2.1. Establishing Demarcation Criteria
The PASC tool for the family of
Filoviridae at NCBI can be accessed through [
6] (a screen shot is shown in
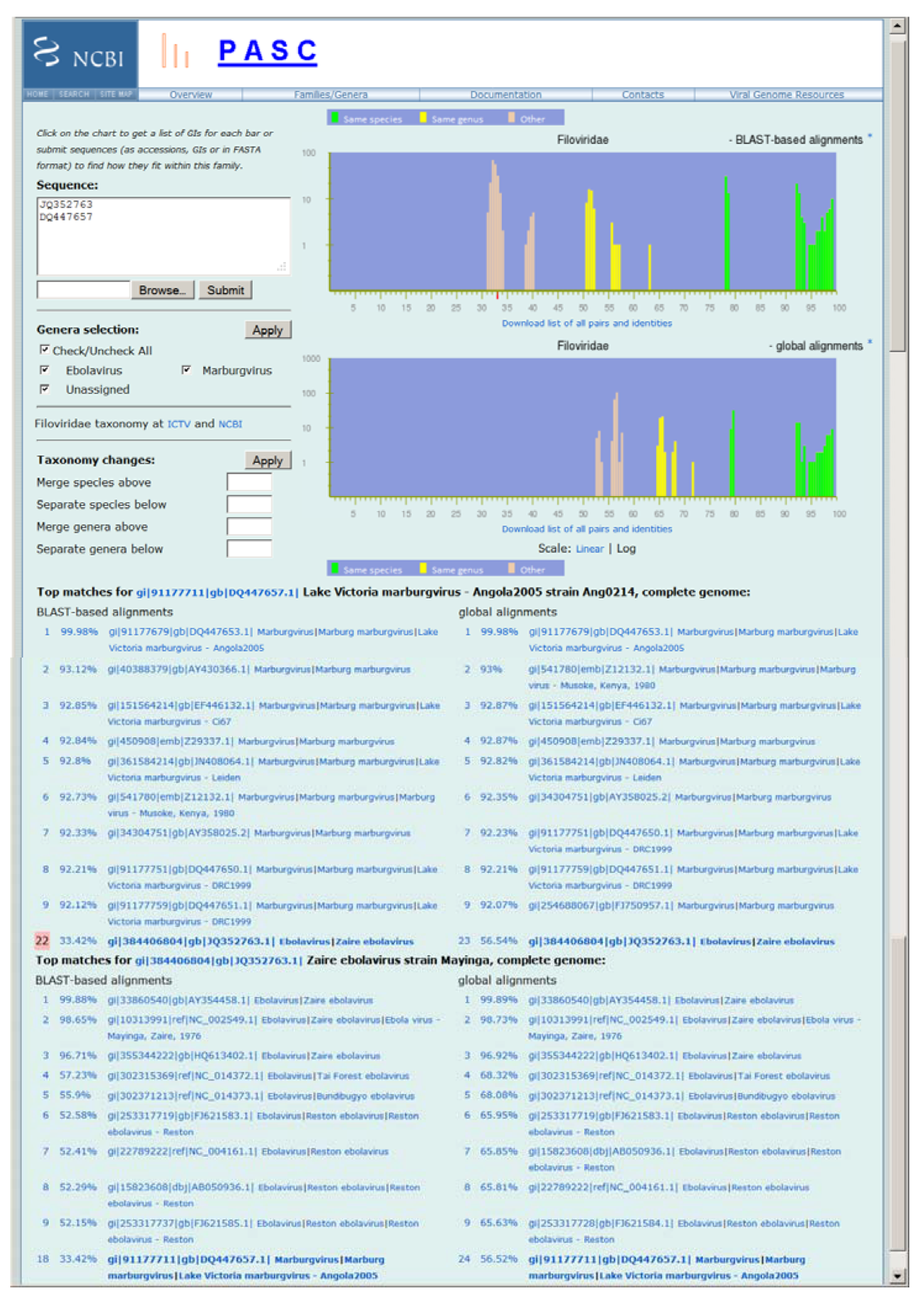
Figure 1). It currently contains 52 complete genome sequences available in GenBank. The upper right plot in
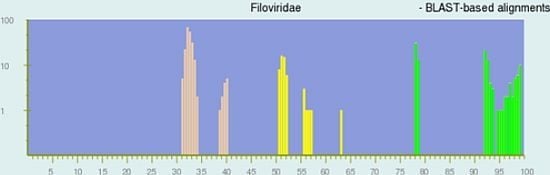
Figure 1 displays the pairwise identity distribution of 28 non-redundant genomes calculated by the BLAST-based alignment, while the lower right plot shows the pairwise identity distribution of 27 non-redundant genomes by the global alignment (See
Experimental Section for the alignment methods). The green, yellow and peach bars in the plots represent pairs of genomes that are assigned to the same species, different species but in the same genus, and different genera respectively in the current NCBI Taxonomy Database. PASC works best when the bars of different colors group together and are well separated, which is the case in both plots in
Figure 1. The results clearly indicate that (a) sequence-based classification agrees with the current ICTV taxonomy for the family
Filoviridae; (b) the taxonomic assignments of the filovirus genome sequences in GenBank are accurate; (c) species and genera demarcations can be set for the family.
It can be seen from
Figure 1 that the yellow and peach peaks are different in the two distribution plots. For the BLAST-based alignment plot, the yellow peaks are located between 50% and 64%, and the peach ones are between 30% and 41%; while in the global-alignment plot, they are between 64% and 72%, and between 52% and 58%, respectively. Originally, the Needleman–Wunsch global‑alignment method [
7] was used to calculate the genome identities in PASC [
4]. It was later observed that this method was not optimized for some virus families [
8]. For filoviruses, this is illustrated on
Figure 2, where the dot matrix and text alignments between the genomes of Ebola virus (NC_002549) and Taï Forest virus (NC_014372) are shown (Similar alignments can be obtained by clicking the percentage of identities for the pairs of interest such as those listed in
Figure 1). The identity between the two genomes calculated using the BLAST-based and global alignment is 57% and 68% respectively, neither of which matches the 63% reported by Towner
et al. [
9]. The BLAST-based method calculates percent identity only in the regions of alignment that in the case of filoviruses correspond to the 7 filoviral genes (see the dot matrix view at the upper section in
Figure 2). This method essentially resembles the strategy used to classify bacteriophages based on protein sequence similarities [
10,
11]. The global alignment method forces alignment over the whole length of the sequence (see the dot matrix view at the lower section in
Figure 2). Since, on average, any two random nucleotide sequences of the same size will have an identity of 25%, the identities obtained by the global alignment method are usually inflated, especially for genomes with high divergence. We therefore believe the BLAST‑based alignment result represents the true relationship among genome sequences of filoviruses, and should be used to establish taxa demarcation criteria, which are between 64% and 77% for species, and between 41% and 50% for genera. These demarcation criteria better match those set up by the ICTV
Filoviridae Study Group, which are currently set as 70% for species and 50% for genera [
1]. The demarcation criteria suggested by PASC are in a range rather than precise percentages as in many other viral groups, as the filovirus genomes sequenced so far are not very diverse. When more genome sequences become available, these demarcations will most likely change to narrower ranges or even to a single percentage.
It is important to keep in mind that the demarcations obtained by the BLAST-based alignment in PASC are different from those determined by other algorithms using different datasets and/or different genome regions, and therefore can only be used in the PASC system at NCBI. In another word, one cannot apply the demarcations discussed here to sequence identities calculated by other algorithms such as ClustalX, or using a single gene rather than the whole genome.
Figure 1.
Frequency distribution of pairwise identities for the complete genome sequences of filoviruses, and the application in classifying newly sequenced viruses. The two plots represent results obtained by the BLAST-based alignments and global alignments respectively. The green, yellow and peach bars in the plots represent pairs of genomes that are assigned to the same species, different species but in the same genus, and different genera respectively in the current National Center for Biotechnology Information (NCBI) Taxonomy Database. The top matches for each input genome (JQ352763 and DQ447657) to the existing genomes in GenBank and other input genome are listed, and their pairwise identities are shown. The small red bar on the X-axis of the top plot indicates the percentage of identity of the selected pair (#22 which is highlighted). Not all virus species names listed reflect the most recent International Committee on Taxonomy of Viruses (ICTV) species names.
Figure 1.
Frequency distribution of pairwise identities for the complete genome sequences of filoviruses, and the application in classifying newly sequenced viruses. The two plots represent results obtained by the BLAST-based alignments and global alignments respectively. The green, yellow and peach bars in the plots represent pairs of genomes that are assigned to the same species, different species but in the same genus, and different genera respectively in the current National Center for Biotechnology Information (NCBI) Taxonomy Database. The top matches for each input genome (JQ352763 and DQ447657) to the existing genomes in GenBank and other input genome are listed, and their pairwise identities are shown. The small red bar on the X-axis of the top plot indicates the percentage of identity of the selected pair (#22 which is highlighted). Not all virus species names listed reflect the most recent International Committee on Taxonomy of Viruses (ICTV) species names.
Figure 2.
Dot matrix and text views of pairwise alignment between genome sequences of Ebola virus (NC_002549) and Taï Forest virus (NC_014372), using the BLAST-based and global alignment methods. Not all virus species names listed reflect the most recent ICTV species names.
Figure 2.
Dot matrix and text views of pairwise alignment between genome sequences of Ebola virus (NC_002549) and Taï Forest virus (NC_014372), using the BLAST-based and global alignment methods. Not all virus species names listed reflect the most recent ICTV species names.
2.3. Sub-Species Grouping of Marburgvirus
Although only one species is currently recognized in the genus
Marburgvirus, five major lineages can be found in the phylogenetic analysis of all genomes under the species [
1]. The four more closely related lineages constitute Marburg virus (MARV), and the distant lineage constitutes Ravn virus (RAVV).
The PASC result for the species
Marburg marburgvirus (
Figure 3) supports this division. In
Figure 3, the peak above 97% represents the pairs of viruses within each of the five major lineages; the peak between 91% and 94% represents viruses between the four lineages of MARV; and the peak below 79% represents viruses between RAVV and MARV. Since these peaks are well-separated, PASC can be used to suggest sub-species classifications of newly sequenced marburgviruses.
Similar sub-species patterns are not observed in PASC for ebolaviruses, and the identities of members within each ebolavirus species are above 94% (data not shown), suggesting that intra-species variations of ebolaviruses are less than those of marburgviruses, at least when based on currently available genomes.
Figure 3.
Frequency distribution of pairwise identities from the complete genome sequences of marburgviruses. The three peaks from right to left represent the pairs of viruses within each of the five major lineages of marburgviruses, between the four lineages of Marburg virus (MARV), and between Ravn virus (RAVV) and MARV.
Figure 3.
Frequency distribution of pairwise identities from the complete genome sequences of marburgviruses. The three peaks from right to left represent the pairs of viruses within each of the five major lineages of marburgviruses, between the four lineages of Marburg virus (MARV), and between Ravn virus (RAVV) and MARV.
2.4. Classifying Newly Sequenced Viruses
The use of PASC for classification of viruses with newly sequenced genomes is demonstrated in
Figure 1. The accession numbers of two filovirus genomes, DQ447657 (Marburg virus—Angola2005 isolate Ang0214) and JQ352763 (Ebola virus isolate Mayinga), were entered into the “Sequence” box of the PASC page for the
Filoviridae family, and compared with existing filovirus genomes and with each other.
The nine sequences most similar to DQ447657 are listed in
Figure 1, together with their pairwise identities. The most similar was DQ447653 (Marburg virus—Angola2005 isolate Ang1379c), with a sequence identity of 99.98% by the BLAST-based alignment method. Since this is in the region consisting of green bars, it suggests that DQ447657 and DQ447653 belong to the same species (
i.e., that DQ447657 represents a variant of marburgviruses).
Similarly, a sequence identity of 99.88% between JQ352763 and its closest neighbor, AY354458 (Ebola virus variant Kikwit) suggests that JQ352763 represents a variant of ebolaviruses.
The identity of 33.42% between the two input sequences DQ447657 and JQ352763 is also reported by PASC (see the pair number 22 highlighted in pink background when the number is clicked). Such a report is very useful in determining the relationships among genomes of novel species—if the new genomes are similar enough to each other (i.e., their identity is in the green peaks), they represent the same new species; otherwise, they represent different new species.

{kind=link}
{kind=link}
{kind=link}
{kind=link}


