Methane Flux and Authigenic Carbonate in Shallow Sediments Overlying Methane Hydrate Bearing Strata in Alaminos Canyon, Gulf of Mexico

Abstract
:1. Introduction
2. Methods
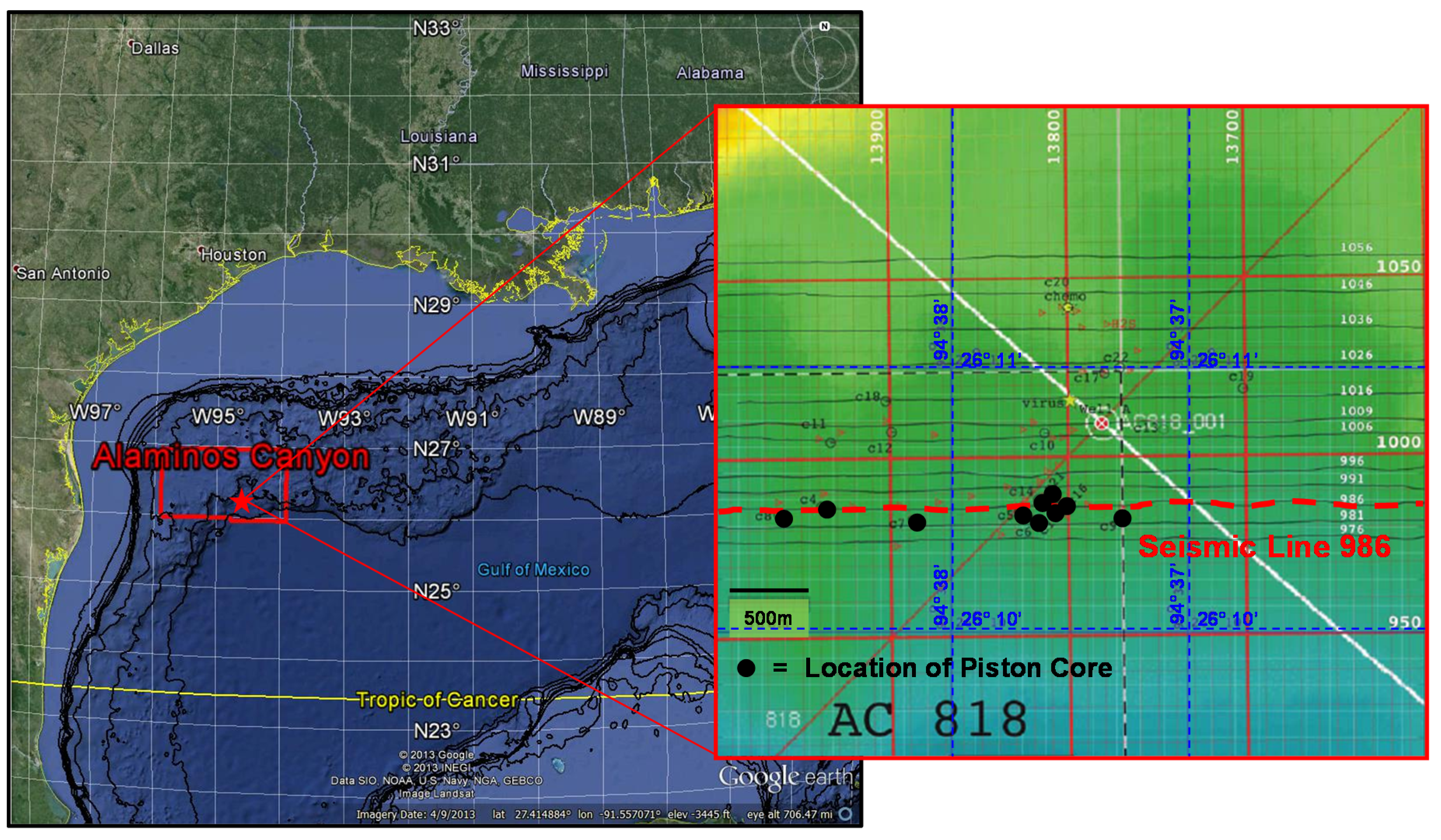
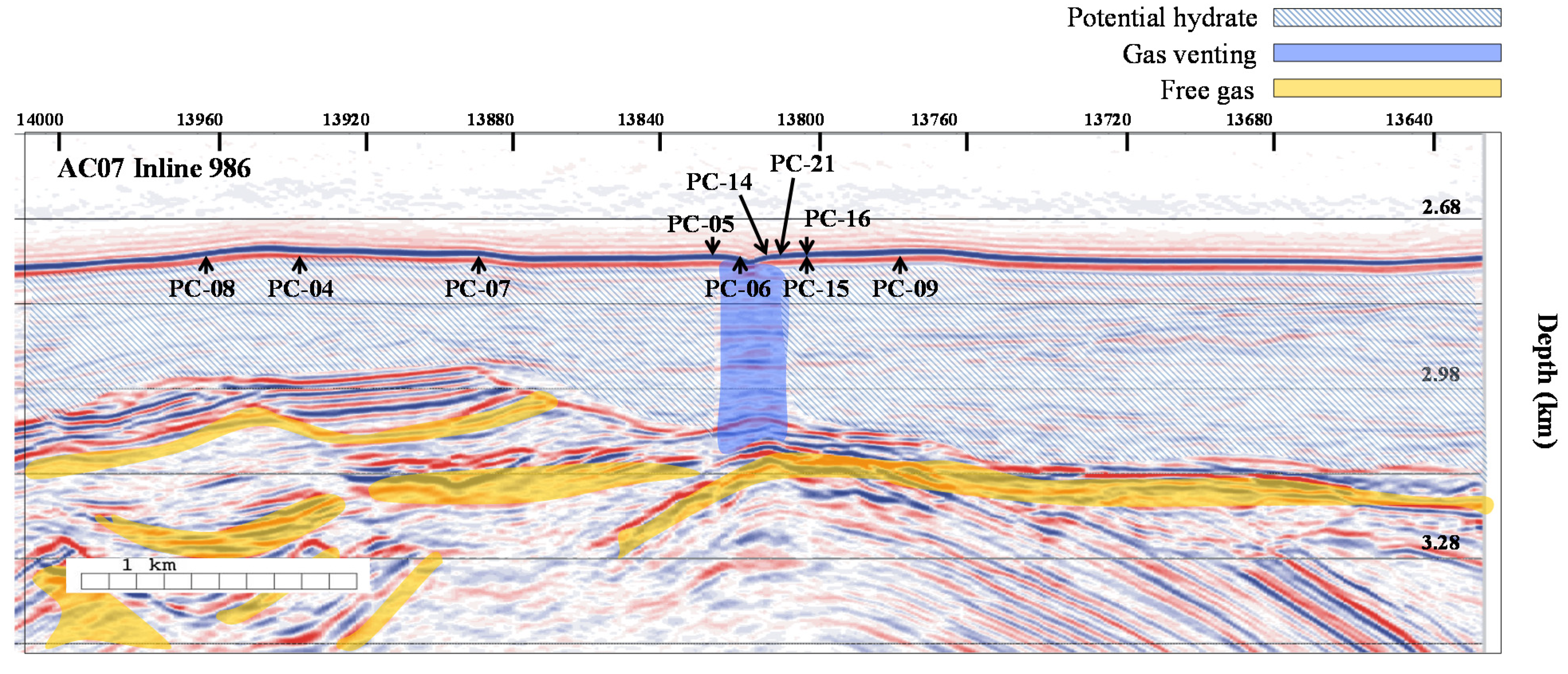
2.1. Study Area

2.2. Sample Collection

2.3. Sample Processing
2.4. Sample and Data Analysis
3. Results
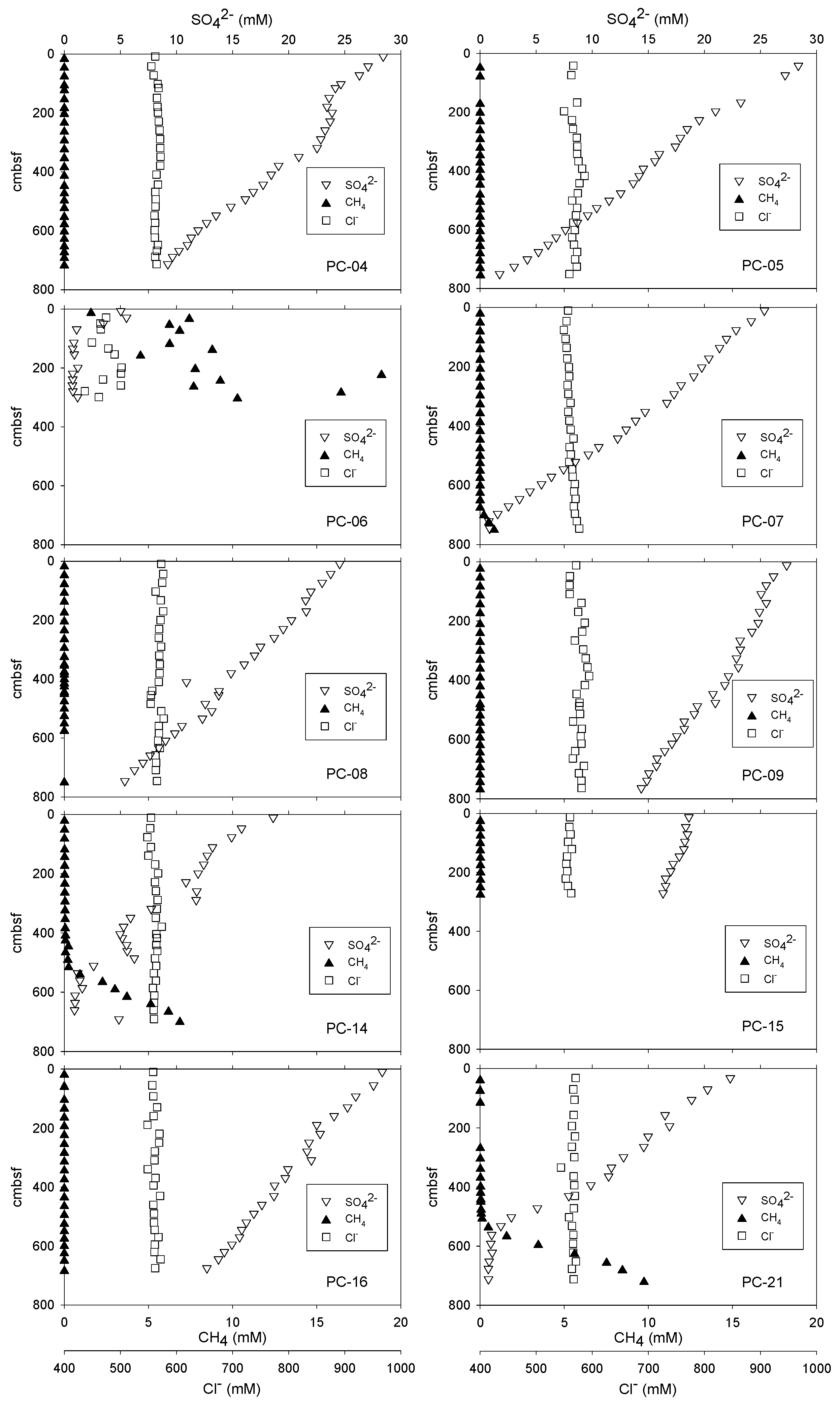
3.1. Sediment Headspace CH4 and Porewater SO42− and Cl− Concentrations

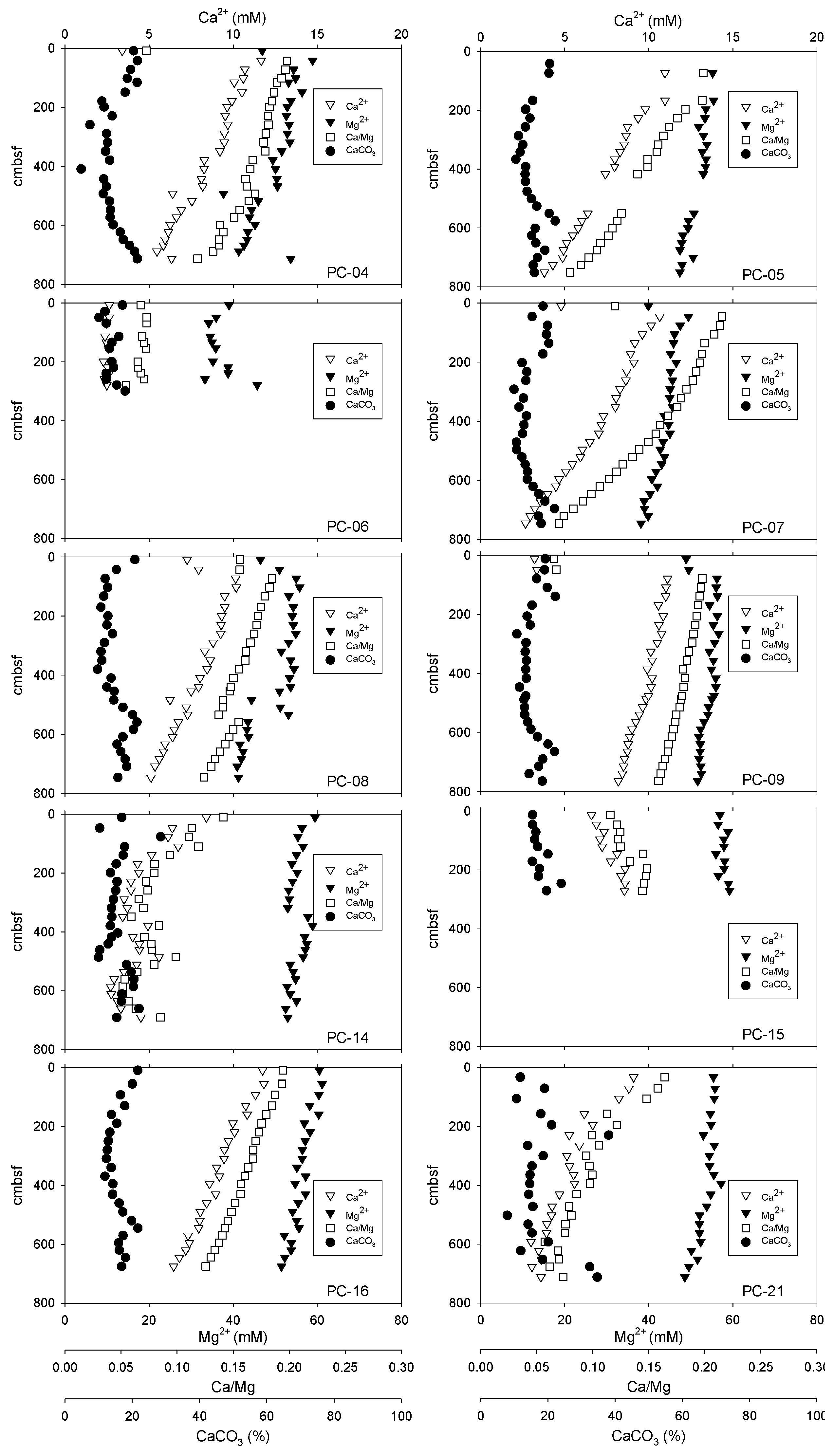
3.2. Porewater Ca2+and Mg2+ Concentrations, Ca/Mg Ratios, and Sediment CaCO3 Content

3.3. Porewater DIC Concentrations and δ13C-DIC

4. Discussion
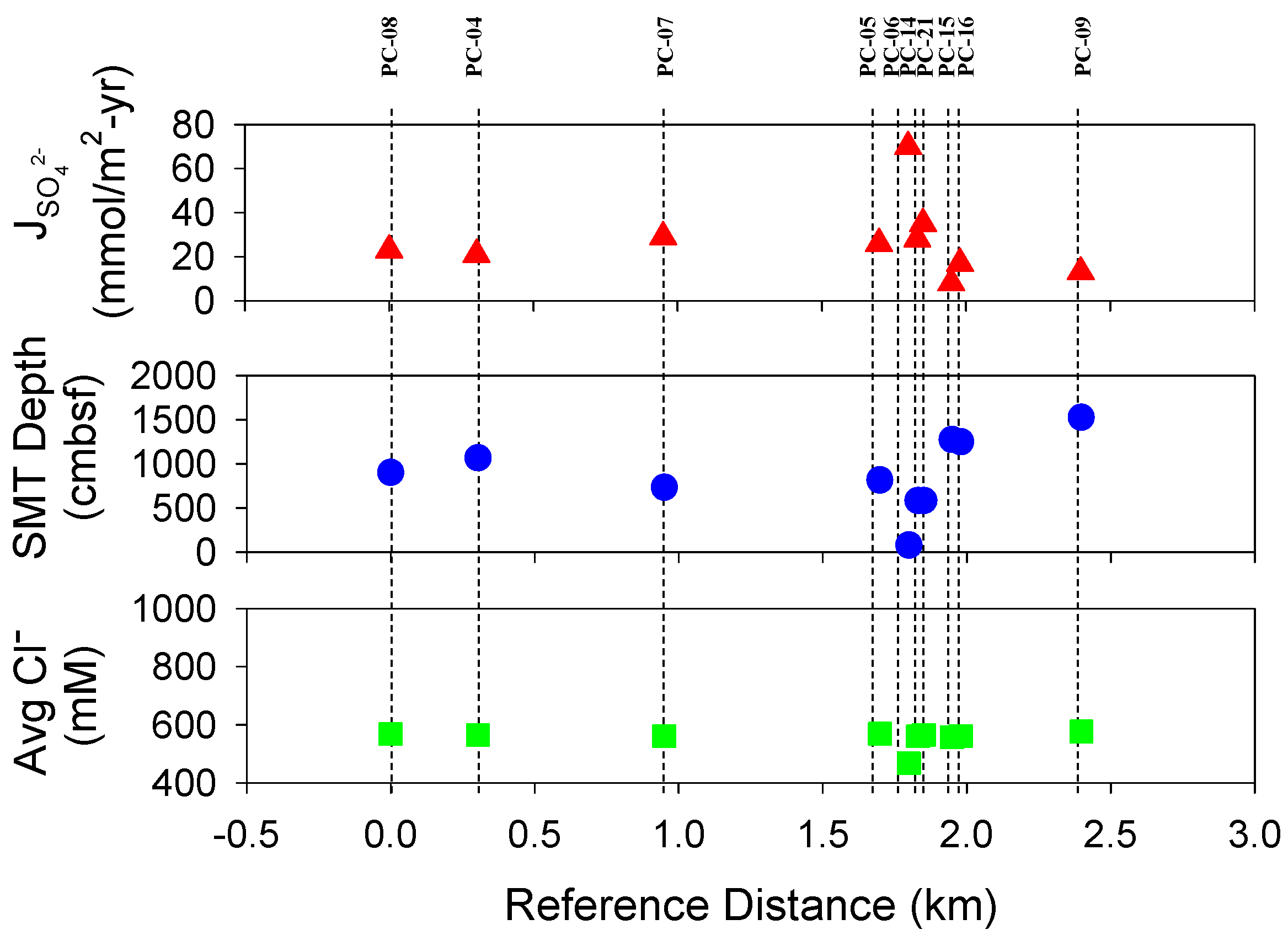
4.1. Estimated SMT Depths and Sulfate Diffusion Rates

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Core # | AC-07 Core # | Distance from Core #1 (AC-07 PC-08) | Core Length (cm) | SMT (cm) | (mmol/m2-yr) | Average Cl− (mM) | Comments |
|---|---|---|---|---|---|---|---|
| [1] | 08 | 0.00 | 759 | 901 | 23 | 568 | Non-linear sulfate profile; SMT deeper than max. core depth |
| [2] | 04 | 0.30 | 718 | 1069 | 21 | 565 | SMT deeper than max. core depth |
| [3] | 07 | 0.95 | 759 | 735 | 29 | 562 | SMT present |
| [4] | 05 | 1.70 | 756 | 816 | 26 | 570 | SMT deeper than max. core depth |
| [5] | 06 | 1.80 | 309 | 80 | 70 | 468 | Non-linear sulfate profile; Solid phase hydrates present, destabilized on recovery |
| [6] | 14 | 1.83 | 706 | 584 | 28 | 561 | Non-linear sulfate profile; SMT present |
| [7] | 21 | 1.85 | 730 | 585 | 35 | 566 | SMT present |
| [8] | 15 | 1.95 | 284 | 1278 | 8 | 558 | SMT deeper than max. core depth |
| [9] | 16 | 1.98 | 690 | 1252 | 17 | 561 | SMT deeper than max. core depth |
| [10] | 09 | 2.40 | 789 | 1528 | 13 | 577 | SMT deeper than max. core depth |

4.2. Diffusive Fluxes of Ca2+, Mg2+, and DIC and Carbon Mass Balance
| AC-07 Core # | (mmol/m2-yr) | JDIC-shallow (mmol/m2-yr) | (mmol/m2-yr) | (mmol/m2-yr) | JDIC-deep * (mmol/m2-yr) | JDIC-net (mmol/m2-yr) |
|---|---|---|---|---|---|---|
| 04 | 21 | −8 | 3 | 8 | – | −17 |
| 05 | 26 | −11 | 5 | 5 | – | −21 |
| 07 | 29 | −12 | 7 | 8 | – | −27 |
| 09 | 13 | −4 | 2 | 3 | – | −9 |
| 16 | 17 | −7 | 4 | 6 | – | −17 |
| 21 | 35 | −22 | 4 | 7 | – | −33 |

| AC-07 Core # | δ13C-DICMIN (‰) | δ13C-CH4MIN (‰) | %DICCH4 = %AOM Contribution to the DIC Pool | %DICcarbonate = %DIC to Carbonate |
|---|---|---|---|---|
| 04 | −45.8 | −65.7 | 68 | 52 |
| 05 | −50.7 | −94.6 | 54 | 38 |
| 07 | −47.1 | −105.5 | 45 | 52 |
| 09 | −27.5 | −62.5 | 44 | 38 |
| 16 | −39.3 | −62.9 | 62 | 59 |
| 21 | −45.5 | −93.9 | 49 | 31 |
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kvenvolden, K.A.; Lorenson, T.D. The global occurrence of natural gas hydrate. Am. Geophys. Union 2001, 124, 3–18. [Google Scholar]
- Borowski, W.S.; Paull, C.K.; Ussler, W., III. Global and local variations of interstitial sulfate gradients in deep-water, continental margin sediments: Sensitivity to underlying methane and gas hydrates. Mar. Geol. 1999, 159, 131–154. [Google Scholar] [CrossRef]
- Coffin, R.B.; Hamdan, L.; Plummer, R.; Smith, J.P.; Gardner, J.; Hagen, R.; Wood, W. Analysis of methane and sulfate flux in methane-charged sediments from the Mississippi Canyon, Gulf of Mexico. Mar. Pet. Geol. 2008, 25, 977–987. [Google Scholar] [CrossRef]
- Hutchinson, D.R.; Hart, P.E.; Ruppel, C.D.; Snyder, F.; Dugan, B. Seismic and thermal Characterization of a Bottom Simulating Reflection in the Northern Gulf of Mexico. In Natural Gas Hydrates: Energy Resources, Potential and Associated Geologic Hazards; Collett, T.S., Johnson, A., Knapp, C., Boswell, R., Eds.; AAPG Special Publication Memoir: Tulsa, OK, USA, 2009; Volume 89, pp. 266–286. [Google Scholar]
- Charlou, J.L.; Donval, J.P.; Fouquet, Y.; Ondreas, H.; Knoery, J.; Cochonat, P.; Levaché, D.; Poirier, Y.; Jean-Baptiste, P.; Fourré, E.; et al. Physical and chemical characterization of gas hydrates and associated methane plumes in the Congo-Angola basin. Chem. Geol. 2004, 205, 405–425. [Google Scholar]
- Sassen, R.; Roberts, H.H.; Carney, R.; Milkov, A.V.; Freitas, D.A.; Lanoil, B.; Zhang, C. Free hydrocarbon gas, gas hydrate, and authigenic minerals in chemosynthetic communities of northern Gulf of Mexico continental slope: Relation to microbial processes. Chem. Geol. 2004, 205, 195–217. [Google Scholar] [CrossRef]
- Paull, C.K.; Matsumoto, R.; Wallace, P.J.; Dillon, W.P. Proceedings of the Ocean Drilling Program: Initial Reports; Ocean Drilling Program: College Station, TX, USA, 1996; Volume 164.
- Xu, W.Y.; Ruppel, C. Predicting the occurrence, distribution, and evolution of methane gas hydrate in porous marine sediments. J. Geophys. Res. 1999, 104, 5081–5096. [Google Scholar] [CrossRef]
- Cooper, A.K.; Hart, P.E. High-resolution seismic-reflection investigation of the northern Gulf of Mexico gas hydrate stability zone. Mar. Pet. Geol. 2003, 19, 1275–1293. [Google Scholar] [CrossRef]
- Bohrmann, G.; Greinert, J.; Suess, E.; Torres, M. Authigenic carbonates from the Cascadia subduction zone and their relation to gas hydrate stability. Geology 1998, 26, 647–650. [Google Scholar] [CrossRef]
- Aloisi, G.; Pierre, C.; Rouchy, J.-M.; Foucher, J.-P.; Woodside, J. Methane-related authigenic carbonates of eastern Mediterranean Sea mud volcanoes and their possible relation to gas hydrate destabilisation. Earth Planet. Sci. Lett. 2000, 184, 321–338. [Google Scholar] [CrossRef]
- Naehr, T.H.; Rodriguez, N.M.; Bohrmann, G.; Paull, C.K.; Botz, R. Methane-derived authigenic carbonates associated with gas-hydrate decomposition and fluid venting above the blake ridge diapir. In Proceedings of the Ocean Drilling Program: Science Results; Paull, C.K., Matsumoto, R., Wallace, P.J., Dillon, W.P., Eds.; Ocean Drilling Program: College Station, TX, USA, 2000; Volume 164, pp. 285–295. [Google Scholar]
- Rodriguez, N.M.; Paull, C.K.; Borowski, W.S. Zonation of authigenic carbonates within gas hydrate-bearing sedimentary sections on the blake ridge: offshore southeastern North America. In Proceedings of the Ocean Drilling Program: Science Results; Paull, C.K., Matsumoto, R., Wallace, P.J., Dillon, W.P., Eds.; Ocean Drilling Program: College Station, TX, USA, 2000; Volume 164, pp. 301–312. [Google Scholar]
- Greinert, J.; Bohrmann, G.; Suess, E. Gas hydrate associated carbonates and methane-venting at hydrate ridge: classification, distribution and origin of authigenic lithologies. In Natural Gas Hydrates: Occurrence, Distribution, and Detection; Paull, C.K., Dillon, W.P., Eds.; AGU Geophysical Monograph, American Geophysical Union: Washington, DC, USA, 2001; Volume 124, pp. 99–113. [Google Scholar]
- Luff, R.; Wallmann, K. Fluid flow, methane fluxes, carbonate precipitation and biogeochemical turnover in gas hydrate-bearing sediments at hydrate ridge, Cascadia margin: Numerical modeling and mass balances. Geochim. Cosmochim. Acta 2003, 67, 3403–3421. [Google Scholar] [CrossRef]
- Moore, T.S.; Murray, R.W.; Kurtz, A.C.; Schrag, D.P. Anaerobic methane oxidation and the formation of dolomite. Earth Planet. Sci. Lett. 2004, 229, 141–154. [Google Scholar] [CrossRef]
- Formolo, M.J.; Lyons, T.W.; Zhang, C.; Kelley, C.; Sassen, R.; Horita, J.; Cole, D.R. Quantifying carbon sources in the formation of authigenic carbonates at gas hydrate sites in the Gulf of Mexico. Chem. Geol. 2004, 205, 253–264. [Google Scholar] [CrossRef]
- Mazzini, A.; Ivanov, M.K.; Parnell, J.; Stadnitskaia, A.; Cronin, B.T.; Poludetkinab, E.; Mazurenko, L.; van Weering, T.C.E. Methane-related authigenic carbonates from the Black Sea: Geochemical characterisation and relation to seeping fluids. Mar. Geol. 2004, 212, 153–181. [Google Scholar] [CrossRef]
- Berelson, W.M.; Prokopenko, M.; Sansone, F.J.; Graham, A.W.; McManus, J.; Bernhard, J.M. Anaerobic diagenesis of silica and carbon in continental margin sediments; discrete zones of TCO2 production. Geochim. Cosmochim. Acta 2005, 69, 4611–4629. [Google Scholar] [CrossRef]
- Snyder, G.T.; Hirutab, A.; Matsumoto, R.; Dickens, G.R.; Tomaru, H.; Takeuchi, R.; Komatsubara, J.; Ishida, Y.; Yua, H. Pore water profiles and authigenic mineralization in shallow marine sediments above the methane-charged system on Umitaka Spur, Japan Sea. Deep Sea Res. II 2007, 54, 1216–1239. [Google Scholar] [CrossRef]
- Ussler, W., III; Paull, C.K. Rates of anaerobic oxidation of methane and authigenic carbonate mineralization in methane-rich deep-sea sediments inferred from models and geochemical profiles. Earth Planet. Sci. Lett. 2008, 266, 271–287. [Google Scholar]
- Boetius, A.; Suess, E. Hydrate Ridge: A natural laboratory for the study of microbial life fueled by methane from near-surface gas hydrates. Chem. Geol. 2004, 205, 291–310. [Google Scholar] [CrossRef]
- Kastner, M.; Elderfield, H.; Martin, J.B.; Suess, E.; Kvenvolden, K.A.; Garrison, R.E. Diagenesis and interstitial-water chemistry at the peruvian continental margin-major constituents and strontium isotopes. In Proceedings of the Ocean Drilling Program: Science Results; Suess, E., von Huene, R., Emeis, K.-C., Bourgois, J., Castaneda, J.C., de Wever, P., Eglinton, G., Garrison, R., Greenberg, M., Paz, E.H., et al., Eds.; Ocean Drilling Program: College Station, TX, USA, 1990; Volume 112, pp. 413–440. [Google Scholar]
- Chatterjee, S.; Dickens, G.R.; Bhatnagar, G.; Chapman, W.G.; Dugan, B.; Snyder, G.T.; Hirasaki, G.J. Pore water sulfate, alkalinity, and carbon isotope profiles in shallow sediment above marine gas hydrate systems: A numerical modeling perspective. J. Geophys. Res. 2011. [Google Scholar] [CrossRef]
- Valentine, D.L. Biogeochemistry and microbial ecology of methane oxidation in anoxic environments: A review. Antonie Van Leeuwenhoek 2002, 81, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Bayon, G.; Pierre, C.; Henderson, G.M.; Etoubleau, J.; Voisset, M.; Canquil, E.; Fouquet, Y. Sr/Ca and Ma/Ca ratios in sediments from the Niger fan: Evidence for past episodes of gas hydrate dissociation. Geophys. Res. Abstr. 2005, 7, 08714:1–08714:2. [Google Scholar]
- Burton, E.A. Controls on marine carbonate cement mineralogy: Review and assessment. Chem. Geol. 1993, 105, 291–310. [Google Scholar] [CrossRef]
- Paull, C.K.; Ussler, W.; Lorenson, T.D.; Winters, W.; Dougherty, J.A. Geochemical constraints on the distribution of gas hydrates in the Gulf of Mexico. Geo-Mar. Lett. 2005, 25, 273–280. [Google Scholar]
- Dickens, G.R.; Snyder, G.T. Interpreting upward methane flux from marine pore water profiles. In Fire in the Ice; National Energy Technology Laboratory, U.S. Department of Energy: Washington, DC, USA, 2009; pp. 7–10. [Google Scholar]
- Joye, S.B.; Boetius, A.; Orcutt, B.N.; Montoya, J.P.; Schulz, H.N.; Erickson, M.J.; Lugo, S.K. The anaerobic oxidation of methane and sulfate reduction in sediments from Gulf of Mexico cold seeps. Chem. Geol. 2004, 205, 219–238. [Google Scholar] [CrossRef]
- Kvenvolden, K.A. Gas hydrates-geological perspective and global change. Rev. Geophys. 1993, 31, 173–187. [Google Scholar] [CrossRef]
- Sloan, E.D. Clathrate Hydrates of Natural Gasses, 2nd ed.; Marcel Dekker, Inc.: New York, NY, USA, 1998; p. 705. [Google Scholar]
- Trudgill, B.; Rowan, M.; Fiduk, J.; Weimer, P.; Gale, P.; Korn, B.; Phair, R.; Gafford, W.; Roberts, G.; Dobbs, S. The Perdido Fold Belt, northwestern Gulf of Mexico, Part 1: Structural geology, evolution, and regional implications. Am. Assoc. Pet. Geol. Bull. 1999, 83, 88–113. [Google Scholar]
- Boswell, R.; Shelander, D.; Lee, M.; Latham, T.; Collett, T.; Guerin, G.; Moridis, G.; Reagan, M.; Goldberg, D. Occurrence of gas hydrate in Oligocene Frio sand: Alaminos Canyon Block 818: Northern Gulf of Mexico. Mar. Pet. Geol. 2009, 26, 1499–1512. [Google Scholar] [CrossRef]
- Fiduk, J.; Weimer, P.; Trudgill, B.; Rowan, M.; Gale, P.; Phair, R.; Korn, B.; Roberts, G.; Gafford, W.; Lowe, R.; et al. The Perdido Fold Belt, northwestern Gulf of Mexico, Part 2: Seismic stratigraphy and petroleum systems. Am. Assoc. Pet. Geol. Bull. 1999, 83, 578–612. [Google Scholar]
- Meyer, D.; Zarra, L.; Rains, D.; Meltz, B.; Hall, T. Emergence of the lower Tertiary Wilcox trend in the deepwater Gulf of Mexico. World Oil 2005, 226, 72–77. [Google Scholar]
- Cordes, E.; Carney, S.; Hourdez, S.; Carney, R.; Brooks, J.; Fisher, C. Cold seeps of the deep Gulf of Mexico: Community structure and biogeographic comparisons to Atlantic equatorial belt seep communities. Deep Sea Res. 1 Oceanogr. Res. Pap. 2007, 54, 637–653. [Google Scholar] [CrossRef]
- Roberts, H.; Fisher, C.; Brooks, J.; Bernard, B.; Carney, R.; Cordes, E.; Shedd, W.; Hunt, J.; Joye, S.; MacDonald, I.; et al. Exploration of the deep gulf of mexico slope using dsv alvin: site selection and geologic character. In Proceedings of the 57th Annual Convention of the Gulf Coast Association of Geological Societies (GCAGS), Corpus Christi, TX, USA, 21–23 October 2007.
- Coffin, R.B.; Hamdan, L.J.; Smith, J.P.; Rose, K.; Downer, R.; Edsall, D.; Gardner, J.; Hagen, R.; Wood, W. Geochemical Evaluation of Deep Sediment Hydrate Deposits on Alaminos Canyon, Block 818, Texas-Louisiana Shelf; Cruise Report for UNOLS Ship R/V CAPE HATTERAS Cruise NRL AC-07, U.S. Naval Research Laboratory: Washington, DC, USA, 2008; p. 173. [Google Scholar]
- Hoehler, T.M.; Borowski, W.S.; Alperin, M.J.; Rodriguez, N.M.; Paull, C.K. Model stable isotope and radiocarbon characterization of anaerobic methane oxidation in gas hydrate-bearing sediments of the blake ridge. In Proceedings of the Ocean Drilling Program: Science Results; Paull, C.K., Matsumoto, R., Wallace, P.J., Dillon, W.P., Eds.; Ocean Drilling Program: College Station, TX, USA, 2000; Volume 164, pp. 79–85. [Google Scholar]
- Boehme, S.E.; Blair, N.E.; Chanton, J.P.; Martens, C.S. A mass balance of 13C and 12C in an organic-rich methane-producing marine sediment. Geochim. Cosmochim. Acta 1996, 60, 3835–3848. [Google Scholar] [CrossRef]
- Plummer, R.E.; Pohlman, J.W.; Coffin, R.B. Compound-specific stable carbon isotope analysis of low-concentration complex hydrocarbon mixtures from natural gas hydrate systems. In Proceedings of the EOS American Geophysical Union (AGU) Fall Meeting Supplement, San Francisco, CA, USA, 5–9 December 2005; Volume 86.
- Coffin, R.B.; Hamdan, L.; Smith, J.P.; Rose, P.S.; Plummer, R.E.; Yoza, B.A.; Pecher, I.; Montgomery, M.T. The contribution of vertical methane flux to shallow sediment carbon pools across the Porangahau Ridge, New Zealand. Energies 2014, 7, 5332–5356. [Google Scholar] [CrossRef]
- Borowski, W.S.; Paull, C.K.; Ussler, W., III. Marine porewater sulfate profiles indicate in situ methane flux from underlying gas hydrate. Geology 1996, 24, 655–658. [Google Scholar] [CrossRef]
- Berner, R.A. An idealized model of dissolved sulfate distribution in recent sediments. Geochim. Cosmochim. Acta 1964, 28, 1497–1503. [Google Scholar] [CrossRef]
- Berner, R.A. Sulfate reduction and the rate of deposition of marine sediments. Earth Planet. Sci. Lett. 1978, 37, 492–498. [Google Scholar] [CrossRef]
- Iversen, N.; Jørgensen, B.B. Diffusion coefficients of sulfate and methane in marine sediments: Influence of porosity. Geochim. Cosmochim. Acta 1992, 57, 571–578. [Google Scholar] [CrossRef]
- Kastner, M.; Torres, M.; Solomon, E.; Spivack, A.J. Marine pore fluid profiles of dissolved sulfate: Do they reflect in situ methane fluxes? In Fire in the Ice; National Energy Technology Laboratory, U.S. Department of Energy: Washington, DC, USA, 2008; pp. 6–8. [Google Scholar]
- Sivan, O.; Schrag, D.P.; Murray, R.W. Rates of methanogenesis and methanotrophy in deep-sea sediments. Geobiology 2007, 5, 141–151. [Google Scholar] [CrossRef]
- Atkins, P.W. Physical Chemistry; Oxford University Press: Oxford, UK, 1978; p. 1022. [Google Scholar]
- Nauhaus, K.; Albrecht, M.; Elvert, M.; Boetius, A.; Widdel, F. In vitro cell growth of marine archaeal-bacterial consortia during anaerobic oxidation of methane with sulfate. Environ. Microbiol. 2007, 9, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Cağatay, M.N.; Borowski, W.S.; Ternois, Y.G. Factors affecting the diagenesis of Quaternary sediments at ODP Leg 172 sites in western north Atlantic. Chem. Geol. 2001, 175, 467–484. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Smith, J.P.; Coffin, R.B. Methane Flux and Authigenic Carbonate in Shallow Sediments Overlying Methane Hydrate Bearing Strata in Alaminos Canyon, Gulf of Mexico. Energies 2014, 7, 6118-6141. https://doi.org/10.3390/en7096118
Smith JP, Coffin RB. Methane Flux and Authigenic Carbonate in Shallow Sediments Overlying Methane Hydrate Bearing Strata in Alaminos Canyon, Gulf of Mexico. Energies. 2014; 7(9):6118-6141. https://doi.org/10.3390/en7096118
Chicago/Turabian StyleSmith, Joseph P., and Richard B. Coffin. 2014. "Methane Flux and Authigenic Carbonate in Shallow Sediments Overlying Methane Hydrate Bearing Strata in Alaminos Canyon, Gulf of Mexico" Energies 7, no. 9: 6118-6141. https://doi.org/10.3390/en7096118