Biology of Porcine Parvovirus (Ungulate parvovirus 1)

1
Institute for Veterinary Medical Research, Centre for Agricultural Research, Hungarian Academy of Sciences, 1143 Budapest, Hungary
2
Ceva-Phylaxia Zrt., 1107 Budapest, Hungary
3
INRS-Institut Armand-Frappier, Université du Québec, Québec, QC H7V 1B7, Canada
*
Author to whom correspondence should be addressed.
Viruses 2017, 9(12), 393; https://doi.org/10.3390/v9120393
Submission received: 1 December 2017
/
Revised: 17 December 2017
/
Accepted: 18 December 2017
/
Published: 20 December 2017
(This article belongs to the Special Issue Protoparvoviruses: Friends or Foes?)
{kind=link}
{kind=link}
Abstract
:Porcine parvovirus (PPV) is among the most important infectious agents causing infertility in pigs. Until recently, it was thought that the virus had low genetic variance, and that prevention of its harmful effect on pig fertility could be well-controlled by vaccination. However, at the beginning of the third millennium, field observations raised concerns about the effectiveness of the available vaccines against newly emerging strains. Subsequent investigations radically changed our view on the evolution and immunology of PPV, revealing that the virus is much more diverse than it was earlier anticipated, and that some of the “new” highly virulent isolates cannot be neutralized effectively by antisera raised against “old” PPV vaccine strains. These findings revitalized PPV research that led to significant advancements in the understanding of early and late viral processes during PPV infection. Our review summarizes the recent results of PPV research and aims to give a comprehensive update on the present understanding of PPV biology.
1. Introduction
PPV (Porcine Parvovirus (Ungulate parvovirus 1 in the Protoparvirus genus) was first recognized as a member of the Parvoviridae family and causative agent of SMEDI syndrome (stillbirths, mummification, embryonic death, and infertility) at the end of the 1960s [1,2,3]. Although SMEDI had been described a few years earlier [4], the causative agent of the disease was first erroneously identified as a picornavirus. Since its discovery, PPV has remained a constant worldwide problem of the pig industry, still being one of the most common and important infectious agents of infertility. In this review, we concentrate on the results of the last 15 years and try to give an overview of the research that has supplied relevant information about the biology and containment of the virus.
2. Pathogenesis
In most cases, PPV infection alone does not cause clinical symptoms in non-pregnant adult pigs or piglets. Strain virulence is defined by the severity of the reproductive failure it can cause [5].
The outcome of the infection in the fetus varies with the progression of gestation in sows. Experimental and epidemiological studies indicate that PPV infection during the first half of pregnancy can lead to reproductive failure [6,7,8,9,10,11]. Immunocompetent fetuses infected after day 70 of gestation develop an antibody response and usually survive the infection. Vertical transmission to the fetus takes 12 to 18 days after the initial infection of dams by the natural (oral) route [7,8] and somewhat less time by intramuscular injection. Consequently, infection of sows after 56 days of gestation usually does not cause damage to the fetus [12] (Figure 1).
Besides the timing, the genetic makeup of the virus has a determining effect on the result of the fetal infection. Low pathogenic and vaccine strains (e.g., NADL-2 and MSV) cannot cross the placental barrier as efficiently as highly pathogenic strains (e.g., Kresse and 27a) [5,13], so their harmful effect on gestation cannot be detected as frequently as that of the highly pathogenic strains. However, direct injection of NADL-2 into the amnionic fluid can lead to fetal death [14,15].
One of the unanswered questions about PPV-induced reproductive failure is how the virus passes the placental barrier. No evidence was found about the replication of the virus in the uterine epithelium or in the trophectoderm, so the “replicate through the barrier” theory seems improbable. Alternatively, it was suggested that PPV invades the fetus in or on the surface of maternal macrophages. This is hypothesized given that a high number of macrophages reside within the endometrium/placenta during the entire period of gestation, and monocytes and peritoneal macrophages were shown to phagocytize NADL-2 (though they do not support its replication) [16]. In fact, any direct evidence for macrophages crossing from mother to fetus is also missing.
Experimental infections of pregnant sows indicate that more than 10,000 times as much of strain NADL-2 is needed to reach the conceptus transplacentally as of virulent NADL-8 to establish infection [14]. Applications of nucleic acid detection methods revealed significant differences in the distribution and the quantity of viral DNA in embryos infected with different strains. Kresse and NADL-8 were detected by in situ hybridization from the liver, while only Kresse was detected from brain and spleen [17]. Highly virulent 27a was found in high titer (>1011 copy/106 cell) in all 10 organs examined by real-time PCR, while less virulent field (143a) and vaccine strains (NADL-2 and MSV) showed limited tissue distribution and were mainly detected in the kidneys, with much lower titers (~103 copy/106 cell) [15]. These observations suggest that strain differences in tissue specificity, and consequently, in infection-initiating capability in the embryo, might also play a role in the outcome of fetal infection.
3. Virus-Cell Interaction
The in vivo target cells of PPV in neonates and older animals are difficult to determine. The virus can replicate in activated lymphocytes, cannot replicate in blood monocytes, and the results concerning replication ability in macrophages are inconsistent [18,19]. Based on PCR experiments, PPV can propagate in the cells of heart, lung, kidney, spleen, endometrium, and small intestine [20,21]. However, PCR cannot distinguish between the viruses that are produced in the organs, and viruses that are transported there by the vascular system. This may explain the controversial results of detecting PPV in the lymphoid nodes [20,21].
As the first step of entry, the virion binds to terminal sialic acid moieties of the glycoproteins on the cell surface. Both O- and N-linked sialic acids are used for attachment as proved by the rebuilding of neuraminidase-digested sialic acid moieties (digested by (2,3)-O-sialyltransferase and (2,3)-N-sialyltransferase) [22]. Studying entry by inhibitors revealed that besides clathrin-mediated endocytosis and macropinocytosis, a third unknown entry mechanism is likely to be involved in PPV penetration, while the caveolae route does not play any role [22]. PPV has a tendency to aggregate below pH 8. Single particles strongly prefer entry by clathrin-mediated endocytosis, whereas aggregates clearly favor macropinocytosis [22].
The endosomal translocation to the late endosomes or lysosomes, and acidification between 2 and 10 h p.i. seems to be crucial for effective PPV infection [22]. Acidification likely contributes to the externalization of the unique part of the capsidprotein 1 (VP1up) that is normally internalized in mature protoparvovirus particles [23,24]. The VP1up contains a phospholipase A2 (PLA2) domain that plays a crucial role in viral release from the endosomes to the cytoplasm, acting via enzymatic digestion of the membrane phospholipids that destabilize the endosomal membranes [24,25]. This biomimicry of host cellular processes is not unique to parvoviruses. For instance, caliciviruses use cholic acid to activate acid sphingomyelinase (ASM), which cleaves sphingomyelin on the inner leaflet of endosome membranes and produce ceramide. Increased ceramide destabilizes membrane integrity by forming channels or causing lipid flip-flop. This allows the virus to escape [26]. As co-precipitation and inhibitor studies show, ubiquitination and interaction with proteasomes in the cytoplasm are indispensable steps for effective PPV infection, despite the fact that protease degradation of the capsids could not be demonstrated during entry. In the movement of viral particles towards the nucleus, both the microtubule and the actin networks are involved. Microtubules are crucial in the first 8 to 10 h of the infection, suggesting that they have a role in the endosomal transport of PPV to the perinuclear region. Actin activity is needed later (up to 12 to 16 h p.i.) for productive infection, and it is most probably necessary not only for the transport of incoming viruses, but also for the nuclear transport of the newly synthesized proteins [22].
From the five linear basic clusters identified in the VP1up, only three were confirmed to serve as nuclear localization signals (NLS) [27]. The first, located at the amino terminal of the VP1up (from third to ninth amino acid (aa)) is a classic Pat7 NLS, while the other two (between the 122nd and 137th aa) comprise a classic bipartite NLS. Both NLSs are essential for viral replication. Their mutations do not affect viral assembly but abolish productive infection, strongly suggesting that they are responsible for the nuclear transport of the incoming virion in the early phase of infection [27]. Just like in the case of minute virus of mice (MVM), the VP2 of PPV assembles into trimers in the cytoplasm [27,28]. A non-linear nuclear localization motif (NLM) is comprised of four amino acids (K272, K275, K487, and R576) (Figure 2D) on the inner surface of the trimers, and is recognized and transported to the nucleus by the import machinery of the cells, where they assemble to a capsid. It seems that in all protoparvoviruses, both VP1 NLS and VP2 NLM are internalized in the nucleus during viral assembly, and become inaccessible for transporter proteins [27]. Interestingly, MVM was shown to be actively transported out of the nucleus [29]. It is therefore tempting to speculate that the presence of NLM or NLS on the particle would interfere with such transport, and that would be the reason for the concealment of these signals. However, in the case of PPV (and many other protoparvoviruses), there was no sign of any vesicular transport of the assembled virions towards the cell membrane, leaving the question about the functional role of this topological phenomenon open.
Most of the known mutations influencing the biological feature of PPV were found on the capsid protein. The only documented exception is the I481L mutation in the nonstructural 1 (NS1) protein of the NADL-2 strain, which, together with the N348H change of VP2, contributed to the ability of the P2 strain to cause a cytopathic effect and replicate with high titer in the canine cell line A72 [30] (Figure 2A).
The genomes of NADL-2 and Kresse differ by 13 nucleotides (nt) and a 127-nt repeated sequence near the right-end hairpin. There are only six aa differences between the structural proteins of the two viruses, of which five are also present in other virulent field strains (I215T, D378G, H383Q, S436P and R565K). Three of these (D378G, H383Q and S436P) localizing on the capsid surface was enough to abolish NADL-2 replication in primary bovine testis cells (TV) and reduce titer and cytopathic effect to the level of Kresse in cell lines with porcine origin (PT and PFT) [31]. Later, S436P was found not to be involved in tissue tropism in vitro [32]. Since threonine can be detected in position 436 in virulent (27a) and avirulent (143a) strains, this raises the possibility that mutations of aa 378 and 383 of VP2 (Figure 2B) might be enough to modify the pathogenicity of PPV in vivo [5].
More detailed studies with chimeras verified that mainly the capsid proteins determine viral replication efficiency in both porcine and bovine cell lines. However, an interaction between the NS1 protein of the VP coding region and the noncoding repeated sequence may occur and may influence viral replication, most probably through the low-affinity NS1 binding sites scattered throughout the genome. The NADL-2 capsid sequence and repeat region are favorable compared to that of Kresse regarding viral fitness and replication efficiency in vitro [32,33]. However, this is obviously not the case in vivo.
PPV infection facilitates the accumulation of total cellular p53 as early as 3 h p.i. in PK-15 cells (origin form porcine kidney). In the infected cells, p53 activates caspase-9 and caspase-3 through the mitochondria-mediated apoptotic pathway, releasing cytochrome c from the mitochondria [34]. The ratio of apoptotic cells can reach 50% [34,35] in YL strain-infected swein testis (ST) and PK-15 cells at the late phase of infection (60 h p.i.), while in Kresse-infected PT cells, the number of apoptotic cells remains below 14% (as indicated by the number of pyknotic and karyorrhectic cells). Swelling of the infected nuclei, early cell membrane failure (as shown by propidium-iodide uptake), rapid lactate dehydrogenase and free viral DNA release, all point toward necrosis as the main form of cell death in PT cells during Kresse infection [36]. Even subtle mutations of the PPV capsid can modify interactions with host factors and can change the cytopathic effect of the virus [31,33]. Although it is difficult to compare the results of the different experiments that investigated the cytopathic effect of PPV infection, the emerging picture is that it can activate the necrotic and/or apoptotic pathways just like other protoparvoviruses [37], and the actual outcome of the infection largely depends on the viral strain and the cell type. Nevertheless, early cell disintegration accelerates the release and the spread of PPV in vitro. These processes must have highly significant effects on the virus life cycle in vivo, because a small alternatively translated protein (SATp) has evolved in PPV (and other protoparvoviruses) to facilitate quick cell lysis and virus release. The protein is expressed by a leaky scanning mechanism from the same mRNA as VP2, and its start codon is seven nucleotides downstream of the VP2 initiation codon. SATp is an endoplasmic reticulum (ER) resident short membrane protein (68aa) that contains a single membrane-spanning helix and accelerates cell death and viral spread [38]. PPV infection induces an unfolded protein response (UPR) in infected PT cells, regardless of the presence or absence of SATp. It also leads to the activation of the anti-apoptotic, reversible ER stress marker Xbp1 (from 12–14 h p.i.). The marker Xbp1 most probably delays cell death to allow viral synthetic processes to be completed. However, in a later phase, the presence of SATp accelerates cell death by making ER stress irreversible, as shown by the higher rate of expression and the nuclear localization of CHOP (from 22 h p.i.). The involvement of severe ER stress in porcine testis (PT) cell necrosis and viral egress was confirmed by the treatment of infected cells by ER stress-inducing chemicals (MG132, dithiothreitol, and thapsigargin), which accelerated the egress and spreading of both the wild-type and the SAT− viruses [36].
4. Genetic Variation and Evolution
Until the beginning of the 2000s, genetic changes of the PPV genome were not studied systematically due to field observations that the virus remains very stable immunologically, and commercial vaccines developed from ancient strains provide full protection against newly emerging PPV variants. Studies in the first decade of the 21st century seemed to reinforce the conventional view, and even suggested that PPV has a more conservative genome than other parvoviruses [39,40,41].
However, systematic studies in the last fifteen years concentrating on the genetic diversity in VP proteins in field isolates from domestic pigs revealed at least seven clusters, with a predominance of the European strains in clusters C and D, and Chinese strains in cluster F [42].
A similar correlation between clustering and geographical distribution of strains isolated from wild boars was not observed. Fourteen viruses from Romanian wild boars could be distributed into five clusters that grouped together with domestic isolates from all over the world [43]. This finding indicates that PPVs of wild boar populations are more diverse than viruses of domestic pigs in the same area. It also suggests that the pressure driving PPV evolution in domestic pig and wild boar population are fundamentally not so different, wild boar- or domestic pig-specific PPV strain clusters have not yet evolved, and/or viruses more or less freely shuttle between subspecies [43]. Field observations and experimental investigations of some of the new highly virulent isolates of cluster D revealed that antisera raised against “old” PPV vaccine strains cannot effectively neutralize these viruses [5,44]. It was hypothesized that these mutants might have emerged by escaping immune pressure forced by vaccination. However, neither in vitro nor in silico studies could find any evidence to support this idea. A decrease in genetic diversity of PPV was observed in the presence of antibodies in tissue culture or in vaccinated herds as modeled from available sequence data. Mutations found on immune selected capsids (NADL-2 I320S, H383Q; Str. Challenge S45T, P436S) were not present on novel variants either [45]. The authors concluded that vaccine failures and non-vaccinated animals (e.g., wild boars) may have a more important impact on the emergence of new phenotypes than vaccinated populations.
No obvious correlation could be observed between clustering (phylogeny) and virulence status of PPV isolates. Closely related highly virulent and less virulent strains can be found in most of the clusters [5,14,42], suggesting that the ability of field strains to effectively cross the placental barrier and kill the fetus has a secondary role on viral spreading and PPV evolution. It seems that other, less studied biological features (e.g., tissue specificity and long-term/high titer shedding), which in certain cases may be directly or indirectly influenced by virulence, determine the fitness of a PPV strain under highly variable field conditions.
The application of molecular clock models in independent investigations predicted that current strains are the result of relatively recent evolutionary events, and main branches started to diverge from each other in the last 10–60 years [42,43,46]. However, it remained elusive how the predicted sole ancestor of the present strains evolved prior to 60 years ago. Another analysis estimated the age of the most recent common ancestor of PPV stains to be around 250 years, and suggested that Western colonialization could have contributed to the spread of PPV [47].
Investigations of the mutation patterns of the NS and VP genes of field strains revealed the contrasting evolution of the two coding regions. The mutation rate of VP is 30–50 times higher (3–5 × 10−4 mutation/nucleotide/year) than that of one of the NS genes (10−5) [46,47]. This, together with the negative difference between non-synonymous and synonymous substitution rates (dN–dS) found in the NS region, suggests that the number of nucleotide changes that allow the sustained functionality of the NS proteins is limited, and that purifying selection determines the evolution of the region. In contrast, earlier investigations suggested that the VP1/VP2 gene evolves under a near-neutral model (drift) [48], but several positions are under positive selection, especially on the outer loops of the capsid that determine cellular interactions and immunogenicity [46,47,48]. In addition, the majority of mutations were found to be in the surface loops [46,48,49,50], among them, 215, 228, 383, 414, 419, and 436 seem to be the main variable sites in the capsid [47]. However, more recent investigations on higher numbers of samples have given a more nuanced picture, finding no mutation hotspots between loops and β-strands [42], and showing that complete VP1 is also under purifying selection [42,43,47].
A three aa surface substitution is characteristic of the members of cluster D (Q228E, E419Q, and S436T), including 27a (Figure 2C). Position 228 is part one of the nine known linear epitopes on VP2 [51]. However, the contribution of these aa’s to the apparent immune escape feature of 27a remains to be studied [5].
It is a widely accepted theory that the main reason behind CpG depletion in small DNA viruses is due to natural selection coming from replicative advantage and/or immune escape [52,53]. The PPV genome is CpG-depleted, yet neither experimental nor in silico investigations can confirm these assumptions in the case of PPV. The lack of measurable biological effect after introducing additional CpGs into the PPV genome argued against the replicative advantage of CpG depletion, and the ascendant distribution of CpGs by position does not support the presence of immunological pressure against CpGs. These data, taken together with the findings that CpG sites are more prone to be mutated than GC or C and G sites in the PPV genome, suggest that mutational pressure, rather than a selective force, is responsible for CpG underrepresentation in the PPV genome [54].
5. Immunity and Prevention
Hardly anything is known about the role of cellular immunity in controlling PPV infection. The only investigation made in this direction found weak cytotoxic T cell (CTL) activity, suggesting that T-cell activation may occur with repeated exposure to the virus, but confirmed that effective clearance of PPV infection is obtained by rapid antibody response in infected pigs [55].
Many immunological studies proved that the presence of neutralizing serum antibodies is a decisive factor in the outcome of the PPV infection [5,44,55,56]. Since the placenta is impermeable to maternal antibodies, neonatal piglets obtain passive protection against PPV1 by taking up maternally derived antibodies (MDA) from the colostrum. A strong correlation can be observed between MDA levels in piglets and the anti-PPV colostrum antibody levels in their dams. The amount of MDA decreases steadily in piglets as their age progresses. Previous studies have shown that MDA levels last up to 14–26 (mean 21) weeks in pigs [7]. Etoh et al. [57] found that a high titer of MDA after birth may persist until the 22nd week, while the low level of MDA can be exhausted within nine weeks. A recent study has demonstrated that maternal protection terminates somewhat sooner [58]. This investigation has found that PPV1-specific antibodies are detectable in 93.6% of the one-week-old piglets after colostrum uptake. At 57 days of age, 35.3% of pigs carry detectable amounts of PPV1-specific antibody, while at 87 days only 1.5% of pigs do the same. These values have been verified by a field survey conducted in a Hungarian herd (Cságola personal communication).
High levels of passive antibodies can prevent infection, while lower levels can reduce dissemination of PPV from infected pigs [56,59]. Nonetheless, in a PPV-affected, unvaccinated herd, sooner or later the majority of the piglets will be infected with the virus by lateral transmission [58] as indicated by the high occurrence of seropositive gilts (86%) [58]. Despite the presence of antibodies at high titer, relatively high virus copy number (>104/mL) can be detected in some animals until day 21 p.i., and viremia can endure until day 42 p.i. [60]. Another systematic experiment indirectly supports this observation, finding that infected pigs can shed the virus in body excretions (e.g., feces, nasal discharge) from the 4th day p.i. for at least 49 days, though the quantity significantly decreases after two weeks [61].
PPV infection activates IFN-γ and IFN-α synthesis [60,62], and the seronegative pigs start antibody production shortly after infection. PPV-specific antibodies can be detected as early as day 6 p.i. [60], and the antibody titer peaks on days 14–21 [3,12,63,64]. An in vitro study demonstrated that the transcription of the potent B cell differentiation factor IL-6 is stimulated through the toll-like receptor 9 (TLR9)-mediated NF-κB signaling pathway in infected cells. It is additionally suggested that IFN-α might also be activated through these pathways during infection. The expression levels of 17 immune-related miRNAs, including miR-10b, miR-20a, miR-19b, miR-181a, miR-146b, and miR-18a were found to be significantly altered in PK-15 cells during PPV infection. These miRNAs were shown earlier to be involved in the regulation of at least six immune response pathways, TLRs and NF-κB among them [65].
The inactivated vaccines used currently are based on NADL-2 (cluster A) and similar strains, and were isolated 40 years ago. These vaccines are effective against homologous infections, but do not prevent infection and virus shedding after challenge with the antigenically heterologous 27a (cluster D) strain [44]. However, they can protect the fetus against disease. Infection of 27a in pigs or the inoculation of the virus into rabbits induced 100- to 1000-fold lower homologous neutralizing antibody titers than heterologous titers against 143a, NADL-2 or MSV strains [5]. Vaccination by inactivated 27a prevented fetal death after homologous virus challenge with PPV-27a. However, a substantial increase in antibody titers was detected after infection, indicating virus replication in the immunized animals [61]. These experiments suggest that 27a has unique immunological features, and although present vaccines are able to prevent disease, inactivated vaccines cannot induce the desired sterile immunity against 27a [61]. Modified live-virus or vector vaccines inducing cellular immunity might be alternative approaches. However, due to the past success of the inactivated vaccines and the complication and cost of licensing genetically modified organisms, only a few attempts were made in this direction. Genetically modified Lactobacillus and pseudorabies virus expressing the PPV VP2 protein were shown to induce neutralizing antibodies against PPV, but in vivo challenge experiments for testing vaccine efficacy are still missing [66,67].
6. Co-Infection with Circovirus
The post-weaning multisystemic wasting syndrome (PMWS) was first identified in western Canada in the 1990s [68]. Although not all Porcine circovirus type 2 (PCV2) infected animals develop disease symptoms, it was demonstrated that PCV-2 alone could induce the clinical signs of PMWS in cesarean-derived, colostrum-deprived or specific pathogen free (SPF) pigs [69,70]. Laboratory experiments and field studies have shown that co-infection with PPV and other pathogens potentiates the effect of PCV-2 in the development of PMWS [71,72,73,74]. However, even simultaneous co-infection of SPF piglets with PPV and PCV-2 [62,75,76] does not necessarily lead to the manifestations of PMWS. The age, management factors, immunological status, and the time of infection can significantly modify the outcome of the co-infection [62]. Early PCV-2 (prenatal or colostral transmission) infection followed by PPV over-infection within a few weeks seems to significantly increase the onset of PMWS in serologically negative animals [75,77]. However, serological analysis of both experimentally-infected and field samples suggests that the presence of serum antibodies against any of the two viruses reduce the risk of developing PMWS [76,78].
PPV might facilitate PCV-2 infection either indirectly, by reducing immunoprotection or/and stimulating virus replication by host cell activation, or directly, by promoting virus DNA replication infecting the same host cells. In fact, both PCV-2 and PPV were shown to replicate in lymphocytes [18,19,79] and detected in (inguinal) lymph nodes during co-infection [75]. However, the indirect involvement of PPV seems more probable given that non-viral immunostimulation of PCV-2-infected animals also promotes PMWS, over-infection with PPV seems more effective to induce the disease than co-infection [62,75,76], and PPV has been associated with transient immunosuppressive effects [17,18,19].
7. Detection and Isolation of PPV
The virus can agglutinate chicken, guinea pig, mouse, human, monkey, rat, and cat erythrocytes. Thus, hemagglutination inhibition assays (HAI) were developed relatively early [64,80] and they are still used in research and practice in some countries [5,44,81,82]. Other serological methods like the serum neutralization (SN) assay or the modified direct complement-fixation (MDCF) test were also utilized earlier, but today the enzyme-linked immunosorbent assay (ELISA) is the most frequently applied test to detect PPV-specific antibodies. A recombinant NS1 protein-based DIVA test (Differentiating Infected from Vaccinated Animals) was also developed to distinguish vaccinated (inactivated vaccine) from infected pigs [83].
Sensitive nucleic acid detection methods can provide accurate information about the presence and the quantity of the virus in the animals. Swab (nasal, rectal, genital tract), blood, kidney, lung, lymph nodes and tissues from abortion material are used most frequently for DNA preparation. Using real-time PCR, a detection limit of 20–500 copies could be achieved [15,84,85,86]. The loop-mediated isothermal amplification (LAMP) assay, nanoPCR, and recombinase polymerase amplification (RPA) assay, were also successfully applied for PPV detection with a very low detection limit (5, 56, 300 copies per reaction respectively) [87,88,89]. However, the application of these methods in the everyday practice is rare or nonexistent. Recent diagnostic developments focus on the detection of several porcine pathogens in one PCR tube, including RNA viruses. PPV can be detected together with the pseudorabies virus, classical swine fever virus, African swine fever virus, porcine reproductive and respiratory syndrome virus, porcine circovirus type 2, or Japanese encephalitis virus in multiplex (reverse transcriptase) PCRs [90,91,92,93,94,95,96].
For the isolation of PPV established cell lines, such as swine testis (ST and PT), pig fallopian tube (PFT) and pig kidney (PK-13, PK-15) cells can be used [2,97]. Though some PPV isolates are able to replicate in Cos7 (African green monkey) [27], KB (human) [98], or A72 (canine) cells [30], porcine cells are much more susceptible than cell cultures originating from other species. Depending on the strain, 500–10,000 packaged genome copies are needed to initiate infection in PT cells [36,38], which seem to be the most sensitive to PPV infection [31]. Contaminated trypsin, derived from the pancreas of PPV-infected pigs, was a major cause of virus transmission into cell cultures before suppliers started to test for the presence of PPV. This was probably also the case for the KBSH strain isolated from KB cells. This strain was one of several parvoviruses recovered from permanent human cell lines [98].
Acknowledgments
The research was supported by NFKI K108607 and NFKI K119381.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cartwrigh, S.; Huck, R. Viruses isolated in association with herd infertility abortions and stillbirths in pigs. Vet. Rec. 1967, 81, 196–197. [Google Scholar]
- Cartwright, S.F.; Lucas, M.; Huck, R. A small haemagglutinating porcine DNA virus: I. Isolation and properties. J. Comp. Pathol. 1969, 79. [Google Scholar] [CrossRef]
- Johnson, R.; Collings, D. Experimental infection of piglets and pregnant gilts with a parvovirus. Vet. Rec. 1969, 85, 446–447. [Google Scholar] [CrossRef] [PubMed]
- Dunne, H.; Gobble, J.; Hokanson, J.; Kradel, D.; Bubash, G. Porcine reproductive failure associated with a newly identified “SMEDI” group of picorna viruses. Am. J. Vet. Res. 1965, 26, 1284–1297. [Google Scholar] [PubMed]
- Zeeuw, E.J.L.; Leinecker, N.; Herwig, V.; Selbitz, H.J.; Truyen, U. Study of the virulence and cross-neutralization capability of recent porcine parvovirus field isolates and vaccine viruses in experimentally infected pregnant gilts. J. Gen. Virol. 2007, 88, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Mengeling, W.; Cutlip, R. Pathogenesis of in utero infection: Experimental infection of five-week-old porcine fetuses with porcine parvovirus. Am. J. Vet. Res. 1975, 36, 1173–1177. [Google Scholar] [PubMed]
- Joo, H.; Donaldson-Wood, C.; Johnson, R. Observations on the pathogenesis of porcine parvovirus infection. Arch. Virol. 1976, 51, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Mengeling, W.L.; Paul, P.S.; Brown, T.T. Transplacental infection and embryonic death following maternal exposure to porcine parvovirus near the time of conception. Arch. Virol. 1980, 65, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Donaldson-Wood, C.; Joo, H.; Johnson, R. The effect on reproductive performance of porcine parvovirus infection in a susceptible pig herd. Vet. Rec. 1977, 100, 237–239. [Google Scholar] [CrossRef] [PubMed]
- Mengeling, W. Prevalence of porcine parvovirus-induced reproductive failure: An abattoir study. J. Am. Vet. Med. Assoc. 1978, 172, 1291–1294. [Google Scholar] [PubMed]
- Mengeling, W.L.; Lager, K.M.; Zimmerman, J.K.; Samarikermani, N.; Beran, G.W. A current assessment of the role of porcine parvovirus as a cause of fetal porcine death. J. Vet. Diagn. Investig. 1991, 3, 33–35. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.; Donaldson-Wood, C.; JOD, H.; Allender, U. Observations on the epidemiology of porcine parvovirus. Aust. Vet. J. 1976, 52, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Kresse, J.I.; Taylor, W.D.; Stewart, W.W.; Eernisse, K.A. Parvovirus infection in pigs with necrotic and vesicle-like lesions. Vet. Microbiol. 1985, 10, 525–531. [Google Scholar] [CrossRef]
- Mengeling, W.L.; Pejsak, Z.; Paul, P.S. Biological assay of attenuated strain NADL-2 and virulent strain NADL-8 of porcine parvovirus. Am. J. Vet. Res. 1984, 45, 2403–2407. [Google Scholar] [PubMed]
- Wilhelm, S.; Zeeuw, E.J.L.; Selbitz, H.J.; Truyen, U. Tissue distribution of two field isolates and two vaccine strains of porcine parvovirus in foetal organs after experimental infection of pregnant sows as determined by real-time PCR. J. Vet. Med. B 2005, 52, 323–326. [Google Scholar] [CrossRef] [PubMed]
- Mengeling, W.; Lager, K.; Vorwald, A. The effect of porcine parvovirus and porcine reproductive and respiratory syndrome virus on porcine reproductive performance. Anim. Reprod. Sci. 2000, 60, 199–210. [Google Scholar] [CrossRef]
- Oraveerakul, K.; Choi, C.S.; Molitor, T.W. Tissue tropisms of porcine parvovirus in swine. Arch. Virol. 1993, 130, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Paul, P.S.; Mengeling, W.L.; Brown, T.T. Replication of Porcine Parvovirus in Peripheral-Blood Lymphocytes, Monocytes, and Peritoneal-Macrophages. Infect. Immun. 1979, 25, 1003–1007. [Google Scholar] [PubMed]
- Harding, M.J.; Molitor, T.W. Porcine parvovirus: Replication in and inhibition of selected cellular functions of swine alveolar macrophages and peripheral blood lymphocytes. Arch. Virol. 1988, 101, 105–117. [Google Scholar] [CrossRef] [PubMed]
- McKillen, J.; Hjertner, B.; Millar, A.; McNeilly, F.; Beldak, S.; Adair, B.; Allan, G. Molecular beacon real-time PCR detection of swine viruses. J. Virol. Methods 2007, 140, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.F.; Zhang, C.F.; Chen, C.M.; Cui, S.J. Real-time PCR to detect and analyze virulent PPV loads in artificially challenged sows and their fetuses. Vet. Microbiol. 2009, 138, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Boisvert, M.; Fernandes, S.; Tijssen, P. Multiple pathways involved in porcine parvovirus cellular entry and trafficking toward the nucleus. J. Virol. 2010, 84, 7782–7792. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; D’Abramo, A.M.; Ticknor, C.M.; Tattersall, P. Controlled conformational transitions in the MVM virion expose the VP1 N-terminus and viral genome without particle disassembly. Virology 1999, 254, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Canaan, S.; Zadori, Z.; Ghomashchi, F.; Bollinger, J.; Sadilek, M.; Moreau, M.E.; Tijssen, P.; Gelb, M.H. Interfacial enzymology of parvovirus phospholipases A2. J. Biol. Chem. 2004, 279, 14502–14508. [Google Scholar] [CrossRef] [PubMed]
- Zádori, Z.; Szelei, J.; Lacoste, M.-C.; Li, Y.; Gariépy, S.; Raymond, P.; Allaire, M.; Nabi, I.R.; Tijssen, P. A viral phospholipase A 2 is required for parvovirus infectivity. Dev. Cell 2001, 1, 291–302. [Google Scholar] [CrossRef]
- Shivanna, V.; Kim, Y.; Chang, K.O. Ceramide formation mediated by acid sphingomyelinase facilitates endosomal escape of caliciviruses. Virology 2015, 483, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Boisvert, M.; Bouchard-Levesque, V.; Fernandes, S.; Tijssen, P. Classic nuclear localization signals and a novel nuclear localization motif are required for nuclear transport of porcine parvovirus capsid proteins. J. Virol. 2014, 88, 11748–11759. [Google Scholar] [CrossRef] [PubMed]
- Riolobos, L.; Reguera, J.; Mateu, M.G.; Almendral, J.M. Nuclear transport of trimeric assembly intermediates exerts a morphogenetic control on the icosahedral parvovirus capsid. J. Mol. Biol. 2006, 357, 1026–1038. [Google Scholar] [CrossRef] [PubMed]
- Bar, S.; Daeffler, L.; Rommelaere, J.; Nuesch, J.P. Vesicular egress of non-enveloped lytic parvoviruses depends on gelsolin functioning. PLoS Pathog. 2008, 4, e1000126. [Google Scholar] [CrossRef] [PubMed]
- Pirtle, E.C. Titration of two porcine respiratory viruses in mammalian cell cultures by direct fluorescent antibody staining. Am. J. Vet. Res. 1974, 35, 249–250. [Google Scholar] [PubMed]
- Bergeron, J.; Hebert, B.; Tijssen, P. Genome organization of the Kresse strain of porcine parvovirus: Identification of the allotropic determinant and comparison with those of NADL-2 and field isolates. J. Virol. 1996, 70, 2508–2515. [Google Scholar] [PubMed]
- Fernandes, S.; Boisvert, M.; Tijssen, P. Genetic elements in the VP region of porcine parvovirus are critical to replication efficiency in cell culture. J. Virol. 2011, 85, 3025–3029. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, S.; Boisvert, M.; Szelei, J.; Tijssen, P. Differential replication of two porcine parvovirus strains in bovine cell lines ensues from initial DNA processing and NS1 expression. J. Gen. Virol. 2014, 95, 910–921. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.L.; Huang, Y.; Du, Q.; Luo, X.M.; Zhang, L.; Zhao, X.M.; Tong, D.W. Porcine parvovirus infection induces apoptosis in PK-15 cells through activation of p53 and mitochondria-mediated pathway. Biochem. Biophys. Res. Commun. 2015, 456, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.M.; Xiang, H.L.; Bai, X.Y.; Fei, N.J.; Huang, Y.; Song, X.J.; Zhang, H.L.; Zhang, L.; Tong, D.W. Porcine parvovirus infection activates mitochondria-mediated apoptotic signaling pathway by inducing ROS accumulation. Virol. J. 2016, 13. [Google Scholar] [CrossRef] [PubMed]
- Meszaros, I.; Toth, R.; Olasz, F.; Tijssen, P.; Zadori, Z. The SAT Protein of Porcine Parvovirus Accelerates Viral Spreading through Induction of Irreversible Endoplasmic Reticulum Stress. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.Y.; Qiu, J. Parvovirus infection-induced cell death and cell cycle arrest. Future Virol. 2010, 5, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Zadori, Z.; Szelei, J.; Tijssen, P. SAT: A late NS protein of porcine parvovirus. J. Virol. 2005, 79, 13129–13138. [Google Scholar] [CrossRef] [PubMed]
- Lukashov, V.V.; Goudsmit, J. Evolutionary relationships among parvoviruses: Virus-host coevolution among autonomous primate parvoviruses and links between adeno-associated and avian parvoviruses. J. Virol. 2001, 75, 2729–2740. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Bueno, A.; Villarreal, L.P.; Almendral, J.M. Parvovirus variation for disease: A difference with RNA viruses? Curr. Top. Microbiol. Immunol. 2006, 299, 349–370. [Google Scholar] [PubMed]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Streck, A.F.; Canal, C.W.; Truyen, U. Molecular epidemiology and evolution of porcine parvoviruses. Infect. Genet. Evol. 2015, 36, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Cadar, D.; Dan, A.; Tombacz, K.; Lorincz, M.; Kiss, T.; Becskei, Z.; Spinu, M.; Tuboly, T.; Csagola, A. Phylogeny and evolutionary genetics of porcine parvovirus in wild boars. Infect. Genet. Evol. 2012, 12, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Jóźwik, A.; Manteufel, J.; Selbitz, H.-J.; Truyen, U. Vaccination against porcine parvovirus protects against disease, but does not prevent infection and virus shedding after challenge infection with a heterologous virus strain. J. Gen. Virol. 2009, 90, 2437–2441. [Google Scholar] [CrossRef] [PubMed]
- Streck, A.F.; Homeier, T.; Foerster, T.; Truyen, U. Population dynamics and in vitro antibody pressure of porcine parvovirus indicate a decrease in variability. J. Gen. Virol. 2013, 94, 2050–2055. [Google Scholar] [CrossRef] [PubMed]
- Streck, A.F.; Bonatto, S.L.; Homeier, T.; Souza, C.K.; Goncalves, K.R.; Gava, D.; Canal, C.W.; Truyen, U. High rate of viral evolution in the capsid protein of porcine parvovirus. J. Gen. Virol. 2011, 92, 2628–2636. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Tao, Y.; Cui, J.; Suo, S.; Cong, Y.; Tijssen, P. Phylogeny and evolution of porcine parvovirus. Virus Res. 2013, 178, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Shangjin, C.; Cortey, M.; Segales, J. Phylogeny and evolution of the NS1 and VP1/VP2 gene sequences from porcine parvovirus. Virus Res. 2009, 140, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Martins Soares, R.; Cortez, A.; Heinemann, M.B.; Sakamoto, S.M.; Martins, V.G.; Bacci, M., Jr.; De Campos Fernandes, F.M.; Richtzenhain, L.J. Genetic variability of porcine parvovirus isolates revealed by analysis of partial sequences of the structural coding gene VP2. J. Gen. Virol. 2003, 84, 1505–1515. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, P.; Ritzmann, M.; Selbitz, H.J.; Heinritzi, K.; Truyen, U. VP1 sequences of German porcine parvovirus isolates define two genetic lineages. J. Gen. Virol. 2006, 87, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Kamstrup, S.; Langeveld, J.; Botner, A.; Nielsen, J.; Schaaper, W.M.M.; Boshuizen, R.S.; Casal, J.I.; Hojrup, P.; Vela, C.; Meloen, R.; et al. Mapping the antigenic structure of porcine parvovirus at the level of peptides. Virus Res. 1998, 53, 163–173. [Google Scholar] [CrossRef]
- Hoelzer, K.; Shackelton, L.A.; Parrish, C.R. Presence and role of cytosine methylation in DNA viruses of animals. Nucleic Acids Res. 2008, 36, 2825–2837. [Google Scholar] [CrossRef] [PubMed]
- Shackelton, L.A.; Parrish, C.R.; Holmes, E.C. Evolutionary basis of codon usage and nucleotide composition bias in vertebrate DNA viruses. J. Mol. Evol. 2006, 62, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Toth, R.; Meszaros, I.; Stefancsik, R.; Bartha, D.; Balint, A.; Zadori, Z. CpG Distribution and Methylation Pattern in Porcine Parvovirus. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Ladekjaer-Mikkelsen, A.S.; Nielsen, J. A longitudinal study of cell-mediated immunity in pigs infected with porcine parvovirus. Viral Immunol. 2002, 15, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Paul, P.S.; Mengeling, W.L.; Brown, T.T. Effect of Vaccinal and Passive-Immunity on Experimental-Infection of Pigs with Porcine Parvovirus. Am. J. Vet. Res. 1980, 41, 1368–1371. [Google Scholar] [PubMed]
- Etoh, M.; Morishita, E.; Ochiai, M.; Watanabe, Y. Transitional antibodies and spontaneous infection in the porcine (Sus scrofa) parvo viral infections. Jpn. J. Swine Husb. Res. (Japan) 1979, 16, 237–239. [Google Scholar]
- Gava, D.; Souza, C.K.; Mores, T.J.; Argenti, L.E.; Streck, A.F.; Canal, C.W.; Bortolozzo, F.P.; Wentz, I. Dynamics of vanishing of maternally derived antibodies of Ungulate protoparvovirus 1 suggests an optimal age for gilts vaccination. Trop. Anim. Health Prod. 2017, 49, 1085–1088. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Fujisaki, Y. Immunizing effects of inactivated porcine parvovirus vaccine on piglets. Bull. Natl. Inst. Anim. Health 1976, 16, 81. [Google Scholar]
- Opriessnig, T.; Gerber, P.F.; Matzinger, S.R.; Meng, X.J.; Halbur, P.G. Markedly different immune responses and virus kinetics in littermates infected with porcine circovirus type 2 or porcine parvovirus type 1. Vet. Immunol. Immunopathol. 2017, 191, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Foerster, T.; Streck, A.F.; Speck, S.; Selbitz, H.-J.; Lindner, T.; Truyen, U. An inactivated whole-virus porcine parvovirus vaccine protects pigs against disease but does not prevent virus shedding even after homologous virus challenge. J. Gen. Virol. 2016, 97, 1408–1413. [Google Scholar] [CrossRef] [PubMed]
- Steiner, E.; Balmelli, C.; Gerber, H.; Summerfield, A.; McCullough, K. Cellular adaptive immune response against porcine circovirus type 2 in subclinically infected pigs. BMC Vet. Res. 2009, 5. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.; Collings, D. Transplacental infection of piglets with a porcine parvovirus. Res. Vet. Sci. 1971, 12, 570–572. [Google Scholar] [PubMed]
- Cartwright, S.F.; Lucas, M.; Huck, R. A small haemagglutinating porcine DNA virus: II. Biological and serological studies. J. Comp. Pathol. 1971, 81, 145–155. [Google Scholar] [CrossRef]
- Li, X.; Zhu, L.; Liu, X.; Sun, X.; Zhou, Y.; Lang, Q.; Li, P.; Cai, Y.; Qiao, X.; Xu, Z. Differential expression of micrornas in porcine parvovirus infected porcine cell line. Virol. J. 2015, 12, 128. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.G.; Cui, L.C.; Tian, C.Y.; Zhang, G.C.; Huo, G.C.; Tang, L.J.; Li, Y.J. Immunogenicity of Recombinant Classic Swine Fever Virus CD8(+) T Lymphocyte Epitope and Porcine Parvovirus VP2 Antigen Coexpressed by Lactobacillus casei in Swine via Oral Vaccination. Clin. Vaccine Immunol. 2011, 18, 1979–1986. [Google Scholar] [CrossRef] [PubMed]
- Hong, Q.; Qian, P.; Li, X.M.; Yu, X.L.; Chen, H.C. A recombinant pseudorabies virus co-expressing capsid proteins precursor P1-2A of FMDV and VP2 protein of porcine parvovirus: A trivalent vaccine candidate. Biotechnol. Lett. 2007, 29, 1677–1683. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.; Hassard, L.; Clark, E.; Harding, J.; Allan, G.; Willson, P.; Strokappe, J.; Martin, K.; McNeilly, F.; Meehan, B. Isolation of circovirus from lesions of pigs with postweaning multisystemic wasting syndrome. Can. Vet. J. 1998, 39, 44–51. [Google Scholar] [PubMed]
- Krakowka, S.; Ellis, J.A.; Meehan, B.; Kennedy, S.; McNeilly, F.; Allan, G. Viral wasting syndrome of swine: Experimental reproduction of postweaning multisystemic wasting syndrome in gnotobiotic swine by coinfection with porcine circovirus 2 and porcine parvovirus. Vet. Pathol. 2000, 37, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Rose, N.; Larour, G.; Le Diguerher, G.; Eveno, E.; Jolly, J.P.; Blanchard, P.; Oger, A.; Le Dimna, M.; Jestin, A.; Madec, F. Risk factors for porcine post-weaning multisystemic wasting syndrome (PMWS) in 149 French farrow-to-finish herds. Prev. Vet. Med. 2003, 61, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Allan, G.M.; Kennedy, S.; McNeilly, F.; Foster, J.C.; Ellis, J.A.; Krakowka, S.J.; Meehan, B.M.; Adair, B.M. Experimental reproduction of severe wasting disease by co-infection of pigs with porcine circovirus and porcine parvovirus. J. Comp. Pathol. 1999, 121, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.; Chae, C. Distribution of porcine parvovirus in porcine circovirus 2-infected pigs with postweaning multisystemic wasting syndrome as shown by in-situ hybridization. J. Comp. Pathol. 2000, 123, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.A.; Bratanich, A.; Clark, E.G.; Allan, G.; Meehan, B.; Haines, D.M.; Harding, J.; West, K.H.; Krakowka, S.; Konoby, C.; et al. Coinfection by porcine circoviruses and porcine parvovirus in pigs with naturally acquired postweaning multisystemic wasting syndrome. J. Vet. Diagn. Investig. 2000, 12, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, S.; Moffett, D.; McNeilly, F.; Meehan, B.; Ellis, J.; Krakowka, S.; Allan, G.M. Reproduction of lesions of postweaning multisystemic wasting syndrome by infection of conventional pigs with porcine circovirus type 2 alone or in combination with porcine parvovirus. J. Comp. Pathol. 2000, 122, 9–24. [Google Scholar] [CrossRef] [PubMed]
- Ha, Y.; Lee, Y.H.; Ahn, K.K.; Kim, B.; Chae, C. Reproduction of postweaning multisystemic wasting syndrome in pigs by prenatal porcine circovirus 2 infection and postnatal porcine parvovirus infection or immunostimulation. Vet. Pathol. 2008, 45, 842–848. [Google Scholar] [CrossRef] [PubMed]
- Ostanello, F.; Caprioli, A.; di Francesco, A.; Battilani, M.; Sala, G.; Sarli, G.; Mandrioli, L.; McNeilly, F.; Allan, G.M.; Prosperi, S. Experimental infection of 3-week-old conventional colostrum-fed pigs with porcine circovirus type 2 and porcine parvovirus. Vet. Microbiol. 2005, 108, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Ha, Y.; Shin, J.H.; Chae, C. Colostral transmission of porcine circovirus 2 (PCV-2): Reproduction of post-weaning multisystemic wasting syndrome in pigs fed milk from PCV-2-infected sows with post-natal porcine parvovirus infection or immunostimulation. J. Gen. Virol. 2010, 91, 1601–1608. [Google Scholar] [CrossRef] [PubMed]
- Grau-Roma, L.; Stockmarr, A.; Kristensen, C.S.; Enoe, C.; Lopez-Soria, S.; Nofrarias, M.; Bille-Hansen, V.; Hjulsager, C.K.; Sibila, M.; Jorsal, S.E.; et al. Infectious risk factors for individual postweaning multisystemic wasting syndrome (PMWS) development in pigs from affected farms in Spain and Denmark. Res. Vet. Sci. 2012, 93, 1231–1240. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, R.E.; Nauwynck, H.J.; McNeilly, F.; Allan, G.M.; Pensaert, M.B. Porcine circovirus 2 infection in swine foetuses inoculated at different stages of gestation. Vet. Microbiol. 2001, 83, 169–176. [Google Scholar] [CrossRef]
- Joo, H.S.; Donaldson-Wood, C.R.; Johnson, R.H. A standardised haemagglutination inhibition test for porcine parvovirus antibody. Aust. Vet. J. 1976, 52, 422–424. [Google Scholar] [CrossRef] [PubMed]
- Jeoung, H.Y.; Lim, S.I.; Kim, J.J.; Cho, Y.Y.; Kim, Y.K.; Song, J.Y.; Hyun, B.H.; An, D.J. Serological prevalence of viral agents that induce reproductive failure in South Korean wild boar. BMC Vet. Res. 2015, 11. [Google Scholar] [CrossRef] [PubMed]
- Phuong, C.T.B.; Le, H.T.M. Prevalence of Antibodies to Porcine Parvovirus in Swine in Hanoi and Its Vicinity. In Proceedings of the Conference on Agriculture Development in the Context of International Integration: Opportunities and Challenges, Hanoi, Vietnam, 7–8 December 2016; p. 101. [Google Scholar]
- Qing, L.; Lv, J.; Li, H.; Tan, Y.; Hao, H.; Chen, Z.; Zhao, J.; Chen, H. The recombinant nonstructural polyprotein NS1 of porcine parvovirus (PPV) as diagnostic antigen in ELISA to differentiate infected from vaccinated pigs. Vet. Res. Commun. 2006, 30, 175–190. [Google Scholar] [CrossRef] [PubMed]
- Song, C.P.; Zhu, C.; Zhang, C.F.; Cui, S.J. Detection of porcine parvovirus using a taqman-based real-time pcr with primers and probe designed for the NS1 gene. Virol. J. 2010, 7. [Google Scholar] [CrossRef] [PubMed]
- Streck, A.F.; Hergemoller, F.; Ruster, D.; Speck, S.; Truyen, U. A TaqMan qPCR for quantitation of Ungulate protoparvovirus 1 validated in several matrices. J. Virol. Methods 2015, 218, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.Q.; Cai, X.Q.; Lin, Z.X.; Li, X.L.; Yue, Q.Y.; Li, R.; Zhu, X.Q. Rapid and specific detection of porcine parvovirus using real-time PCR and High Resolution Melting (HRM) analysis. BMC Vet. Res. 2015, 11. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.T.; Zhang, J.; Yang, S.H.; Ma, L.N.; Ma, Y.P.; Liu, X.T.; Cai, X.P.; Zhang, Y.G.; Liu, Y.S. Rapid detection of porcine parvovirus DNA by sensitive loop-mediated isothermal amplification. J. Virol. Methods 2009, 158, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Wang, Z.; Ma, X.; Liu, J.; Cui, S. A sensitive and specific nanoparticle-assisted PCR assay for rapid detection of porcine parvovirus. Lett. Appl. Microbiol. 2014, 58, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Qin, X.D.; Zhang, W.; Li, Y.M.; Zhang, Z.D. Rapid and specific detection of porcine parvovirus by isothermal recombinase polymerase amplification assays. Mol. Cell. Probes 2016, 30, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.G.; Chen, G.D.; Huang, Y.; Ding, L.; Li, Z.C.; Chang, C.D.; Wang, C.Y.; Tong, D.W.; Liu, H.J. Development of multiplex PCR for simultaneous detection of six swine DNA and RNA viruses. J. Virol. Methods 2012, 183, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.L.; Wang, Y.B.; Li, M.F.; Chen, H.Y.; Guo, X.P.; Geng, J.W.; Wang, Z.Y.; Wei, Z.Y.; Cui, B.A. Simultaneous detection of porcine parvovirus and porcine circovirus type 2 by duplex real-time PCR and amplicon melting curve analysis using SYBR Green. J. Virol. Methods 2013, 187, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.B.; Wu, H.G.; Jiang, Y.H.; Opriessnig, T.; Zheng, X.W.; Mo, Y.C.; Yang, Z.Q. Development of an EvaGreen-based multiplex real-time PCR assay with melting curve analysis for simultaneous detection and differentiation of six viral pathogens of porcine reproductive and respiratory disorder. J. Virol. Methods 2014, 208, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.G.; Rao, P.B.; Jiang, Y.H.; Opriessnig, T.; Yang, Z.Q. A sensitive multiplex real-time PCR panel for rapid diagnosis of viruses associated with porcine respiratory and reproductive disorders. Mol. Cell. Probes 2014, 28, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.Y.; Liu, Z.J.; Wang, W.C.; Tang, D.Y.; Liang, H.Y.; Liu, Z. Establishment and application of a multiplex PCR for rapid and simultaneous detection of six viruses in swine. J. Virol. Methods 2014, 208, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.X.; Xie, Z.X.; Xie, L.J.; Deng, X.W.; Xie, Z.Q.; Luo, S.S.; Liu, J.B.; Pang, Y.S.; Khan, M.I. Simultaneous detection of eight swine reproductive and respiratory pathogens using a novel GeXP analyser-based multiplex PCR assay. J. Virol. Methods 2015, 224, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Lin, X.Y.; Nie, F.P.; Yang, Z.X.; Yao, X.P.; Li, G.L.; Wu, X.L.; Ren, M.S.; Wang, Y. Simultaneous typing of seven porcine pathogens by multiplex PCR with a GeXP analyser. J. Virol. Methods 2016, 232, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Heldt, C.L.; Hernandez, R.; Mudiganti, U.; Gurgel, P.V.; Brown, D.T.; Carbonell, R.G. A colorimetric assay for viral agents that produce cytopathic effects. J. Virol. Methods 2006, 135, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Siegl, G.; Hallauer, C.; Novak, A. Paroviruses as contaminants of permanent human cell lines. IV. Multiplication of KBSH-virus in KB-cells. Arch. Gesamte Virusforsch. 1971, 36, 351–362. [Google Scholar] [CrossRef]
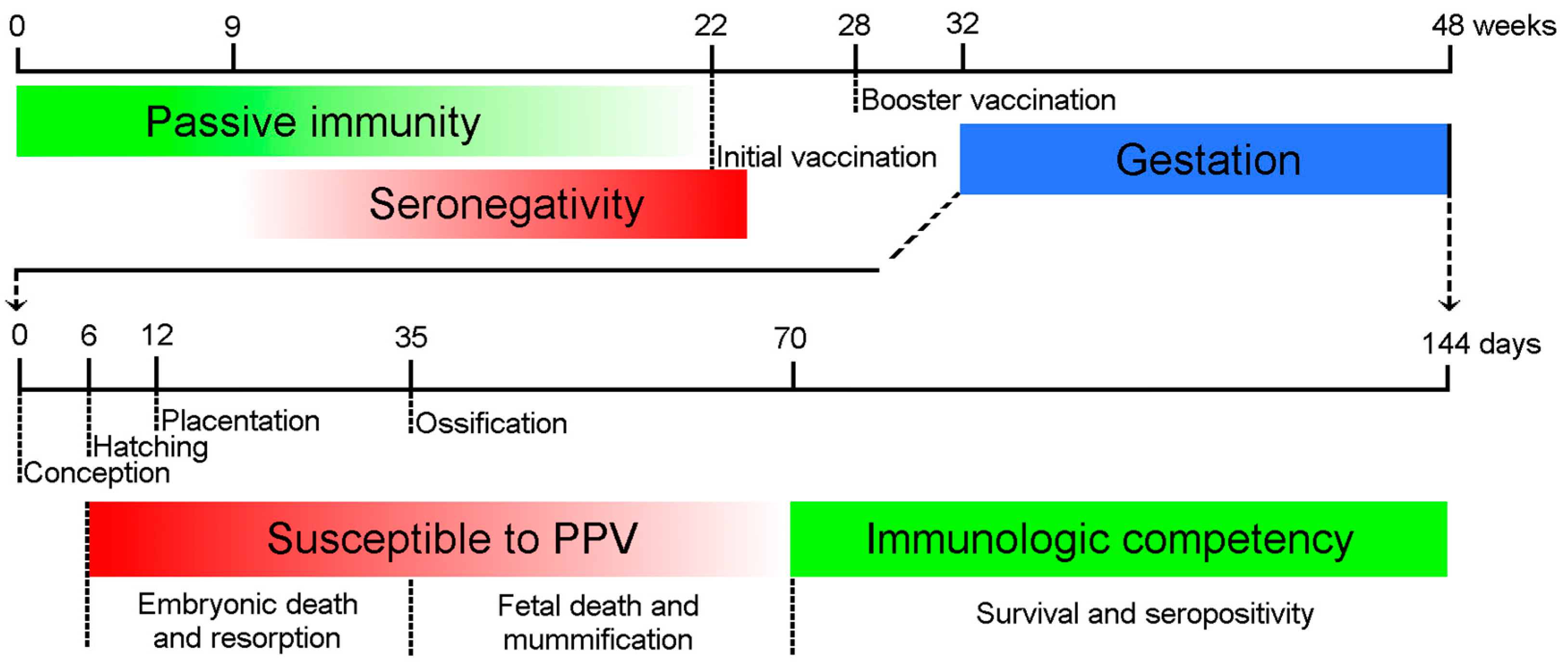
Figure 1.
Major time points of Porcine Parvovirus (PPV) infection and immune response. The maternal antibodies protect the piglets passively (green line) until 9–22 weeks of age. The animals should be inoculated first after the depletion of maternal antibodies, and long-term immunity is maintained by booster vaccination. The embryos are susceptible to the infection (red line) until the Development of immune competence around day 70 day of gestation (green line). The consequences of the intrauterine infection depend on the time of the infection.
Figure 1.
Major time points of Porcine Parvovirus (PPV) infection and immune response. The maternal antibodies protect the piglets passively (green line) until 9–22 weeks of age. The animals should be inoculated first after the depletion of maternal antibodies, and long-term immunity is maintained by booster vaccination. The embryos are susceptible to the infection (red line) until the Development of immune competence around day 70 day of gestation (green line). The consequences of the intrauterine infection depend on the time of the infection.
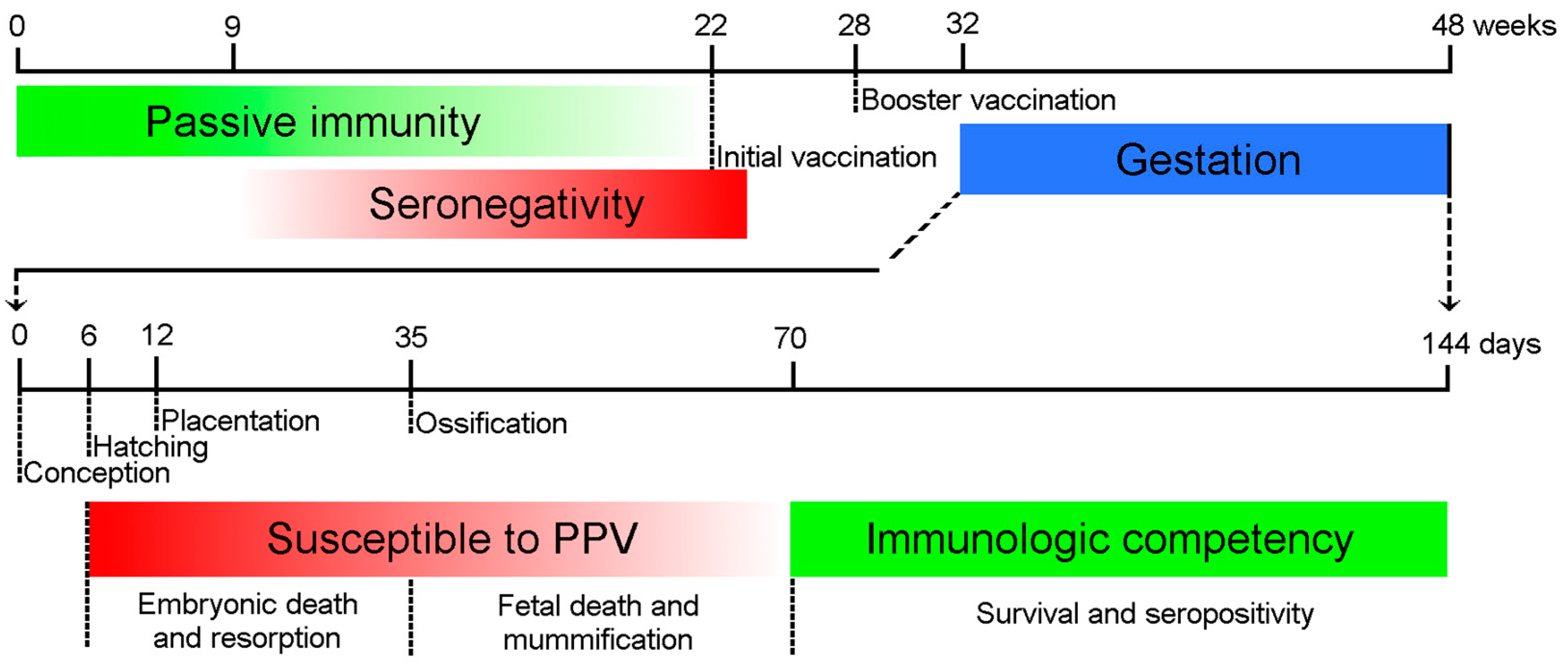
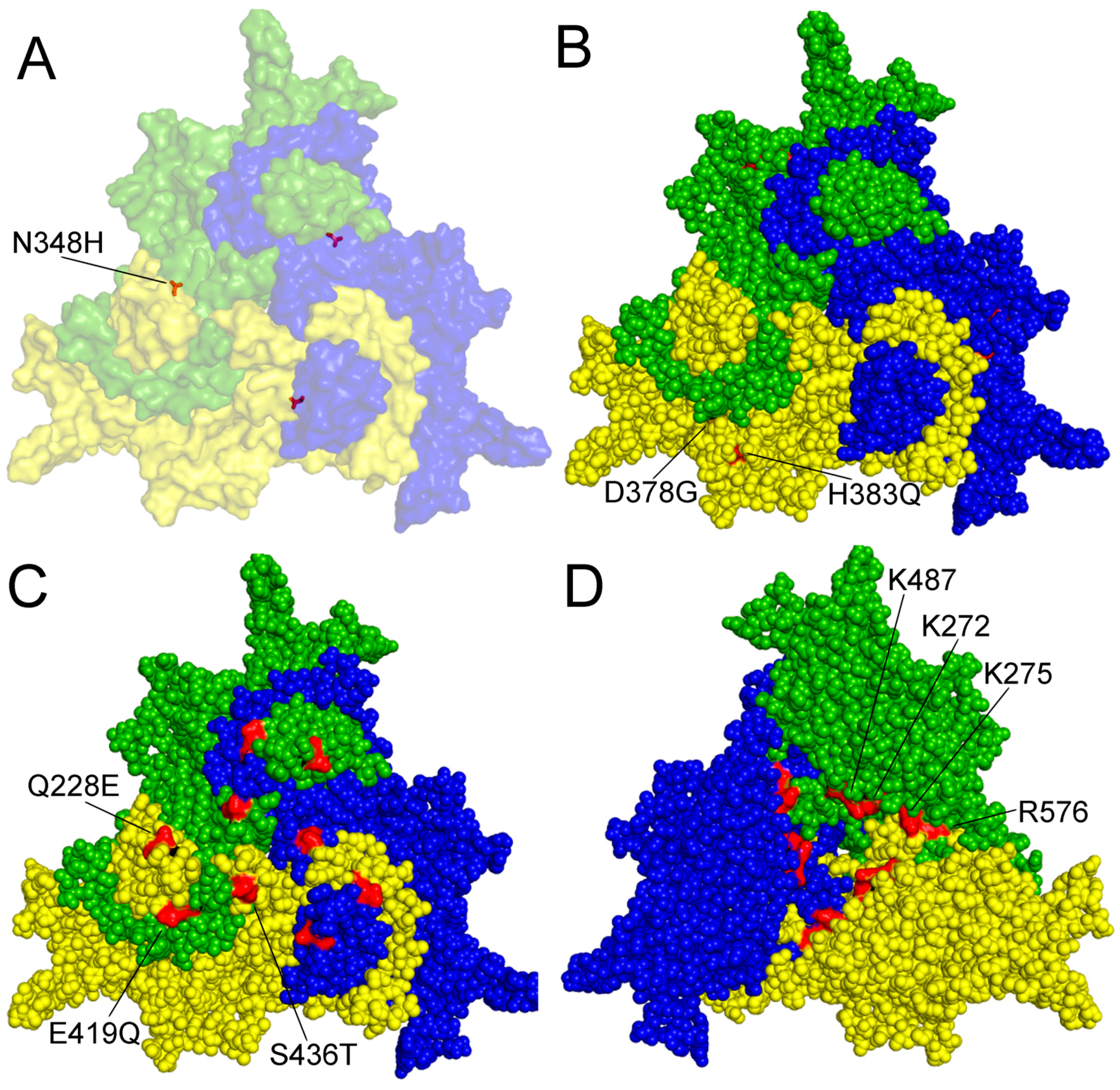
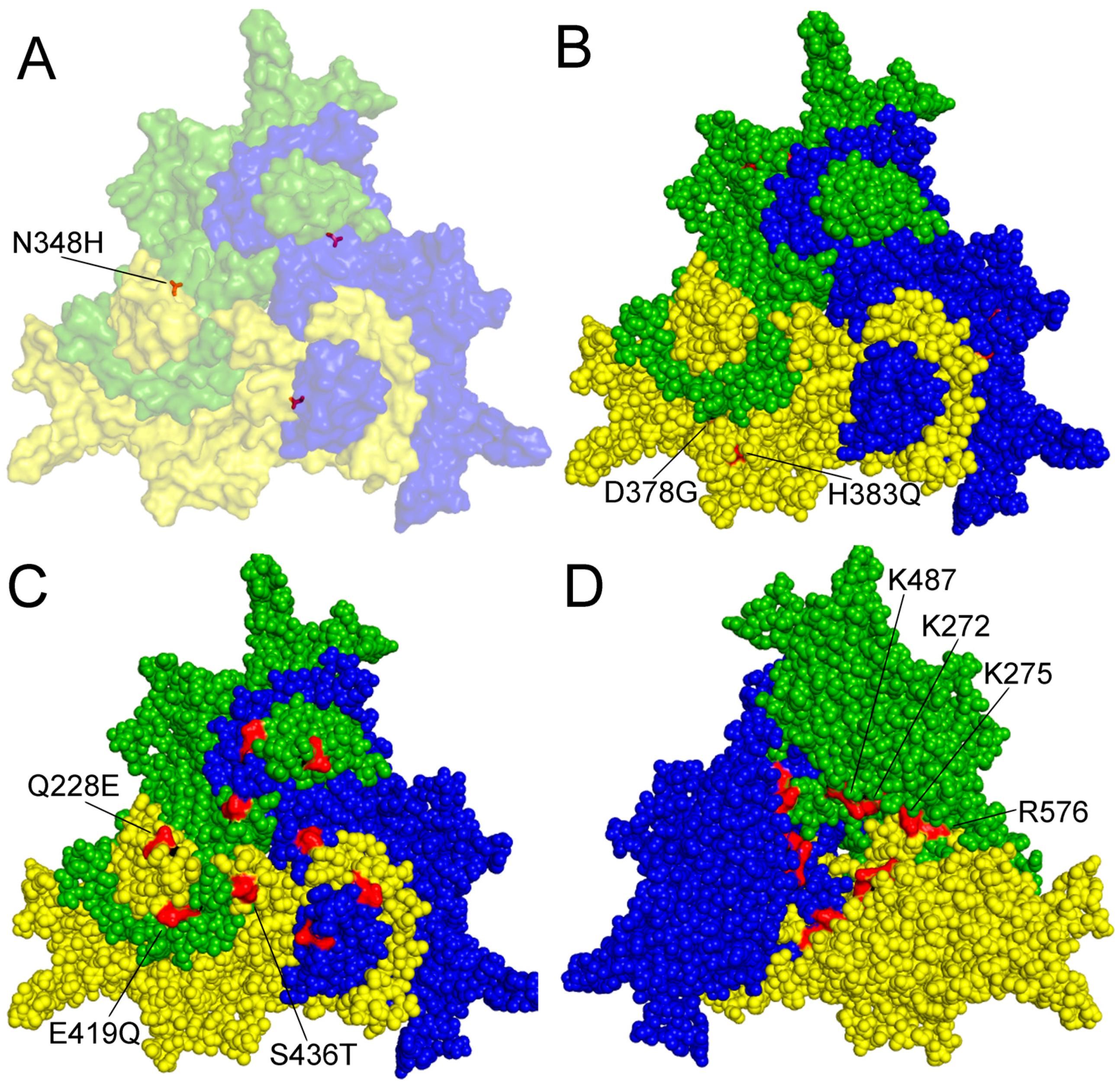
Figure 2.
Amino acid residues with known function on the surface of a capsid protein 2 (VP2) trimer of PPV. A–C outer surface is shown. (A) amino acid (aa) 348 highlighted contributing to P2 replication in the canine cell line A72; (B) aa 378 and 383 are involved in tissue tropism and probably in virulence; (C) aa 228E, 419Q and 436T are characteristic of the members of the D cluster; (D) aa K272, K275, K487 and R576 form a nuclear localization motif on the inner surface of the trimer. Numbering is presented according to NADL-2 VP2 sequence, amino acid changes labeled by commonly used colloquial nomenclature. Trimer was generated by Viper program and visualized by Polivew.
Figure 2.
Amino acid residues with known function on the surface of a capsid protein 2 (VP2) trimer of PPV. A–C outer surface is shown. (A) amino acid (aa) 348 highlighted contributing to P2 replication in the canine cell line A72; (B) aa 378 and 383 are involved in tissue tropism and probably in virulence; (C) aa 228E, 419Q and 436T are characteristic of the members of the D cluster; (D) aa K272, K275, K487 and R576 form a nuclear localization motif on the inner surface of the trimer. Numbering is presented according to NADL-2 VP2 sequence, amino acid changes labeled by commonly used colloquial nomenclature. Trimer was generated by Viper program and visualized by Polivew.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mészáros, I.; Olasz, F.; Cságola, A.; Tijssen, P.; Zádori, Z. Biology of Porcine Parvovirus (Ungulate parvovirus 1). Viruses 2017, 9, 393. https://doi.org/10.3390/v9120393
AMA Style
Mészáros I, Olasz F, Cságola A, Tijssen P, Zádori Z. Biology of Porcine Parvovirus (Ungulate parvovirus 1). Viruses. 2017; 9(12):393. https://doi.org/10.3390/v9120393
Chicago/Turabian StyleMészáros, István, Ferenc Olasz, Attila Cságola, Peter Tijssen, and Zoltán Zádori. 2017. "Biology of Porcine Parvovirus (Ungulate parvovirus 1)" Viruses 9, no. 12: 393. https://doi.org/10.3390/v9120393
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.