D471G Mutation in LCMV-NP Affects Its Ability to Self-associate and Results in a Dominant Negative Effect in Viral RNA Synthesis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
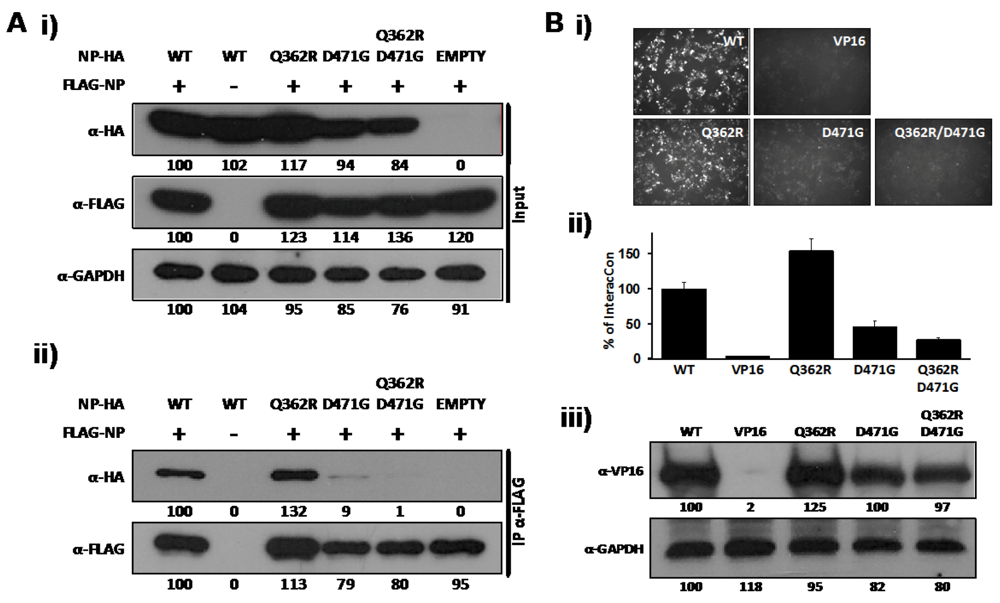
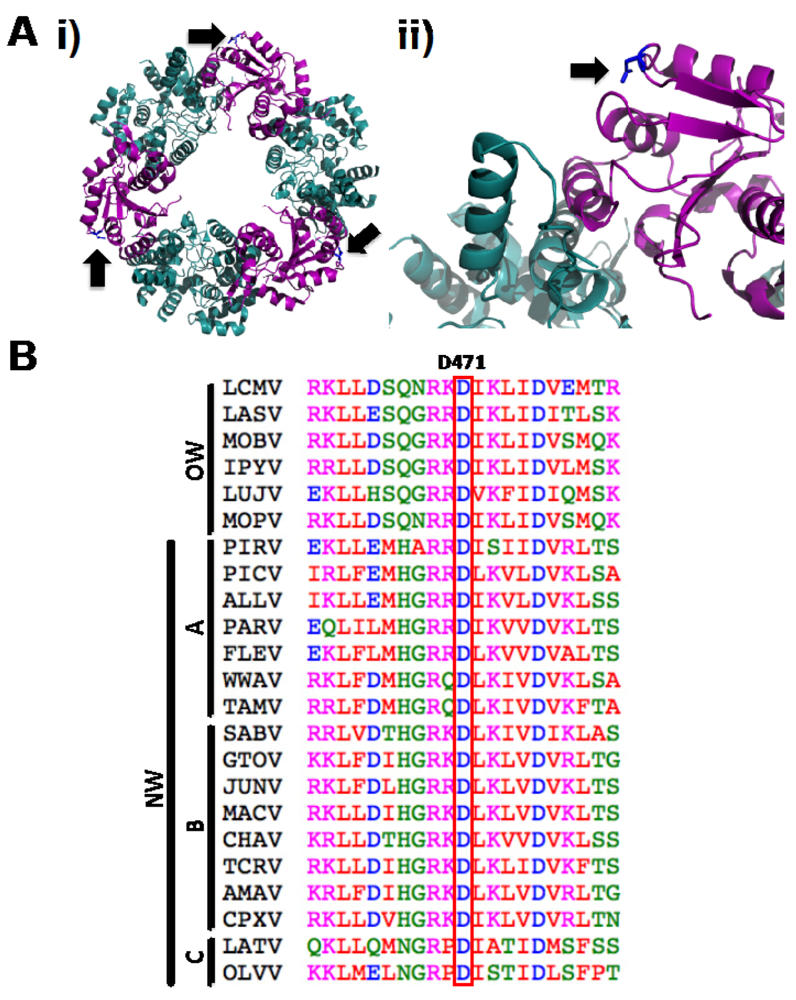
2.1. Identification of a Specific Amino Acid Residue with Critical Role in LCMV-NP Self-association
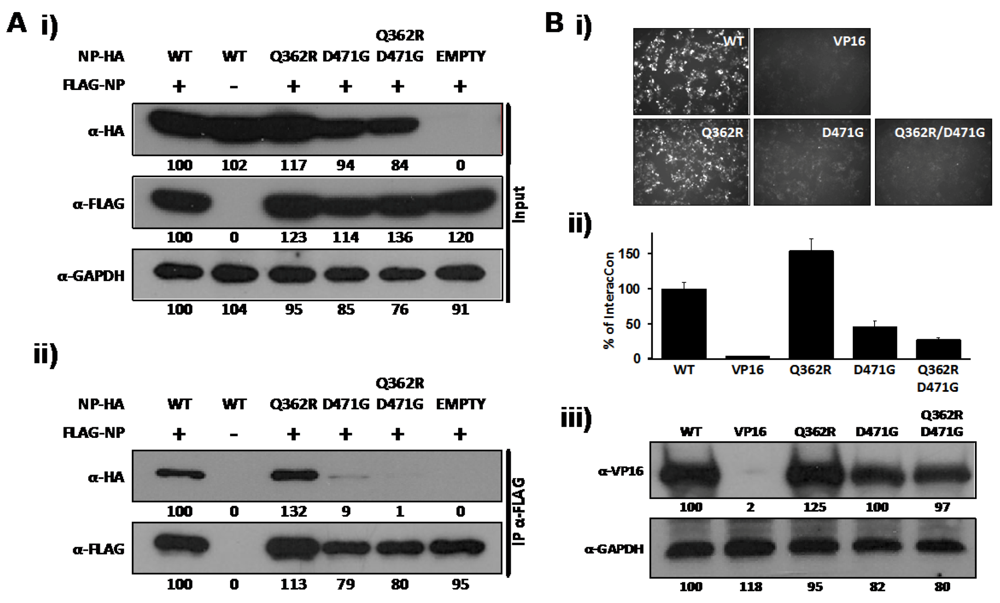
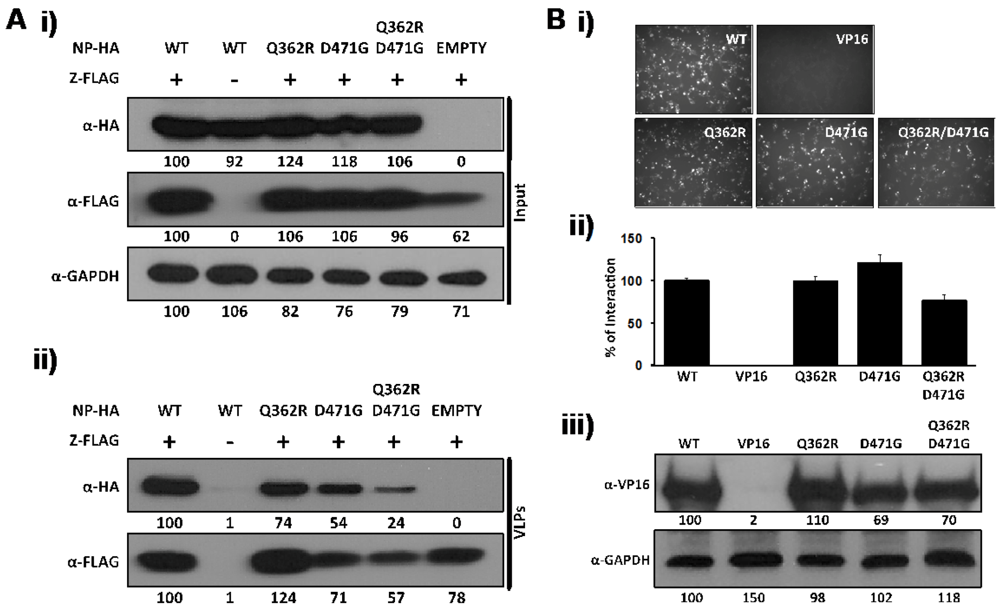
2.2. Role of D471 in LCMV NP-Z Interaction
2.3. Role of NP Self-association in Virus RNA Replication and Gene Transcription

2.4. Role of NP-self- association in the Anti-IFN-I Activity of NP

2.5. Characterization of Double-stranded (ds)RNA Binding Properties of LCMV-NP
2.6. Assessing the Functional Impact of Other Single Amino Acid Substitutions at Position D471 in LCMV-NP

2.7. Assessing the Dominant Negative Phenotype of LCMV-NP D471G
3. Experimental Section
3.1. Cells and Viruses
3.2. Plasmids
3.3. Co-immunoprecipitation (Co-IP)
3.4. Virus-Like Particle (VLP) Assay
3.5. Minigenome (MG) Assay
3.6. Mammalian Two-hybrid (M2H) Assay
3.7. Double-stranded (ds)RNA Pull Down Assay
3.8. Type I Interferon (IFN-I) Reporter Assay
3.9. Protein Gel Electrophoresis and Western Blot (WB) Analysis
3.10. Generation of NP-expressing Stable Cell Lines
3.11. Virus Growth Kinetics and Titrations
4. Conclusions and Discussion
Acknowledgments
Conflict of Interest
References and Notes
- Buchmeier, M.J.; Peters, C.J.; de la Torre, J.C. Arenaviridae: The viruses and their replication. In Fields virology, Fifth; Fields B. N.;, Knipe, D. M.; Howley P., M., Eds.; 2007; Vol. 2, pp. 1792–1827. [Google Scholar]
- Bowen, M.D.; Peters, C.J.; Nichol, S.T. Phylogenetic analysis of the Arenaviridae: patterns of virus evolution and evidence for cospeciation between arenaviruses and their rodent hosts. Mol Phylogenet. Evol. 1997, 8, 301–316. [Google Scholar] [CrossRef]
- McCormick, J.B.; Fisher-Hoch, S.P. Lassa Fever. In Arenaviruses I; Oldstone M., B., Ed.; Springer-Verlag: Berlin, Heidelberg, New York, 2002; Vol. 262, pp. 75–110. [Google Scholar]
- Peters, C.J. Human infection with Arenaviruses in the Americas. In Arenaviruses I; Oldstone M., B., Ed.; Springer-Verlag: Berlin, Heidelberg, New York, 2002; Vol. 262, pp. 65–74. [Google Scholar]
- Charrel, R.N.; de Lamballerie, X. Arenaviruses other than Lassa virus. Antiviral. Res. 2003, 57, 89–100. [Google Scholar] [CrossRef]
- Gunther, S.; Lenz, O. Lassa virus. Crit. Rev. Clin. Lab. Sci. 2004, 41, 339–390. [Google Scholar] [CrossRef]
- Isaacson, M. Viral hemorrhagic fever hazards for travelers in Africa. Clin. Infect. Dis. 2001, 33, 1707–1712. [Google Scholar] [CrossRef]
- Seinbergas, M.M.; Cibiriene, I.K.; Sverdlov Iu, M. [Main syndrome of prenatal infection caused by lymphocytic choriomeningitis virus - hydrocephalus and chorioretinal degeneration]. Pediatriia 1976, 7–11. [Google Scholar]
- Ackermann, R.; Korver, G.; Turss, R.; Wonne, R.; Hochgesand, P. [Prenatal infection with the virus of lymphocytic choriomeningitis: report of two cases (author's transl)]. Dtsch. Med. Wochenschr. 1974, 99, 629–632. [Google Scholar] [CrossRef]
- Fischer, S.A.; Graham, M.B.; Kuehnert, M.J.; Kotton, C.N.; Srinivasan, A.; Marty, F.M.; Comer, J.A.; Guarner, J.; Paddock, C.D.; DeMeo, D.L.; Shieh, W.J.; Erickson, B.R.; Bandy, U.; DeMaria, A., Jr.; Davis, J.P.; Delmonico, F.L.; Pavlin, B.; Likos, A.; Vincent, M.J.; Sealy, T.K.; Goldsmith, C.S.; Jernigan, D.B.; Rollin, P.E.; Packard, M.M.; Patel, M.; Rowland, C.; Helfand, R.F.; Nichol, S.T.; Fishman, J.A.; Ksiazek, T.; Zaki, S.R. Transmission of lymphocytic choriomeningitis virus by organ transplantation. N. Engl. J. Med. 2006, 354, 2235–2249. [Google Scholar]
- Palacios, G.; Druce, J.; Du, L.; Tran, T.; Birch, C.; Briese, T.; Conlan, S.; Quan, P.L.; Hui, J.; Marshall, J.; Simons, J.F.; Egholm, M.; Paddock, C.D.; Shieh, W.J.; Goldsmith, C.S.; Zaki, S.R.; Catton, M.; Lipkin, W.I. A new arenavirus in a cluster of fatal transplant-associated diseases. N. Engl. J. Med. 2008, 358, 991–998. [Google Scholar]
- Kilgore, P.E.; Ksiazek, T.G.; Rollin, P.E.; Mills, J.N.; Villagra, M.R.; Montenegro, M.J.; Costales, M.A.; Paredes, L.C.; Peters, C.J. Treatment of Bolivian hemorrhagic fever with intravenous ribavirin. Clin. Infect. Dis. 1997, 24, 718–722. [Google Scholar]
- McCormick, J.B.; King, I.J.; Webb, P.A.; Scribner, C.L.; Craven, R.B.; Johnson, K.M.; Elliott, L.H.; Belmont-Williams, R. Lassa fever. Effective therapy with ribavirin. N. Engl. J. Med. 1986, 314, 20–26. [Google Scholar] [CrossRef]
- McKee, K.T., Jr.; Huggins, J.W.; Trahan, C.J.; Mahlandt, B.G. Ribavirin prophylaxis and therapy for experimental argentine hemorrhagic fever. Antimicrob. Agents Chemother. 1988, 32, 1304–1309. [Google Scholar] [CrossRef]
- Oldstone, M.B. Biology and pathogenesis of lymphocytic choriomeningitis virus infection. In Arenaviruses; Oldstone M., B., Ed.; 2002; Vol. 263, pp. 83–118. [Google Scholar]
- Zinkernagel, R.M. Lymphocytic choriomeningitis virus and immunology. Curr. Top. Microbiol. Immunol. 2002, 263, 1–5. [Google Scholar] [CrossRef]
- Meyer, B.J.; de la Torre, J.C.; Southern, P.J. Arenaviruses: genomic RNAs, transcription, and replication. Curr. Top Microbiol. Immunol. 2002, 262, 139–157. [Google Scholar]
- Eichler, R.; Strecker, T.; Kolesnikova, L.; ter Meulen, J.; Weissenhorn, W.; Becker, S.; Klenk, H.D.; Garten, W.; Lenz, O. Characterization of the Lassa virus matrix protein Z: electron microscopic study of virus-like particles and interaction with the nucleoprotein (NP). Virus Res. 2004, 100, 249–255. [Google Scholar] [CrossRef]
- Neuman, B.W.; Adair, B.D.; Burns, J.W.; Milligan, R.A.; Buchmeier, M.J.; Yeager, M. Complementarity in the supramolecular design of arenaviruses and retroviruses revealed by electron cryomicroscopy and image analysis. J. Virol. 2005, 79, 3822–3830. [Google Scholar] [CrossRef]
- Perez, M.; Craven, R.C.; de la Torre, J.C. The small RING finger protein Z drives arenavirus budding: implications for antiviral strategies. Proc. Natl. Acad. Sci. USA 2003, 100, 12978–12983. [Google Scholar]
- de la Torre, J.C. Molecular and cell biology of the prototypic arenavirus LCMV: implications for understanding and combating hemorrhagic fever arenaviruses. Ann. N.Y. Acad. Sci. 2009, 1171 Suppl 1, E57–64. [Google Scholar] [CrossRef]
- Lenz, O.; ter Meulen, J.; Klenk, H.D.; Seidah, N.G.; Garten, W. The Lassa virus glycoprotein precursor GP-C is proteolytically processed by subtilase SKI-1/S1P. Proc. Natl. Acad. Sci. USA 2001, 98, 12701–12705. [Google Scholar]
- Pinschewer, D.D.; Perez, M.; de la Torre, J.C. Role of the virus nucleoprotein in the regulation of lymphocytic choriomeningitis virus transcription and RNA replication. J. Virol. 2003, 77, 3882–3887. [Google Scholar] [CrossRef]
- Lee, K.J.; Novella, I.S.; Teng, M.N.; Oldstone, M.B.; de La Torre, J.C. NP and L proteins of lymphocytic choriomeningitis virus (LCMV) are sufficient for efficient transcription and replication of LCMV genomic RNA analogs. J. Virol. 2000, 74, 3470–3477. [Google Scholar]
- Martinez-Sobrido, L.; Zuniga, E.I.; Rosario, D.; Garcia-Sastre, A.; de la Torre, J.C. Inhibition of the type I interferon response by the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J. Virol. 2006, 80, 9192–9199. [Google Scholar] [CrossRef]
- Martinez-Sobrido, L.; Giannakas, P.; Cubitt, B.; Garcia-Sastre, A.; de la Torre, J.C. Differential inhibition of type I interferon induction by arenavirus nucleoproteins. J. Virol. 2007, 81, 12696–12703. [Google Scholar] [CrossRef]
- Pythoud, C.; Rodrigo, W.W.; Pasqual, G.; Rothenberger, S.; Martinez-Sobrido, L.; de la Torre, J.C.; Kunz, S. Arenavirus Nucleoprotein Targets Interferon Regulatory Factor-Activating Kinase IKK{varepsilon}. J. Virol. 2012, 86, 7728–7738. [Google Scholar]
- Martinez-Sobrido, L.; Emonet, S.; Giannakas, P.; Cubitt, B.; Garcia-Sastre, A.; de la Torre, J.C. Identification of amino acid residues critical for the anti-interferon activity of the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J. Virol. 2009, 83, 11330–11340. [Google Scholar] [CrossRef]
- Qi, X.; Lan, S.; Wang, W.; Schelde, L.M.; Dong, H.; Wallat, G.D.; Ly, H.; Liang, Y.; Dong, C. Cap binding and immune evasion revealed by Lassa nucleoprotein structure. Nature 2010, 468, 779–783. [Google Scholar] [CrossRef]
- Hastie, K.M.; Kimberlin, C.R.; Zandonatti, M.A.; MacRae, I.J.; Saphire, E.O. Structure of the Lassa virus nucleoprotein reveals a dsRNA-specific 3' to 5' exonuclease activity essential for immune suppression. Proc. Natl. Acad. Sci. USA 2011, 108, 2396–2401. [Google Scholar]
- Rodrigo, W.W.; Ortiz-Riano, E.; Pythoud, C.; Kunz, S.; de la Torre, J.C.; Martinez-Sobrido, L. Arenavirus nucleoproteins prevent activation of nuclear factor kappa B. J. Virol. 2012, 86, 8185–8197. [Google Scholar]
- Borrow, P.; Martinez-Sobrido, L.; de la Torre, J.C. Inhibition of the type I interferon antiviral response during arenavirus infection. Viruses 2010, 2, 2443–2480. [Google Scholar] [CrossRef]
- Ortiz-Riano, E.; Cheng, B.Y.; de la Torre, J.C.; Martinez-Sobrido, L. The C-terminal region of lymphocytic choriomeningitis virus nucleoprotein contains distinct and segregable functional domains involved in NP-Z interaction and counteraction of the type I interferon response. J. Virol. 2011, 85, 13038–13048. [Google Scholar] [CrossRef]
- Ortiz-Riano, E.; Cheng, B.Y.; de la Torre, J.C.; Martinez-Sobrido, L. Self-association of lymphocytic choriomeningitis virus nucleoprotein is mediated by its N-terminal region and is not required for its anti-interferon function. J. Virol. 2012, 86, 3307–3317. [Google Scholar] [CrossRef]
- Levingston Macleod, J.M.; D'Antuono, A.; Loureiro, M.E.; Casabona, J.C.; Gomez, G.A.; Lopez, N. Identification of two functional domains within the arenavirus nucleoprotein. J. Virol. 2011, 85, 2012–2023. [Google Scholar] [CrossRef]
- Shtanko, O.; Imai, M.; Goto, H.; Lukashevich, I.S.; Neumann, G.; Watanabe, T.; Kawaoka, Y. A role for the C terminus of Mopeia virus nucleoprotein in its incorporation into Z protein-induced virus-like particles. J. Virol. 2010, 84, 5415–5422. [Google Scholar] [CrossRef]
- Hastie, K.M.; Liu, T.; Li, S.; King, L.B.; Ngo, N.; Zandonatti, M.A.; Woods, V.L., Jr.; de la Torre, J.C.; Saphire, E.O. Crystal structure of the Lassa virus nucleoprotein-RNA complex reveals a gating mechanism for RNA binding. Proc. Natl. Acad. Sci. USA 2011, 108, 19365–19370. [Google Scholar]
- Perez, M.; Greenwald, D.L.; de la Torre, J.C. Myristoylation of the RING finger Z protein is essential for arenavirus budding. J. Virol. 2004, 78, 11443–11448. [Google Scholar] [CrossRef]
- Strecker, T.; Eichler, R.; Meulen, J.; Weissenhorn, W.; Dieter Klenk, H.; Garten, W.; Lenz, O. Lassa virus Z protein is a matrix protein and sufficient for the release of virus-like particles [corrected]. J. Virol. 2003, 77, 10700–10705. [Google Scholar] [CrossRef]
- Strecker, T.; Maisa, A.; Daffis, S.; Eichler, R.; Lenz, O.; Garten, W. The role of myristoylation in the membrane association of the Lassa virus matrix protein Z. Virol. J. 2006, 3, 93. [Google Scholar] [CrossRef]
- Lee, K.J.; Perez, M.; Pinschewer, D.D.; de la Torre, J.C. Identification of the lymphocytic choriomeningitis virus (LCMV) proteins required to rescue LCMV RNA analogs into LCMV-like particles. J. Virol. 2002, 76, 6393–6397. [Google Scholar] [CrossRef]
- Pinschewer, D.D.; Perez, M.; de la Torre, J.C. Dual role of the lymphocytic choriomeningitis virus intergenic region in transcription termination and virus propagation. J. Virol. 2005, 79, 4519–4526. [Google Scholar]
- Rodrigo, W.W.; de la Torre, J.C.; Martinez-Sobrido, L. Use of single-cycle infectious lymphocytic choriomeningitis virus to study hemorrhagic fever arenaviruses. J. Virol. 2011, 85, 1684–1695. [Google Scholar]
- Rose, K.M.; Elliott, R.; Martinez-Sobrido, L.; Garcia-Sastre, A.; Weiss, S.R. Murine coronavirus delays expression of a subset of interferon-stimulated genes. J. Virol. 2010, 84, 5656–5669. [Google Scholar]
- Chen, G.; Ward, B.M.; Yu, K.H.; Chinchar, V.G.; Robert, J. Improved knockout methodology reveals that frog virus 3 mutants lacking either the 18K immediate-early gene or the truncated vIF-2alpha gene are defective for replication and growth in vivo. J. Virol. 2011, 85, 11131–11138. [Google Scholar] [CrossRef]
- Emonet, S.F.; Garidou, L.; McGavern, D.B.; de la Torre, J.C. Generation of recombinant lymphocytic choriomeningitis viruses with trisegmented genomes stably expressing two additional genes of interest. Proc. Natl. Acad. Sci. USA 2009, 106, 3473–3478. [Google Scholar]
- Capul, A.A.; de la Torre, J.C. A cell-based luciferase assay amenable to high-throughput screening of inhibitors of arenavirus budding. Virology 2008, 382, 107–114. [Google Scholar] [CrossRef]
- Flick, R.; Pettersson, R.F. Reverse genetics system for Uukuniemi virus (Bunyaviridae): RNA polymerase I-catalyzed expression of chimeric viral RNAs. J. Virol. 2001, 75, 1643–1655. [Google Scholar] [CrossRef]
- Martinez-Sobrido, L.; Cadagan, R.; Steel, J.; Basler, C.F.; Palese, P.; Moran, T.M.; Garcia-Sastre, A. Hemagglutinin-pseudotyped green fluorescent protein-expressing influenza viruses for the detection of influenza virus neutralizing antibodies. J. Virol. 2010, 84, 2157–2163. [Google Scholar] [CrossRef]
- Nguyen, D.N.; Kim, P.; Martinez-Sobrido, L.; Beitzel, B.; Garcia-Sastre, A.; Langer, R.; Anderson, D.G. A novel high-throughput cell-based method for integrated quantification of type I interferons and in vitro screening of immunostimulatory RNA drug delivery. Biotechnol. Bioeng. 2009, 103, 664–675. [Google Scholar] [CrossRef]
- Battegay, M.; Cooper, S.; Althage, A.; Banziger, J.; Hengartner, H.; Zinkernagel, R.M. Quantification of lymphocytic choriomeningitis virus with an immunological focus assay in 24- or 96-well plates. J. Virol. Methods 1991, 33, 191–198. [Google Scholar] [CrossRef]
- Albertini, A.A.; Wernimont, A.K.; Muziol, T.; Ravelli, R.B.; Clapier, C.R.; Schoehn, G.; Weissenhorn, W.; Ruigrok, R.W. Crystal structure of the rabies virus nucleoprotein-RNA complex. Science 2006, 313, 360–363. [Google Scholar]
- Tawar, R.G.; Duquerroy, S.; Vonrhein, C.; Varela, P.F.; Damier-Piolle, L.; Castagne, N.; MacLellan, K.; Bedouelle, H.; Bricogne, G.; Bhella, D.; Eleouet, J.F.; Rey, F.A. Crystal structure of a nucleocapsid-like nucleoprotein-RNA complex of respiratory syncytial virus. Science 2009, 326, 1279–1283. [Google Scholar]
- Ng, A.K.; Zhang, H.; Tan, K.; Li, Z.; Liu, J.H.; Chan, P.K.; Li, S.M.; Chan, W.Y.; Au, S.W.; Joachimiak, A.; Walz, T.; Wang, J.H.; Shaw, P.C. Structure of the influenza virus A H5N1 nucleoprotein: implications for RNA binding, oligomerization, and vaccine design. FASEB J. 2008, 22, 3638–3647. [Google Scholar] [CrossRef]
- Rudolph, M.G.; Kraus, I.; Dickmanns, A.; Eickmann, M.; Garten, W.; Ficner, R. Crystal structure of the borna disease virus nucleoprotein. Structure 2003, 11, 1219–1226. [Google Scholar] [CrossRef]
- Green, T.J.; Zhang, X.; Wertz, G.W.; Luo, M. Structure of the vesicular stomatitis virus nucleoprotein-RNA complex. Science 2006, 313, 357–360. [Google Scholar]
- Labarga, A.; Valentin, F.; Anderson, M.; Lopez, R. Web services at the European bioinformatics institute. Nucleic Acids Res. 2007, 35, W6–11. [Google Scholar] [CrossRef]
- Perez, M.; de la Torre, J.C. Characterization of the genomic promoter of the prototypic arenavirus lymphocytic choriomeningitis virus. J. Virol. 2003, 77, 1184–1194. [Google Scholar]
- Ghiringhelli, P.D.; Rivera-Pomar, R.V.; Lozano, M.E.; Grau, O.; Romanowski, V. Molecular organization of Junin virus S RNA: complete nucleotide sequence, relationship with other members of the Arenaviridae and unusual secondary structures. J. Gen. Virol. 1991, 72 ( Pt 9), 2129–2141. [Google Scholar]
- Hovmoller, S.; Zhou, T.; Ohlson, T. Conformations of amino acids in proteins. Acta Crystallogr. D. Biol. Crystallogr. 2002, 58, 768–776. [Google Scholar] [CrossRef]
- Kerber, R.; Rieger, T.; Busch, C.; Flatz, L.; Pinschewer, D.D.; Kummerer, B.M.; Gunther, S. Cross-species analysis of the replication complex of Old World arenaviruses reveals two nucleoprotein sites involved in L protein function. J. Virol. 2011, 85, 12518–12528. [Google Scholar] [CrossRef]
- Watanabe, S.; Noda, T.; Kawaoka, Y. Functional mapping of the nucleoprotein of Ebola virus. J. Virol. 2006, 80, 3743–3751. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ortiz-Riaño, E.; Cheng, B.Y.H.; Torre, J.C.d.l.; Martínez-Sobrido, L. D471G Mutation in LCMV-NP Affects Its Ability to Self-associate and Results in a Dominant Negative Effect in Viral RNA Synthesis. Viruses 2012, 4, 2137-2161. https://doi.org/10.3390/v4102137
Ortiz-Riaño E, Cheng BYH, Torre JCdl, Martínez-Sobrido L. D471G Mutation in LCMV-NP Affects Its Ability to Self-associate and Results in a Dominant Negative Effect in Viral RNA Synthesis. Viruses. 2012; 4(10):2137-2161. https://doi.org/10.3390/v4102137
Chicago/Turabian StyleOrtiz-Riaño, Emilio, Benson Y.H. Cheng, Juan C. de la Torre, and Luis Martínez-Sobrido. 2012. "D471G Mutation in LCMV-NP Affects Its Ability to Self-associate and Results in a Dominant Negative Effect in Viral RNA Synthesis" Viruses 4, no. 10: 2137-2161. https://doi.org/10.3390/v4102137