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids; Clarendon Press: Oxford, UK, 1987. [Google Scholar]
- Shaw, D.E.; Dror, R.O.; Salmon, J.K.; Grossman, J.P.; Mackenzie, K.M.; Bank, J.A.; Young, C.; Deneroff, M.M.; Batson, B.; Bowers, K.J.; et al. Millisecond-Scale Molecular Dynamics Simulations on Anton. In Proceedings the Conference on High Performance Computing Networking, Storage and Analysis, Portland, OR, USA, 14–20 November 2009; pp. 1–11.
- Shaw, D.E.; Maragakis, P.; Lindorff-Larsen, K.; Piana, S.; Dror, R.O.; Eastwood, M.P.; Bank, J.A.; Jumper, J.M.; Salmon, J.K.; Shan, Y. Atomic-Level characterization of the structural dynamics of proteins. Science 2010, 330, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Shaw, D.E. A fast, scalable method for the parallel evaluation of distance-limited pairwise particle interactions. J. Comput. Chem. 2005, 26, 1318. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; Klepeis, J.L.; Eastwood, M.P.; Dror, R.O.; Shaw, D.E. Gaussian split Ewald: A fast Ewald mesh method for molecular simulation. J. Chem. Phys. 2005, 122. [Google Scholar] [CrossRef]
- Stone, J.E.; Phillips, J.C.; Freddolino, P.L.; Hardy, D.J.; Trabuco, L.G.; Schulten, K. Accelerating molecular modeling applications with graphics processors. J. Comput. Chem. 2007, 28, 2618–2640. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N-log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Brown, M.W.; Kohlmeyer, A.; Plimpton, S.J.; Tharrington, A.N. Implementing molecular dynamics on hybrid high performance computers–Particle–particle particle-mesh. Comp. Phys. Commun. 2012, 183, 449–459. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tuckerman, M.E.; Tobias, D.J.; Klein, M.L. Explicit reversible integrators for extended systems dynamics. Mol. Phys. 1996, 87, 1117–1157. [Google Scholar] [CrossRef]
- Trott, C.R. LAMMPSCUDA—A new GPU accelerated Molecular Dynamics Simulations Package and Its Application to Ion-Conducting Glasses. Ph.D. Thesis, University of Technology Ilmenau, Germany, 2011. [Google Scholar]
- Kohlmayer, A. Implementation of Multi-level parallelism in LAMMPS for improved scaling on petaflops supercomputers. In Presented at 2011 TMS Annual Meeting, San Diego, CA, USA, 27 February–3 March 2011.
- Brode, S.; Ahlrichs, R. An optimized MD program for the vector computer cyber-205. Comput. Phys. Commun. 1986, 42, 51. [Google Scholar] [CrossRef]
- Tse, J.S.; Klein, M.L.; McDonald, I.R. Computer simulation studies of the structure. I. Clathrate hydrates of methane, tetrafluoromethane, cyclopropane, and ethylene oxide. J. Chem. Phys. 1984, 81, 6146–6153. [Google Scholar] [CrossRef]
- McMullan, R.K.; Jeffrey, G.A. Polyhedral clathrate hydrate. IX. Structure of ethylene oxide hydrate. J. Chem. Phys. 1965, 42, 2725–2732. [Google Scholar] [CrossRef]
- Bernal, J.D.; Fowler, R.H. A theory of water and ionic solution, with particular reference to hydrogen and hydroxyl ions. J. Chem. Phys. 1933, 1, 515–547. [Google Scholar] [CrossRef]
- English, N.J. Molecular dynamics simulations of liquid water using various long-range electrostatics techniques. Mol. Phys. 2006, 103, 243–253. [Google Scholar] [CrossRef]
- English, N.J.; MacElroy, J.M.D. Structural and dynamical properties of methane clathrate hydrates. J. Comput. Chem. 2003, 24, 1569–1581. [Google Scholar] [CrossRef] [PubMed]
- Sloan, E.D. Fundamental principles and applications of natural gas hydrates. Nature 2003, 426, 353. [Google Scholar] [CrossRef] [PubMed]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Varini, N.; English, N.J.; Trott, C.R. Molecular Dynamics Simulations of Clathrate Hydrates on Specialised Hardware Platforms. Energies 2012, 5, 3526-3533. https://doi.org/10.3390/en5093526
Varini N, English NJ, Trott CR. Molecular Dynamics Simulations of Clathrate Hydrates on Specialised Hardware Platforms. Energies. 2012; 5(9):3526-3533. https://doi.org/10.3390/en5093526
Chicago/Turabian StyleVarini, Nicola, Niall J. English, and Christian R. Trott. 2012. "Molecular Dynamics Simulations of Clathrate Hydrates on Specialised Hardware Platforms" Energies 5, no. 9: 3526-3533. https://doi.org/10.3390/en5093526