The “Connection” Between HIV Drug Resistance and RNase H


Abstract
:
1. Introduction
2. Reverse Transcriptase Inhibitors and Drug Resistance Mutations in the pol Domain
3. Mechanisms of NRTI Resistance Associated with the pol Domain

4. Mechanisms of NRTI Resistance Associated with the cn and rh Domains
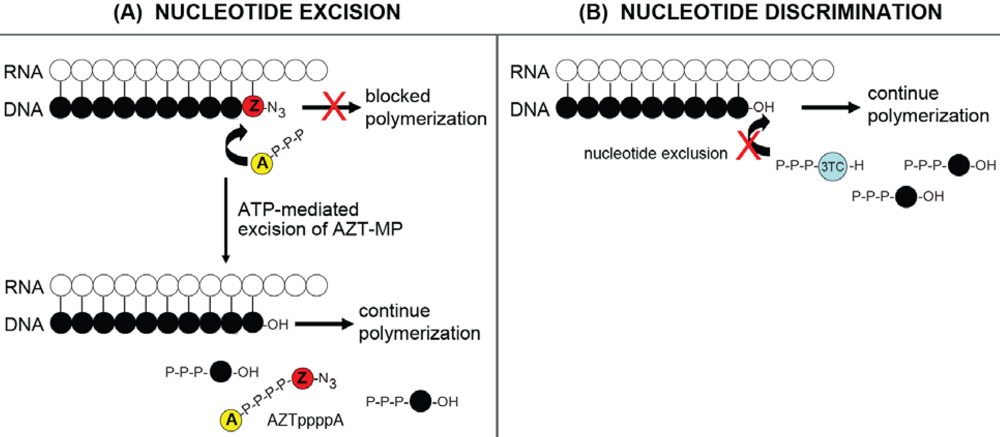
4.1. RNase H-dependent Mechanism for NRTI resistance: Balance between Nucleotide Excision and RNase H Activity


4.2. RNase H-independent Mechanisms of NRTI Resistance
5. Mechanisms of NNRTI Resistance Associated with the pol Domain
6. Mechanisms of NNRTI Resistance Associated with the cn and rh Domains
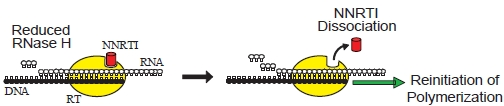
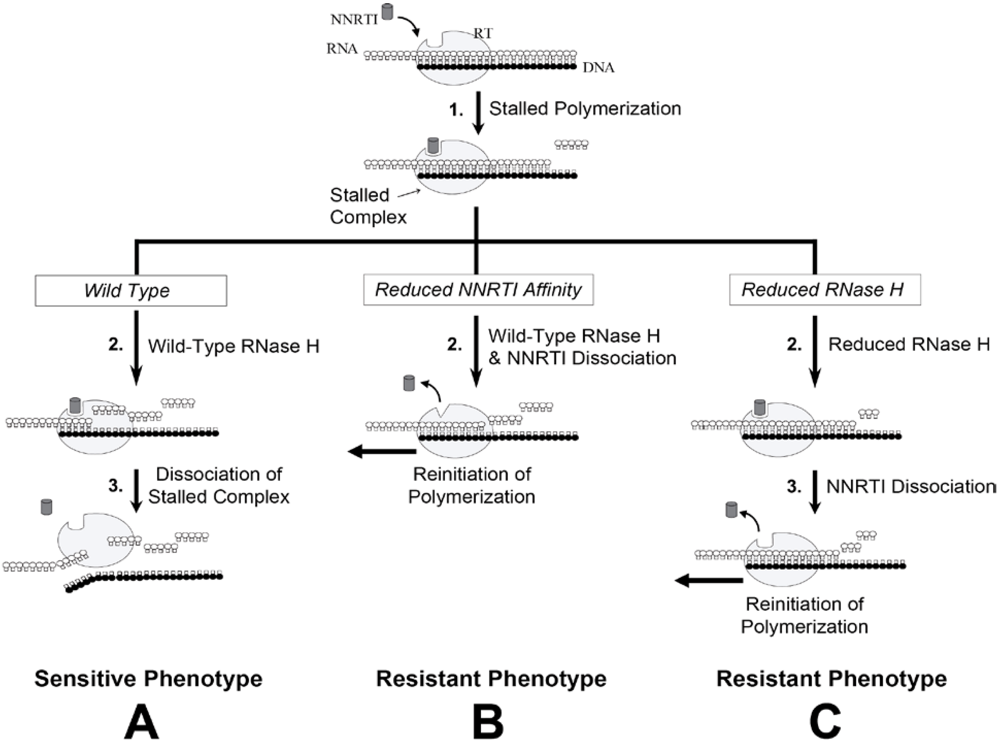
6.1. RNase H-Dependent Mechanism of NNRTI Resistance


6.2. RNase H-independent Mechanisms of NNRTI Resistance Associated with C-terminal Domain Mutations
7. Prevalence of C-terminal Mutations in Treatment-naïve and Treatment-experienced Patients

7.1. Prevalence of cn Mutations in Patient Databases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C-terminal domain mutation | No. of sequences containing a C-terminal domain mutation a | No. of sequences containing a C-terminal domain mutation and no RTI b mutations | No. of sequences containing both a C-terminal domain mutation and ≥1 RTI mutation | Probability of having a C-terminal domain mutation with ≥1 RTI mutation c |
|---|---|---|---|---|
| Connection Subdomain | ||||
| E312Q | 79/6035 (1.3%) | 36/3397 (1.1%) | 43/2638 (1.6%) | *P = 0.0350 |
| Y318F | 48/5983 (0.8%) | 0/3366 (0%) | 48/2617 (1.8%) | *P < 0.000001 |
| G333D | 47/5086 (0.9%) | 21/2864 (0.7%) | 26/2222 (1.2%) | P =0.0717 |
| G333E | 446/5086 (8.8%) | 221/2864 (7.7%) | 225/2222 (10.1%) | *P = 0.0016 |
| G335C | 30/4905 (0.9%) | 14/2711 (0.5%) | 16/2194 (0.7%) | P = 0.2211 |
| G335D | 79/4905 (1.6%) | 37/2711 (1.4%) | 42/2194 (1.9%) | P = 0.0802 |
| N348I | 180/3189 (5.6%) | 5/1213 (0.4%) | 175/1976 (8.9%) | *P < 0.000001 |
| A360V | 128/3147 (4.1%) | 17/1203 (1.4%) | 111/1944 (5.7%) | *P < 0.000001 |
| V365I | 169/3140 (5.4%) | 36/1202 (3.0%) | 133/1938 (6.9%) | *P = 0.000001 |
| T369I | 19/3115 (0.6%) | 1/1195 (0.08%) | 18/1920 (0.9%) | *P = 0.0013 |
| A371V | 518/3112 (16.6%) | 47/1194 (3.9%) | 471/1918 (24.6%) | *P < 0.000001 |
| A376S | 320/3111 (10.3%) | 87/1194 (7.2%) | 233/1917 (12.2%) | *P = 0.000006 |
| E399D | 475/2968 (16%) | 178/1072 (16.6%) | 297/1896 (15.7%) | P = 0.7656 |
| A400T | 455/1616 (28.2%) | 205/628 (32.6%) | 250/988 (25.3%) | P = 1.00 |
| RNase H Domain | ||||
| Q509L | 2/507 (0.4%) | 2/304 (0.7%) | 0/203 (0%) | P = 1.00 |
| a Data from the Stanford HIV Drug Resistance Database, Detailed RT Mutation Profile Program, as of April 2010. | ||||
| b Major and minor RTI mutations were those defined by the Stanford database. Major NRTI mutations (http://hivdb. stanford.edu/pages/documentPage/NRTI_mutationClassification.html) included 41L, 65R/N, 67N, deletion D67, insertion at T69, 69D, 70R/E/G, 74I/V, 75T/A/M, 115F, 151M/L, 184V/I, 210W, and 215Y/F. Minor NRTI mutations included 41 not L, 44D/A, 62V, 67 not deletion or N, 69 not insertion or D, 70 not R/E/G, 74 not I/V, 75 not T/A/M, 77L, 115 not F, 116Y, 118I, 151 not M/L, 184 not V/I, 210 not W, 215 not Y/F, 219 Q/E/N/R/W, 333D/E, and 348I. Major NNRTI mutations (http://hivdb.stanford.edu/pages/documentPage/NNRTI_mutation Classification.html) included 100I, 101E/P, 103N/S/T/H, 106A/M, 179F, 181C/I/V, 188C/H/L, 190A/S/E/Q/T/C/V, 230L, and 236L. Minor NNRTI mutations included 90I, 98G, 100 not I, 101 Q/H/N, 103 not N/S/T/H, 106 not A/M/I/L, 108I, 138K, 179D/E, 181 not C/I/V, 188 not C/H/L, 190 not A/S/E/Q/T/C/V, 225H, 227C/L, 234I, 236 not L, 238N/T, 318F and 348I. The word “not” refers to all mutations at that position except the following mutation(s). | ||||
| c Two proportions statistics were performed by comparing the number of C-terminal domain mutations with at least one RTI mutation (for example, 43 for E312Q) to the total number of sequences containing at least one RTI mutation (2638 of 6035), against the number of C-terminal domain mutations without an RTI mutation (for example, 36 for E312Q) to the total number of sequences without an RTI mutation (3397 of 6035). | ||||
7.2. Prevalence of rh Mutations in Patient Databases
7.3. Role of C-terminal Domain Mutations in Clinical Outcome
7.4. Selection of C-terminal Domain Mutations in HIV-1 Infected Patients
8. Conclusions
Acknowledgments
References
- Quagliarello, V. The Acquired Immunodeficiency Syndrome: current status. Yale J. Biol. Med. 1982, 55, 443–452. [Google Scholar] [PubMed]
- Gottlieb, M.S.; Schroff, R.; Schanker, H.M.; Weisman, J.D.; Fan, P.T.; Wolf, R.A.; Saxon, A. Pneumocystis carinii pneumonia and mucosal candidiasis in previously healthy homosexual men: evidence of a new acquired cellular immunodeficiency. N. Engl. J. Med. 1981, 305, 1425–1431. [Google Scholar] [CrossRef] [PubMed]
- Friedman-Kien, A.; Laubenstein, L.; Marmor, M.; Hymes, K.; Green, J.; Ragaz, A.; Gottlieb, J.; Muggia, F.; Demopoulos, R.; Weintraub, M.; Williams, D. Kaposi's sarcoma and Pneumocystis pneumonia among homosexual men--New York City and California. MMWR Morb. Mortal. Wkly. Rep. 1981, 30, 305–308. [Google Scholar] [PubMed]
- Mitsuya, H.; Weinhold, K.J.; Furman, P.A.; St Clair, M.H.; Lehrman, S.N.; Gallo, R.C.; Bolognesi, D.; Barry, D.W.; Broder, S. 3'-Azido-3'-deoxythymidine (BW A509U): an antiviral agent that inhibits the infectivity and cytopathic effect of human T-lymphotropic virus type III/lymphadenopathy-associated virus in vitro. Proc. Natl. Acad. Sci. U. S. A. 1985, 82, 7096–7100. [Google Scholar] [CrossRef] [PubMed]
- di Marzo Veronese, F.; Copeland, T.D.; DeVico, A.L.; Rahman, R.; Oroszlan, S.; Gallo, R.C.; Sarngadharan, M.G. Characterization of highly immunogenic p66/p51 as the reverse transcriptase of HTLV-III/LAV. Science 1986, 231, 1289–1291. [Google Scholar] [PubMed]
- Lowe, D.M.; Aitken, A.; Bradley, C.; Darby, G.K.; Larder, B.A.; Powell, K.L.; Purifoy, D.J.; Tisdale, M.; Stammers, D.K. HIV-1 reverse transcriptase: crystallization and analysis of domain structure by limited proteolysis. Biochemistry 1988, 27, 8884–8889. [Google Scholar] [CrossRef] [PubMed]
- Telesnitsky, A.; Goff, S.P. Reverse Transcriptase and the Generation of Retroviral DNA. In Retroviruses; Coffin J.M.;, Hughes, S.H.; Varmus, H., Eds.; Cold Spring Harbor Laboratory Press: Plainview, NY, USA, 1997. [Google Scholar]
- Hirsch, M.S.; Gunthard, H.F.; Schapiro, J.M.; Brun-Vezinet, F.; Clotet, B.; Hammer, S.M.; Johnson, V.A.; Kuritzkes, D.R.; Mellors, J.W.; Pillay, D.; Yeni, P.G.; Jacobsen, D.M.; Richman, D.D. Antiretroviral drug resistance testing in adult HIV-1 infection: 2008 recommendations of an International AIDS Society-USA panel. Clin. Infect. Dis. 2008, 47, 266–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, V.A.; Brun-Vezinet, F.; Clotet, B.; Gunthard, H.F.; Kuritzkes, D.R.; Pillay, D.; Schapiro, J.M.; Richman, D.D. Update of the Drug Resistance Mutations in HIV-1. Top. HIV Med. 2008, 16, 138–145. [Google Scholar] [PubMed]
- Goody, R.S.; Muller, B.; Restle, T. Factors contributing to the inhibition of HIV reverse transcriptase by chain-terminating nucleotides in vitro and in vivo. FEBS Lett. 1991, 291, 1–5. [Google Scholar] [CrossRef]
- Furman, P.A.; Fyfe, J.A.; St Clair, M.H.; Weinhold, K.; Rideout, J.L.; Freeman, G.A.; Lehrman, S.N.; Bolognesi, D.P.; Broder, S.; Mitsuya, H.; et al. Phosphorylation of 3'-azido-3'-deoxythymidine and selective interaction of the 5'-triphosphate with human immunodeficiency virus reverse transcriptase . Proc. Natl. Acad. Sci. U. S. A. 1986, 83, 8333–8337. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. The role of non-nucleoside reverse transcriptase inhibitors (NNRTIs) in the therapy of HIV-1 infection. Antiviral Res. 1998, 38, 153–179. [Google Scholar] [CrossRef] [PubMed]
- Johnson, V.A.; Brun-Vezinet, F.; Clotet, B.; Gunthard, H.F.; Kuritzkes, D.R.; Pillay, D.; Schapiro, J.M.; Richman, D.D. Update of the drug resistance mutations in HIV-1: December 2009. Top. HIV Med. 2009, 17, 138–145. [Google Scholar] [PubMed]
- Hanna, G.J.; Johnson, V.A.; Kuritzkes, D.R.; Richman, D.D.; Brown, A.J.; Savara, A.V.; Hazelwood, J.D.; D'Aquila, R.T. Patterns of resistance mutations selected by treatment of human immunodeficiency virus type 1 infection with zidovudine, didanosine, and nevirapine. J. Infect. Dis. 2000, 181, 904–911. [Google Scholar] [CrossRef] [PubMed]
- Marcelin, A.G.; Delaugerre, C.; Wirden, M.; Viegas, P.; Simon, A.; Katlama, C.; Calvez, V. Thymidine analogue reverse transcriptase inhibitors resistance mutations profiles and association to other nucleoside reverse transcriptase inhibitors resistance mutations observed in the context of virological failure. J. Med. Virol. 2004, 72, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Yahi, N.; Tamalet, C.; Tourres, C.; Tivoli, N.; Ariasi, F.; Volot, F.; Gastaut, J.A.; Gallais, H.; Moreau, J.; Fantini, J. Mutation patterns of the reverse transcriptase and protease genes in human immunodeficiency virus type 1-infected patients undergoing combination therapy: survey of 787 sequences. J. Clin. Microbiol. 1999, 37, 4099–4106. [Google Scholar] [PubMed]
- Perno, C.F.; Svicher, V.; Ceccherini-Silberstein, F. Novel drug resistance mutations in HIV: recognition and clinical relevance. AIDS Rev. 2006, 8, 179–190. [Google Scholar] [PubMed]
- De Corte, B.L. From 4,5,6,7-tetrahydro-5-methylimidazo[4,5,1-jk](1,4)benzodiazepin-2(1H)-one (TIBO) to etravirine (TMC125): fifteen years of research on non-nucleoside inhibitors of HIV-1 reverse transcriptase. J. Med. Chem. 2005, 48, 1689–1696. [Google Scholar] [CrossRef] [PubMed]
- Boyer, P.L.; Sarafianos, S.G.; Arnold, E.; Hughes, S.H. Selective excision of AZTMP by drug-resistant human immunodeficiency virus reverse transcriptase. J. Virol. 2001, 75, 4832–4842. [Google Scholar] [CrossRef] [PubMed]
- Meyer, P.R.; Matsuura, S.E.; Mian, A.M.; So, A.G.; Scott, W.A. A mechanism of AZT resistance: an increase in nucleotide-dependent primer unblocking by mutant HIV-1 reverse transcriptase. Mol. Cell 1999, 4, 35–43. [Google Scholar] [CrossRef]
- Meyer, P.R.; Matsuura, S.E.; So, A.G.; Scott, W.A. Unblocking of chain-terminated primer by HIV-1 reverse transcriptase through a nucleotide-dependent mechanism. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 13471–13476. [Google Scholar] [CrossRef] [PubMed]
- Arion, D.; Kaushik, N.; McCormick, S.; Borkow, G.; Parniak, M.A. Phenotypic mechanism of HIV-1 resistance to 3'-azido-3'-deoxythymidine (AZT): increased polymerization processivity and enhanced sensitivity to pyrophosphate of the mutant viral reverse transcriptase. Biochemistry 1998, 37, 15908–15917. [Google Scholar] [CrossRef] [PubMed]
- Naeger, L.K.; Margot, N.A.; Miller, M.D. ATP-dependent removal of nucleoside reverse transcriptase inhibitors by human immunodeficiency virus type 1 reverse transcriptase. Antimicrob. Agents Chemother. 2002, 46, 2179–2184. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.J.; Meyer, P.R.; Asthana, D.; Ashman, M.R.; Scott, W.A. Intracellular substrates for the primer-unblocking reaction by human immunodeficiency virus type 1 reverse transcriptase: detection and quantitation in extracts from quiescent- and activated-lymphocyte subpopulations. Antimicrob. Agents Chemother. 2005, 49, 1761–1769. [Google Scholar] [CrossRef] [PubMed]
- Meyer, P.R.; Matsuura, S.E.; Tolun, A.A.; Pfeifer, I.; So, A.G.; Mellors, J.W.; Scott, W.A. Effects of specific zidovudine resistance mutations and substrate structure on nucleotide-dependent primer unblocking by human immunodeficiency virus type 1 reverse transcriptase. Antimicrob. Agents Chemother. 2002, 46, 1540–1545. [Google Scholar] [CrossRef] [PubMed]
- Sarafianos, S.G.; Das, K.; Clark, A.D.; Ding, J.; Boyer, P.L.; Hughes, S.H.; Arnold, E. Lamivudine (3TC) resistance in HIV-1 reverse transcriptase involves steric hindrance with beta-branched amino acids . Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 10027–10032. [Google Scholar] [CrossRef] [PubMed]
- Krebs, R.; Immendorfer, U.; Thrall, S.H.; Wohrl, B.M.; Goody, R.S. Single-step kinetics of HIV-1 reverse transcriptase mutants responsible for virus resistance to nucleoside inhibitors zidovudine and 3-TC. Biochemistry 1997, 36, 10292–10300. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.Y.; Anderson, K.S. Mechanistic studies examining the efficiency and fidelity of DNA synthesis by the 3TC-resistant mutant (184V) of HIV-1 reverse transcriptase. Biochemistry 1999, 38, 9440–9448. [Google Scholar] [CrossRef] [PubMed]
- Deval, J.; Navarro, J.M.; Selmi, B.; Courcambeck, J.; Boretto, J.; Halfon, P.; Garrido-Urbani, S.; Sire, J.; Canard, B. A loss of viral replicative capacity correlates with altered DNA polymerization kinetics by the human immunodeficiency virus reverse transcriptase bearing the K65R and L74V dideoxynucleoside resistance substitutions. J. Biol. Chem. 2004, 279, 25489–25496. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Hoyos, A.J.; Scott, W.A. The Role of Nucleotide Excision by Reverse Transcriptase in HIV Drug Resistance . Viruses 2010, 2, 372–394. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Marchand, B.; Kirby, K.A.; Michailidis, E.; Sarafianos, S.G. Structural Aspects of Drug Resistance and Inhibition of HIV-1 Reverse Transcriptase. Viruses 2010, 2, 606–638. [Google Scholar] [CrossRef] [PubMed]
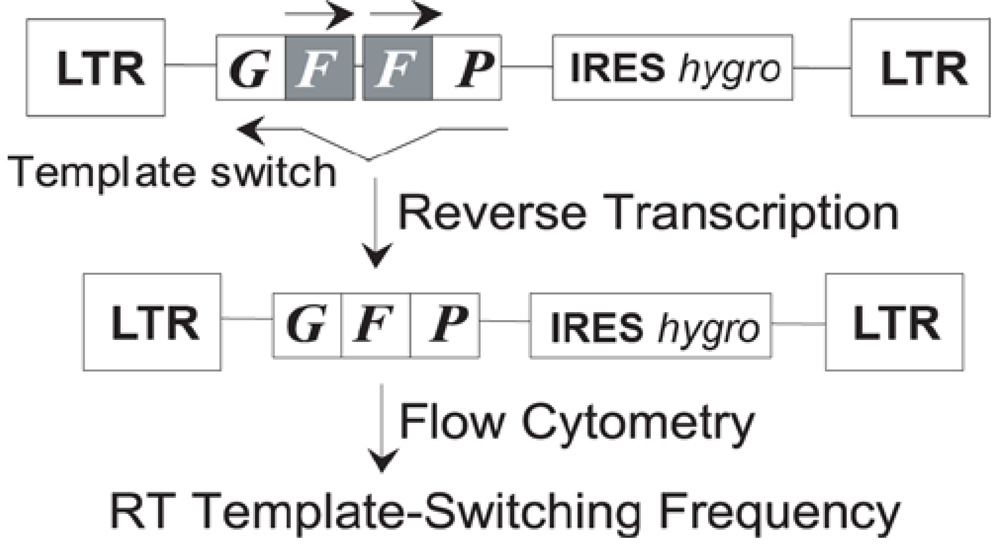
- Nikolenko, G.N.; Svarovskaia, E.S.; Delviks, K.A.; Pathak, V.K. Antiretroviral drug resistance mutations in human immunodeficiency virus type 1 reverse transcriptase increase template-switching frequency. J. Virol. 2004, 78, 8761–8770. [Google Scholar] [CrossRef] [PubMed]
- Nikolenko, G.N.; Delviks-Frankenberry, K.A.; Palmer, S.; Maldarelli, F.; Fivash, M.J.; Coffin, J.M.; Pathak, V.K. Mutations in the connection domain of HIV-1 reverse transcriptase increase 3'-azido-3'-deoxythymidine resistance . Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Delviks-Frankenberry, K.A.; Nikolenko, G.N.; Boyer, P.L.; Hughes, S.H.; Coffin, J.M.; Jere, A.; Pathak, V.K. HIV-1 reverse transcriptase connection subdomain mutations reduce template RNA degradation and enhance AZT excision. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 10943–10948. [Google Scholar] [CrossRef] [PubMed]
- Nikolenko, G.N.; Palmer, S.; Maldarelli, F.; Mellors, J.W.; Coffin, J.M.; Pathak, V.K. Mechanism for nucleoside analog-mediated abrogation of HIV-1 replication: balance between RNase H activity and nucleotide excision. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 2093–2098. [Google Scholar] [CrossRef] [PubMed]
- Tisdale, M.; Schulze, T.; Larder, B.A.; Moelling, K. Mutations within the RNase H domain of human immunodeficiency virus type 1 reverse transcriptase abolish virus infectivity . J. Gen. Virol. 1991, 72 (Pt 1), 59–66. [Google Scholar] [CrossRef] [PubMed]
- Volkmann, S.; Wohrl, B.M.; Tisdale, M.; Moelling, K. Enzymatic analysis of two HIV-1 reverse transcriptase mutants with mutations in carboxyl-terminal amino acid residues conserved among retroviral ribonucleases H. J. Biol. Chem. 1993, 268, 2674–2683. [Google Scholar] [PubMed]
- Wohrl, B.M.; Volkmann, S.; Moelling, K. Mutations of a conserved residue within HIV-1 ribonuclease H affect its exo- and endonuclease activities. J. Mol. Biol. 1991, 220, 801–818. [Google Scholar] [CrossRef] [PubMed]
- Brehm, J.H.; Koontz, D.; Meteer, J.D.; Pathak, V.; Sluis-Cremer, N.; Mellors, J.W. Selection of mutations in the connection and RNase H domains of human immunodeficiency virus type 1 reverse transcriptase that increase resistance to 3'-azido-3'-dideoxythymidine. J. Virol. 2007, 81, 7852–7859. [Google Scholar] [CrossRef] [PubMed]
- Arts, E.J.; Le Grice, S.F. Interaction of retroviral reverse transcriptase with template-primer duplexes during replication. Prog. Nucleic Acid Res. Mol. Biol. 1998, 58, 339–393. [Google Scholar] [PubMed]
- Ding, J.; Das, K.; Hsiou, Y.; Sarafianos, S.G.; Clark, A.D.; Jacobo-Molina, A.; Tantillo, C.; Hughes, S.H.; Arnold, E. Structure and functional implications of the polymerase active site region in a complex of HIV-1 RT with a double-stranded DNA template-primer and an antibody Fab fragment at 2.8 A resolution . J. Mol. Biol. 1998, 284, 1095–1111. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Hughes, S.H.; Arnold, E. Protein-nucleic acid interactions and DNA conformation in a complex of human immunodeficiency virus type 1 reverse transcriptase with a double-stranded DNA template-primer. Biopolymers 1997, 44, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Chopra, R.; Verdine, G.L.; Harrison, S.C. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science 1998, 282, 1669–1675. [Google Scholar] [CrossRef] [PubMed]
- Jacobo-Molina, A.; Ding, J.; Nanni, R.G.; Clark, A.D.; Lu, X.; Tantillo, C.; Williams, R.L.; Kamer, G.; Ferris, A.L.; Clark, P.; et al. Crystal structure of human immunodeficiency virus type 1 reverse transcriptase complexed with double-stranded DNA at 3.0 A resolution shows bent DNA . Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 6320–6324. [Google Scholar] [CrossRef] [PubMed]
- Sarafianos, S.G.; Das, K.; Tantillo, C.; Clark, A.D.; Ding, J.; Whitcomb, J.M.; Boyer, P.L.; Hughes, S.H.; Arnold, E. Crystal structure of HIV-1 reverse transcriptase in complex with a polypurine tract RNA:DNA . EMBO J. 2001, 20, 1449–1461. [Google Scholar] [CrossRef] [PubMed]
- Arion, D.; Sluis-Cremer, N.; Min, K.L.; Abram, M.E.; Fletcher, R.S.; Parniak, M.A. Mutational analysis of Tyr-501 of HIV-1 reverse transcriptase. Effects on ribonuclease H activity and inhibition of this activity by N-acylhydrazones. J. Biol. Chem. 2002, 277, 1370–1374. [Google Scholar] [CrossRef] [PubMed]
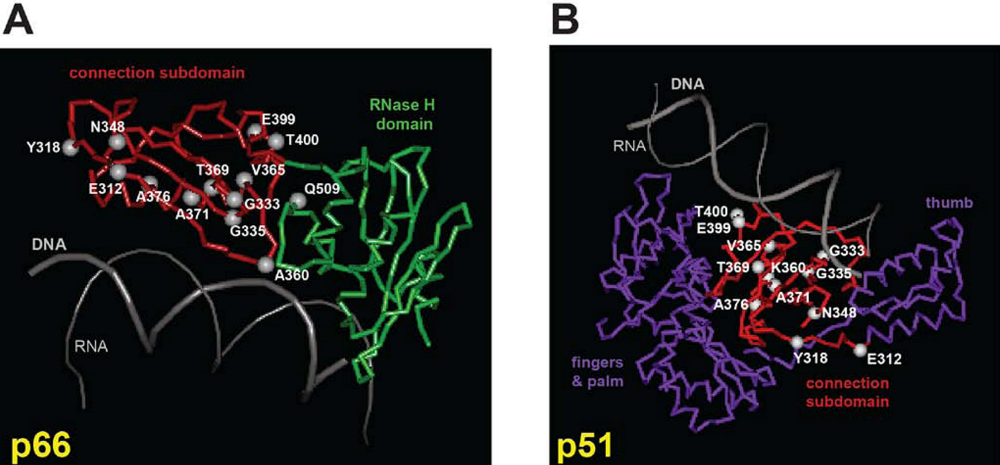
- Julias, J.G.; McWilliams, M.J.; Sarafianos, S.G.; Alvord, W.G.; Arnold, E.; Hughes, S.H. Mutation of amino acids in the connection domain of human immunodeficiency virus type 1 reverse transcriptase that contact the template-primer affects RNase H activity. J. Virol. 2003, 77, 8548–8554. [Google Scholar] [CrossRef] [PubMed]
- Julias, J.G.; McWilliams, M.J.; Sarafianos, S.G.; Arnold, E.; Hughes, S.H. Mutations in the RNase H domain of HIV-1 reverse transcriptase affect the initiation of DNA synthesis and the specificity of RNase H cleavage in vivo. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 9515–9520. [Google Scholar] [CrossRef] [PubMed]
- McWilliams, M.J.; Julias, J.G.; Sarafianos, S.G.; Alvord, W.G.; Arnold, E.; Hughes, S.H. Combining mutations in HIV-1 reverse transcriptase with mutations in the HIV-1 polypurine tract affects RNase H cleavages involved in PPT utilization. Virology 2006, 348, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Rausch, J.W.; Lener, D.; Miller, J.T.; Julias, J.G.; Hughes, S.H.; Le Grice, S.F. Altering the RNase H primer grip of human immunodeficiency virus reverse transcriptase modifies cleavage specificity. Biochemistry 2002, 41, 4856–4865. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.H.; Svarovskaia, E.S.; Barr, R.; Pathak, V.K. Y586F mutation in murine leukemia virus reverse transcriptase decreases fidelity of DNA synthesis in regions associated with adenine-thymine tracts. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 10090–10095. [Google Scholar] [CrossRef] [PubMed]
- Delviks-Frankenberry, K.A.; Nikolenko, G.N.; Barr, R.; Pathak, V.K. Mutations in human immunodeficiency virus type 1 RNase H primer grip enhance 3'-azido-3'-deoxythymidine resistance. J. Virol. 2007, 81, 6837–6845. [Google Scholar] [CrossRef] [PubMed]
- Nikolenko, G.N.; Delviks-Frankenberry, K.A.; Boyer, P.L.; Hughes, S.H.; Coffin, J.M.; Jere, A.; Pathak, V.K. HIV-1 reverse transcriptase connection domain mutations reduce template RNA degradation and enhance NRTI excision . Antivir. Ther. 2008, 13 (Suppl. 3), A60. [Google Scholar] [PubMed]
- Yap, S.H.; Sheen, C.W.; Fahey, J.; Zanin, M.; Tyssen, D.; Lima, V.D.; Wynhoven, B.; Kuiper, M.; Sluis-Cremer, N.; Harrigan, P.R.; Tachedjian, G. N348I in the connection domain of HIV-1 reverse transcriptase confers zidovudine and nevirapine resistance . PLoS Med. 2007, 4, e335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radzio, J.; Yap, S.H.; Tachedjian, G.; Sluis-Cremer, N. N348I in reverse transcriptase provides a genetic pathway for HIV-1 to select thymidine analogue mutations and mutations antagonistic to thymidine analogue mutations. AIDS 2010, 24, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Zelina, S.; Sheen, C.W.; Radzio, J.; Mellors, J.W.; Sluis-Cremer, N. Mechanisms by which the G333D mutation in human immunodeficiency virus type 1 Reverse transcriptase facilitates dual resistance to zidovudine and lamivudine. Antimicrob. Agents Chemother. 2008, 52, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Brehm, J.H.; Mellors, J.W.; Sluis-Cremer, N. Mechanism by which a glutamine to leucine substitution at residue 509 in the ribonuclease H domain of HIV-1 reverse transcriptase confers zidovudine resistance. Biochemistry 2008, 47, 14020–14027. [Google Scholar] [CrossRef] [PubMed]
- Ehteshami, M.; Beilhartz, G.L.; Scarth, B.J.; Tchesnokov, E.P.; McCormick, S.; Wynhoven, B.; Harrigan, P.R.; Gotte, M. Connection domain mutations N348I and A360V in HIV-1 reverse transcriptase enhance resistance to 3'-azido-3'-deoxythymidine through both RNase H-dependent and -independent mechanisms. J. Biol. Chem. 2008, 283, 22222–22232. [Google Scholar] [CrossRef] [PubMed]
- Boyer, P.L.; Sarafianos, S.G.; Arnold, E.; Hughes, S.H. The M184V mutation reduces the selective excision of zidovudine 5'-monophosphate (AZTMP) by the reverse transcriptase of human immunodeficiency virus type 1. J. Virol. 2002, 76, 3248–3256. [Google Scholar] [CrossRef] [PubMed]
- Gotte, M.; Arion, D.; Parniak, M.A.; Wainberg, M.A. The M184V mutation in the reverse transcriptase of human immunodeficiency virus type 1 impairs rescue of chain-terminated DNA synthesis. J. Virol. 2000, 74, 3579–3585. [Google Scholar] [CrossRef] [PubMed]
- Naeger, L.K.; Margot, N.A.; Miller, M.D. Increased drug susceptibility of HIV-1 reverse transcriptase mutants containing M184V and zidovudine-associated mutations: analysis of enzyme processivity, chain-terminator removal and viral replication. Antivir. Ther. 2001, 6, 115–126. [Google Scholar] [PubMed]
- Miranda, L.R.; Gotte, M.; Liang, F.; Kuritzkes, D.R. The L74V mutation in human immunodeficiency virus type 1 reverse transcriptase counteracts enhanced excision of zidovudine monophosphate associated with thymidine analog resistance mutations. Antimicrob. Agents Chemother. 2005, 49, 2648–2656. [Google Scholar] [CrossRef] [PubMed]
- von Wyl, V.; Ehteshami, M.; Symons, J.; Burgisser, P.; Nijhuis, M.; Demeter, L.M.; Yerly, S.; Boni, J.; Klimkait, T.; Schuurman, R.; Ledergerber, B.; Gotte, M.; Gunthard, H.F. Epidemiological and biological evidence for a compensatory effect of connection domain mutation N348I on M184V in HIV-1 reverse transcriptase. J. Infect. Dis. 2010, 201, 1054–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delviks-Frankenberry, K.A.; Nikolenko, G.N.; Maldarelli, F.; Hase, S.; Takebe, Y.; Pathak, V.K. Subtype-Specific Differences in the Human Immunodeficiency Virus Type 1 Reverse Transcriptase Connection Subdomain of CRF01_AE Are Associated with Higher Levels of Resistance to 3'-Azido-3'-Deoxythymidine. J. Virol. 2009, 83, 8502–8513. [Google Scholar] [CrossRef] [PubMed]
- Kohlstaedt, L.A.; Wang, J.; Friedman, J.M.; Rice, P.A.; Steitz, T.A. Crystal structure at 3.5 A resolution of HIV-1 reverse transcriptase complexed with an inhibitor . Science 1992, 256, 1783–1790. [Google Scholar] [PubMed]
- Ren, J.; Esnouf, R.; Garman, E.; Somers, D.; Ross, C.; Kirby, I.; Keeling, J.; Darby, G.; Jones, Y.; Stuart, D.; et al. High resolution structures of HIV-1 RT from four RT-inhibitor complexes . Nat. Struct. Biol. 1995, 2, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Smerdon, S.J.; Jager, J.; Wang, J.; Kohlstaedt, L.A.; Chirino, A.J.; Friedman, J.M.; Rice, P.A.; Steitz, T.A. Structure of the binding site for nonnucleoside inhibitors of the reverse transcriptase of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 3911–3915. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Sarafianos, S.G.; Clark, A.D.; Boyer, P.L.; Hughes, S.H.; Arnold, E. Crystal structures of clinically relevant Lys103Asn/Tyr181Cys double mutant HIV-1 reverse transcriptase in complexes with ATP and non-nucleoside inhibitor HBY 097 . J. Mol. Biol. 2007, 365, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Sarafianos, S.G.; Clark, A.D.; Das, K.; Tuske, S.; Birktoft, J.J.; Ilankumaran, P.; Ramesha, A.R.; Sayer, J.M.; Jerina, D.M.; Boyer, P.L.; Hughes, S.H.; Arnold, E. Structures of HIV-1 reverse transcriptase with pre- and post-translocation AZTMP-terminated DNA . EMBO J. 2002, 21, 6614–6624. [Google Scholar] [CrossRef] [PubMed]
- Hsiou, Y.; Ding, J.; Das, K.; Clark, A.D.; Boyer, P.L.; Lewi, P.; Janssen, P.A.; Kleim, J.P.; Rosner, M.; Hughes, S.H.; Arnold, E. The Lys103Asn mutation of HIV-1 RT: a novel mechanism of drug resistance . J. Mol. Biol. 2001, 309, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Maga, G.; Amacker, M.; Ruel, N.; Hubscher, U.; Spadari, S. Resistance to nevirapine of HIV-1 reverse transcriptase mutants: loss of stabilizing interactions and thermodynamic or steric barriers are induced by different single amino acid substitutions. J. Mol. Biol. 1997, 274, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Spence, R.A.; Anderson, K.S.; Johnson, K.A. HIV-1 reverse transcriptase resistance to nonnucleoside inhibitors. Biochemistry 1996, 35, 1054–1063. [Google Scholar] [CrossRef] [PubMed]
- Sarafianos, S.G.; Marchand, B.; Das, K.; Himmel, D.M.; Parniak, M.A.; Hughes, S.H.; Arnold, E. Structure and function of HIV-1 reverse transcriptase: molecular mechanisms of polymerization and inhibition. J. Mol. Biol. 2009, 385, 693–713. [Google Scholar] [CrossRef] [PubMed]
- Domaoal, R.A.; Demeter, L.M. Structural and biochemical effects of human immunodeficiency virus mutants resistant to non-nucleoside reverse transcriptase inhibitors. Int. J. Biochem. Cell. Biol. 2004, 36, 1735–1751. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Das, K.; Moereels, H.; Koymans, L.; Andries, K.; Janssen, P.A.; Hughes, S.H.; Arnold, E. Structure of HIV-1 RT/TIBO R 86183 complex reveals similarity in the binding of diverse nonnucleoside inhibitors. Nat. Struct. Biol. 1995, 2, 407–415. [Google Scholar] [CrossRef]
- Ren, J.; Nichols, C.E.; Chamberlain, P.P.; Weaver, K.L.; Short, S.A.; Chan, J.H.; Kleim, J.P.; Stammers, D.K. Relationship of potency and resilience to drug resistant mutations for GW420867X revealed by crystal structures of inhibitor complexes for wild-type, Leu100Ile, Lys101Glu, and Tyr188Cys mutant HIV-1 reverse transcriptases. J. Med. Chem. 2007, 50, 2301–2309. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Nichols, C.; Bird, L.; Chamberlain, P.; Weaver, K.; Short, S.; Stuart, D.I.; Stammers, D.K. Structural mechanisms of drug resistance for mutations at codons 181 and 188 in HIV-1 reverse transcriptase and the improved resilience of second generation non-nucleoside inhibitors. J. Mol. Biol. 2001, 312, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Ding, J.; Hsiou, Y.; Clark, A.D.; Moereels, H.; Koymans, L.; Andries, K.; Pauwels, R.; Janssen, P.A.; Boyer, P.L.; Clark, P.; Smith, R.H.; Kroeger Smith, M.B.; Michejda, C.J.; Hughes, S.H.; Arnold, E. Crystal structures of 8-Cl and 9-Cl TIBO complexed with wild-type HIV-1 RT and 8-Cl TIBO complexed with the Tyr181Cys HIV-1 RT drug-resistant mutant . J. Mol. Biol. 1996, 264, 1085–1100. [Google Scholar] [CrossRef] [PubMed]
- Hsiou, Y.; Das, K.; Ding, J.; Clark, A.D.; Kleim, J.P.; Rosner, M.; Winkler, I.; Riess, G.; Hughes, S.H.; Arnold, E. Structures of Tyr188Leu mutant and wild-type HIV-1 reverse transcriptase complexed with the non-nucleoside inhibitor HBY 097: inhibitor flexibility is a useful design feature for reducing drug resistance . J. Mol. Biol. 1998, 284, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Goulden, M.G.; Cammack, N.; Hopewell, P.L.; Penn, C.R.; Cameron, J.M. Selection in vitro of an HIV-1 variant resistant to both lamivudine (3TC) and zidovudine. AIDS 1996, 10, 101–102. [Google Scholar] [CrossRef] [PubMed]
- Kemp, S.D.; Shi, C.; Bloor, S.; Harrigan, P.R.; Mellors, J.W.; Larder, B.A. A novel polymorphism at codon 333 of human immunodeficiency virus type 1 reverse transcriptase can facilitate dual resistance to zidovudine and L-2',3'-dideoxy-3'-thiacytidine. J. Virol. 1998, 72, 5093–5098. [Google Scholar] [PubMed]
- Radzio, J.; Sluis-Cremer, N. Efavirenz accelerates HIV-1 reverse transcriptase ribonuclease H cleavage, leading to diminished zidovudine excision. Mol. Pharmacol. 2008, 73, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Fransen, S.; Paxinos, E.; et al. Infrequent occurrence of mutations in the C-terminal region of reverse transcriptase modulates susceptibility to RT inhibitors . Antivir. Ther. 2006, 11, S143. [Google Scholar]
- Sluis-Cremer, N.; Moore, K.; Radzio, J.; Sonza, S.; Tachedjian, G. N348I in HIV-1 reverse transcriptase decreases susceptibility to tenofovir and etravirine in combination with other resistance mutations. AIDS 2010, 24, 317–319. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Fransen, S.; Paxinos, E.E.; Stawiski, E.; Huang, W.; Petropoulos, C.J. Combinations of mutations in the connection domain of human immunodeficiency virus type 1 reverse transcriptase: assessing the impact on nucleoside and nonnucleoside reverse transcriptase inhibitor resistance. Antimicrob. Agents Chemother. 2010, 54, 1973–1980. [Google Scholar] [CrossRef] [PubMed]
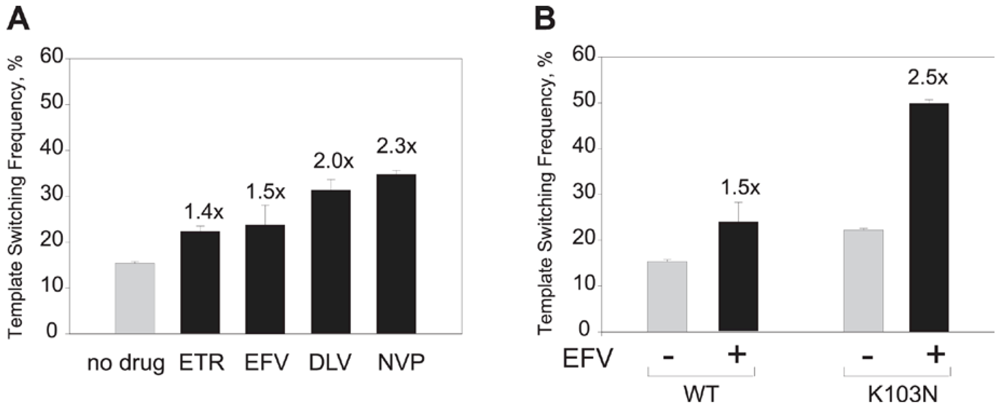
- Nikolenko, G.N.; Delviks-Frankenberry, K.A.; Pathak, V.K. A novel molecular mechanism of dual resistance to nucleoside and nonnucleoside reverse transcriptase inhibitors. J. Virol. 2010, 84, 5238–5249. [Google Scholar] [CrossRef] [PubMed]
- Samuele, A.; Kataropoulou, A.; Viola, M.; Zanoli, S.; La Regina, G.; Piscitelli, F.; Silvestri, R.; Maga, G. Non-nucleoside HIV-1 reverse transcriptase inhibitors di-halo-indolyl aryl sulfones achieve tight binding to drug-resistant mutants by targeting the enzyme-substrate complex. Antiviral Res. 2009, 81, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Zanoli, S.; Gemma, S.; Butini, S.; Brindisi, M.; Joshi, B.P.; Campiani, G.; Fattorusso, C.; Persico, M.; Crespan, E.; Cancio, R.; Spadari, S.; Hubscher, U.; Maga, G. Selective targeting of the HIV-1 reverse transcriptase catalytic complex through interaction with the "primer grip" region by pyrrolobenzoxazepinone non-nucleoside inhibitors correlates with increased activity towards drug-resistant mutants. Biochem. Pharmacol. 2008, 76, 156–168. [Google Scholar] [CrossRef] [PubMed]
- Radi, M.; Maga, G.; Alongi, M.; Angeli, L.; Samuele, A.; Zanoli, S.; Bellucci, L.; Tafi, A.; Casaluce, G.; Giorgi, G.; Armand-Ugon, M.; Gonzalez, E.; Este, J.A.; Baltzinger, M.; Bec, G.; Dumas, P.; Ennifar, E.; Botta, M. Discovery of chiral cyclopropyl dihydro-alkylthio-benzyl-oxopyrimidine (S-DABO) derivatives as potent HIV-1 reverse transcriptase inhibitors with high activity against clinically relevant mutants. J. Med. Chem. 2009, 52, 840–851. [Google Scholar] [CrossRef] [PubMed]
- Tachedjian, G.; Goff, S.P. The effect of NNRTIs on HIV reverse transcriptase dimerization. Curr. Opin. Investig. Drugs 2003, 4, 966–973. [Google Scholar] [PubMed]
- Mulky, A.; Sarafianos, S.G.; Jia, Y.; Arnold, E.; Kappes, J.C. Identification of amino acid residues in the human immunodeficiency virus type-1 reverse transcriptase tryptophan-repeat motif that are required for subunit interaction using infectious virions. J. Mol. Biol. 2005, 349, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Mulky, A.; Vu, B.C.; Conway, J.A.; Hughes, S.H.; Kappes, J.C. Analysis of amino acids in the beta7-beta8 loop of human immunodeficiency virus type 1 reverse transcriptase for their role in virus replication. J. Mol. Biol. 2007, 365, 1368–1378. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, A.; Pandey, N.; Mishra, C.A.; Talele, T.T.; Pandey, V.N. A single deletion at position 134, 135, or 136 in the beta 7-beta 8 loop of the p51 subunit of HIV-1 RT disrupts the formation of heterodimeric enzyme . J. Cell Biochem. 2010, 109, 598–605. [Google Scholar] [PubMed]
- Tachedjian, G.; Orlova, M.; Sarafianos, S.G.; Arnold, E.; Goff, S.P. Nonnucleoside reverse transcriptase inhibitors are chemical enhancers of dimerization of the HIV type 1 reverse transcriptase. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 7188–7193. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, A.; Zelina, S.; Sluis-Cremer, N.; Tachedjian, G. Impact of residues in the nonnucleoside reverse transcriptase inhibitor binding pocket on HIV-1 reverse transcriptase heterodimer stability. Curr. HIV Res. 2008, 6, 130–137. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Benkovic, S. Effect of a thiobenzimidazolone derivative on DNA strand transfer catalyzed by HIV-1 reverse transcriptase. J. Biol. Chem. 1994, 269, 4110–4115. [Google Scholar] [PubMed]
- Palaniappan, C.; Fay, P.J.; Bambara, R.A. Nevirapine alters the cleavage specificity of ribonuclease H of human immunodeficiency virus 1 reverse transcriptase. J. Biol. Chem. 1995, 270, 4861–4869. [Google Scholar] [CrossRef] [PubMed]
- Shaw-Reid, C.A.; Feuston, B.; Munshi, V.; Getty, K.; Krueger, J.; Hazuda, D.J.; Parniak, M.A.; Miller, M.D.; Lewis, D. Dissecting the effects of DNA polymerase and ribonuclease H inhibitor combinations on HIV-1 reverse-transcriptase activities. Biochemistry 2005, 44, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Hang, J.Q.; Li, Y.; Yang, Y.; Cammack, N.; Mirzadegan, T.; Klumpp, K. Substrate-dependent inhibition or stimulation of HIV RNase H activity by non-nucleoside reverse transcriptase inhibitors (NNRTIs). Biochem. Biophys. Res. Commun. 2007, 352, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Pelemans, H.; Esnouf, R.M.; Jonckheere, H.; De Clercq, E.; Balzarini, J. Mutational analysis of Tyr-318 within the non-nucleoside reverse transcriptase inhibitor binding pocket of human immunodeficiency virus type I reverse transcriptase. J. Biol. Chem. 1998, 273, 34234–34239. [Google Scholar] [CrossRef] [PubMed]
- Harrigan, P.R.; Salim, M.; Stammers, D.K.; Wynhoven, B.; Brumme, Z.L.; McKenna, P.; Larder, B.; Kemp, S.D. A mutation in the 3' region of the human immunodeficiency virus type 1 reverse transcriptase (Y318F) associated with nonnucleoside reverse transcriptase inhibitor resistance. J. Virol. 2002, 76, 6836–6840. [Google Scholar] [CrossRef] [PubMed]
- Dau, B.; Ayers, D.; Singer, J.; Harrigan, P.R.; Brown, S.; Kyriakides, T.; Cameron, D.W.; Angus, B.; Holodniy, M. Connection domain mutations in treatment-experienced patients in the OPTIMA trial. J. Acquir. Immune Defic. Syndr. 2010, 54, 160–166. [Google Scholar] [PubMed]
- Hachiya, A.; Kodama, E.N.; Sarafianos, S.G.; Schuckmann, M.M.; Sakagami, Y.; Matsuoka, M.; Takiguchi, M.; Gatanaga, H.; Oka, S. Amino acid mutation N348I in the connection subdomain of human immunodeficiency virus type 1 reverse transcriptase confers multiclass resistance to nucleoside and nonnucleoside reverse transcriptase inhibitors. J. Virol. 2008, 82, 3261–3270. [Google Scholar] [CrossRef] [PubMed]
- Waters, J.M.; O'Neal, W.; White, K.L.; Wakeford, C.; Lansdon, E.B.; Harris, J.; Svarovskaia, E.S.; Miller, M.D.; Borroto-Esoda, K. Mutations in the thumb-connection and RNase H domain of HIV type-1 reverse transcriptase of antiretroviral treatment-experienced patients. Antivir. Ther. 2009, 14, 231–239. [Google Scholar] [PubMed]
- Price, H.; Asboe, D.; Pozniak, A.; Gazzard, B.; Fearnhill, E.; Pillay, D.; Dunn, D. Positive and negative drug selection pressures on the N348I connection domain mutation: new insights from in vivo data. Antivir. Ther. 2010, 15, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.F.; Lengruber, R.B.; Soares, E.A.; Jere, A.; Sprinz, E.; Martinez, A.M.; Silveira, J.; Sion, F.S.; Pathak, V.K.; Soares, M.A. Conservation patterns of HIV-1 RT connection and RNase H domains: identification of new mutations in NRTI-treated patients . PLoS One 2008, 3, e1781. [Google Scholar] [CrossRef] [PubMed]
- Cane, P.A.; Green, H.; Fearnhill, E.; Dunn, D. Identification of accessory mutations associated with high-level resistance in HIV-1 reverse transcriptase. AIDS 2007, 21, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Roquebert, B.; Flandre, P.; Malet, I.; Wirden, M.; ait-arkoub, Z.; Boutonnet, V.; Simon, A.; Katlama, C.; Calvez, V.; Marcelin, A.G. Identification of a Mutation (A400T) in the Connection Domain of the HIV-1 Reverse Transcriptase Associated to Exposure and Resistance to NRTI . 2007, Abstract #596. [Google Scholar] [CrossRef] [PubMed]
- Saeng-Aroon, S.; Tsuchiya, N.; Auwanit, W.; Ayuthaya, P.I.; Pathipvanich, P.; Sawanpanyalert, P.; Rojanawiwat, A.; Kannagi, M.; Ariyoshi, K.; Sugiura, W. Drug-resistant mutation patterns in CRF01_AE cases that failed d4T+3TC+nevirapine fixed-dosed, combination treatment: Follow-up study from the Lampang cohort. Antiviral Res. 2010, 87, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Roquebert, B.; Wirden, M.; Simon, A.; Deval, J.; Katlama, C.; Calvez, V.; Marcelin, A.G. Relationship between mutations in HIV-1 RNase H domain and nucleoside reverse transcriptase inhibitors resistance mutations in naive and pre-treated HIV infected patients. J. Med. Virol. 2007, 79, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Ntemgwa, M.; Wainberg, M.A.; Oliveira, M.; Moisi, D.; Lalonde, R.; Micheli, V.; Brenner, B.G. Variations in reverse transcriptase and RNase H domain mutations in human immunodeficiency virus type 1 clinical isolates are associated with divergent phenotypic resistance to zidovudine. Antimicrob. Agents Chemother. 2007, 51, 3861–3869. [Google Scholar] [CrossRef] [PubMed]
- Hachiya, A.; Shimane, K.; Sarafianos, S.G.; Kodama, E.N.; Sakagami, Y.; Negishi, F.; Koizumi, H.; Gatanaga, H.; Matsuoka, M.; Takiguchi, M.; Oka, S. Clinical relevance of substitutions in the connection subdomain and RNase H domain of HIV-1 reverse transcriptase from a cohort of antiretroviral treatment-naive patients. Antiviral Res. 2009, 82, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Soares, E.A.; Makamche, M.F.; Siqueira, J.D.; Lumngwena, E.; Mbuagbaw, J.; Kaptue, L.; Asonganyi, T.; Seuanez, H.N.; Soares, M.A.; Alemnji, G. Molecular diversity and polymerase gene genotypes of HIV-1 among treatment-naive Cameroonian subjects with advanced disease. J. Clin. Virol. 2010, 48, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Torti, C.; Quiros-Roldan, E.; Monno, L.; Patroni, A.; Saracino, A.; Angarano, G.; Tinelli, C.; Mazzotta, F.; Lo Caputo, S.; Pierotti, P.; Carosi, G. HIV-1 resistance to dideoxynucleoside reverse transcriptase inhibitors: genotypic-phenotypic correlations. J. Acquir. Immune Defic. Syndr. 2004, 36, 1104–1107. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Delviks-Frankenberry, K.A.; Nikolenko, G.N.; Pathak, V.K. The “Connection” Between HIV Drug Resistance and RNase H. Viruses 2010, 2, 1476-1503. https://doi.org/10.3390/v2071476
Delviks-Frankenberry KA, Nikolenko GN, Pathak VK. The “Connection” Between HIV Drug Resistance and RNase H. Viruses. 2010; 2(7):1476-1503. https://doi.org/10.3390/v2071476
Chicago/Turabian StyleDelviks-Frankenberry, Krista A., Galina N. Nikolenko, and Vinay K. Pathak. 2010. "The “Connection” Between HIV Drug Resistance and RNase H" Viruses 2, no. 7: 1476-1503. https://doi.org/10.3390/v2071476