The Influence of Pressure on the Intrinsic Dissolution Rate of Amorphous Indomethacin

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Preparation of Amorphous Indomethacin (IND)
2.2.2. X-ray Powder Diffraction (XRPD)
2.2.3. Intrinsic Dissolution Studies
Dissolution Procedure
UV Analysis
Data Analysis
3. Results and Discussion
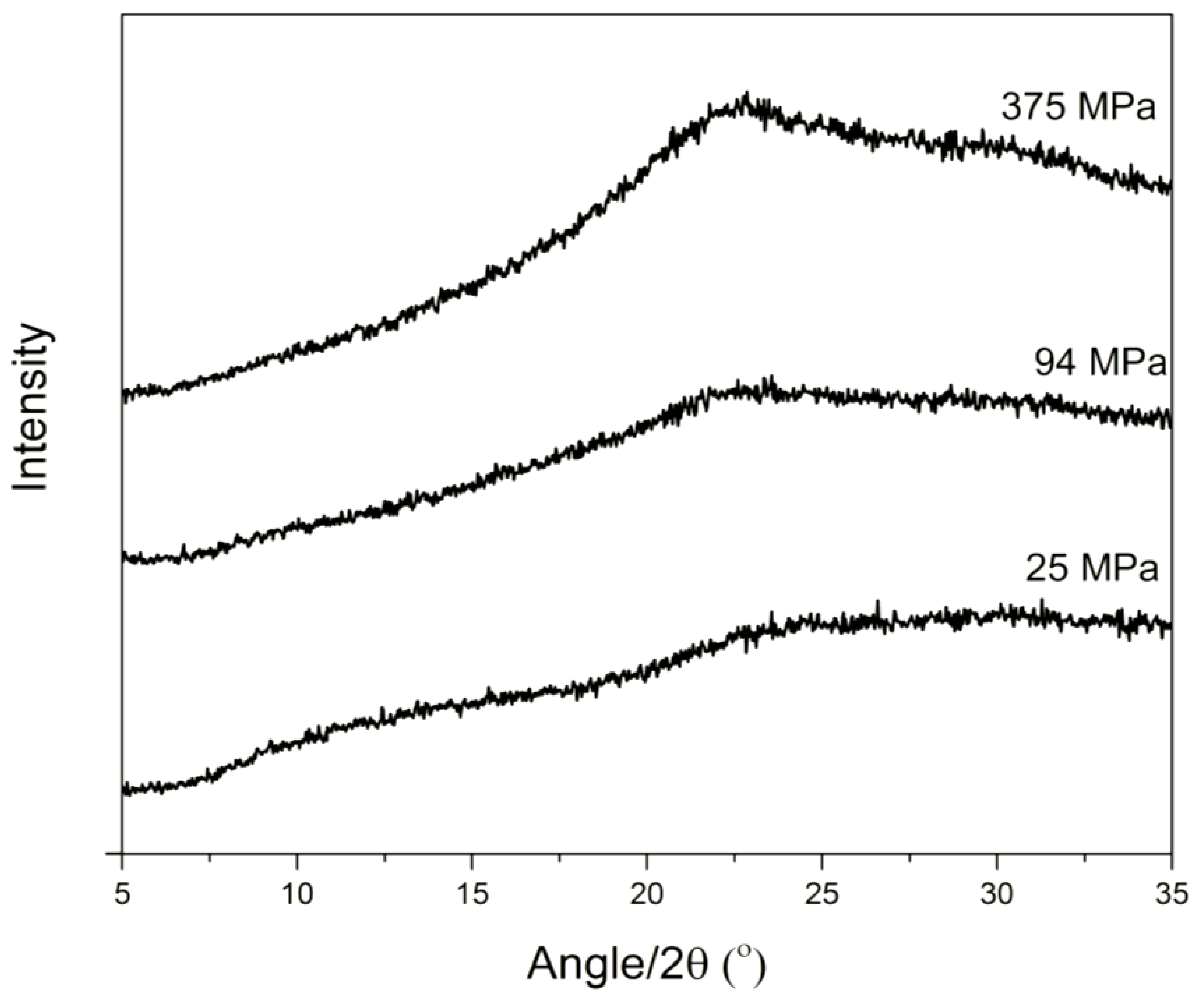
3.1. Solid State Analysis


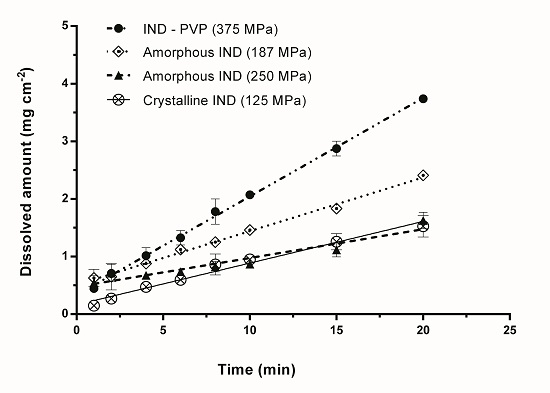
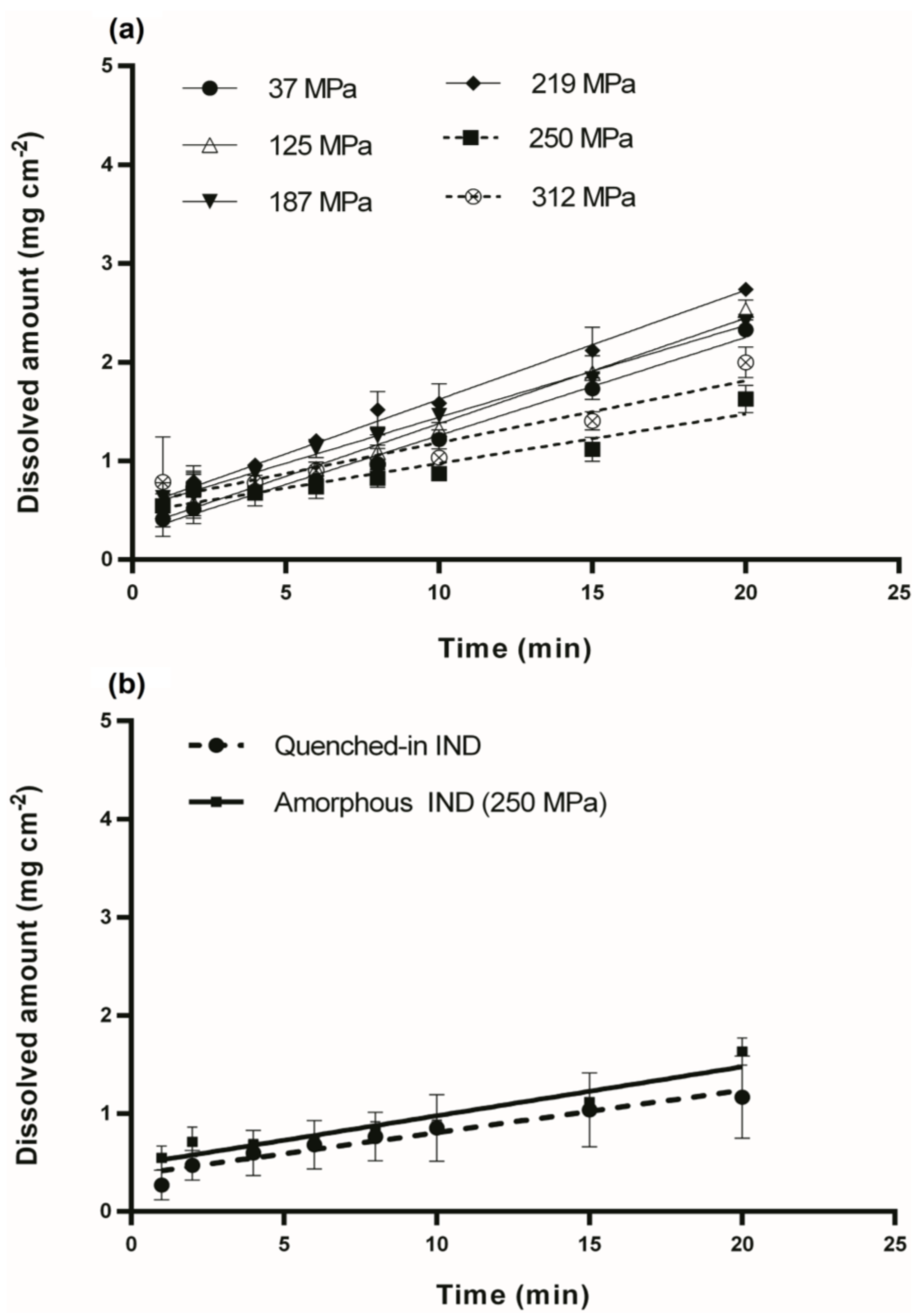
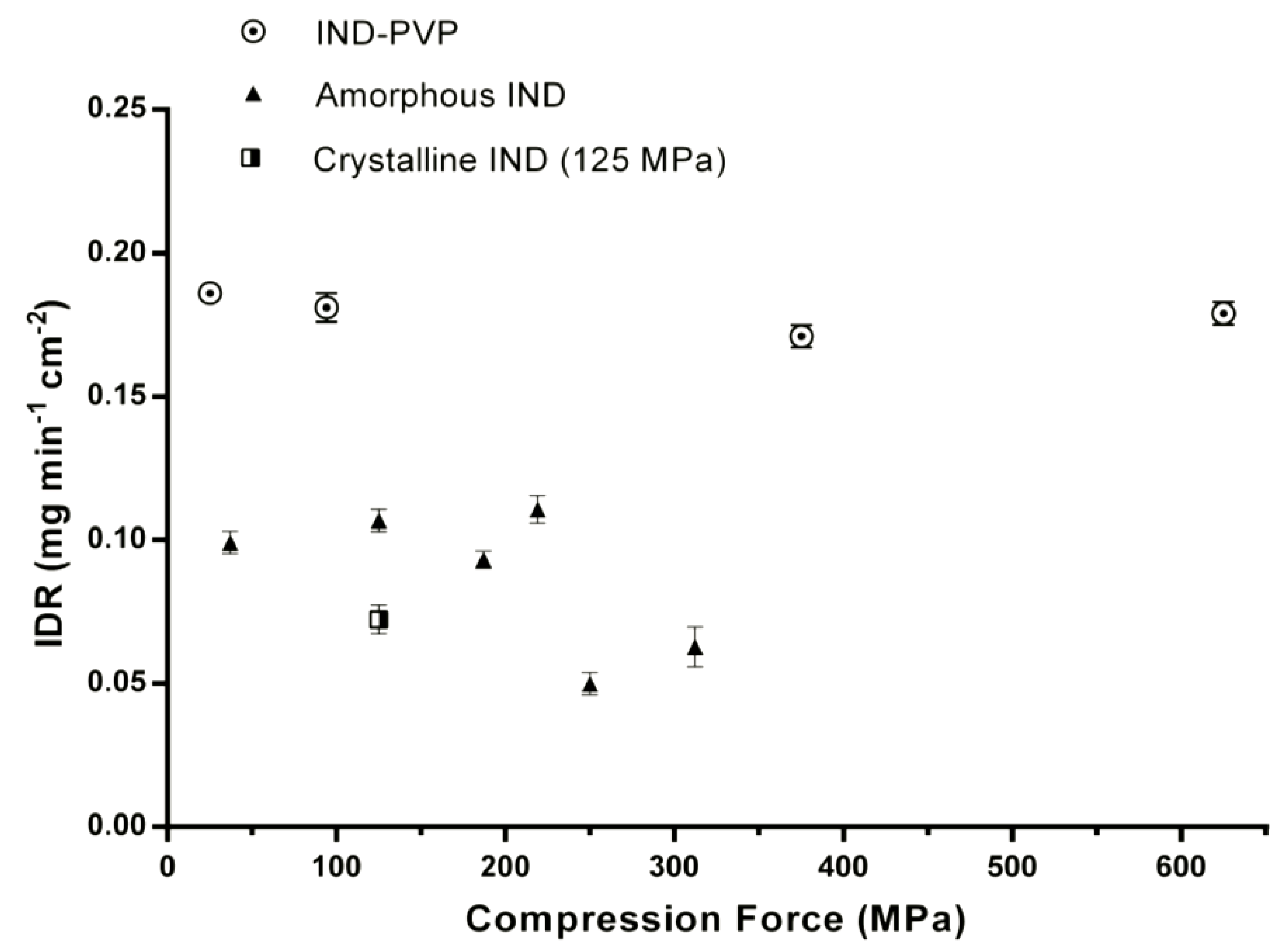
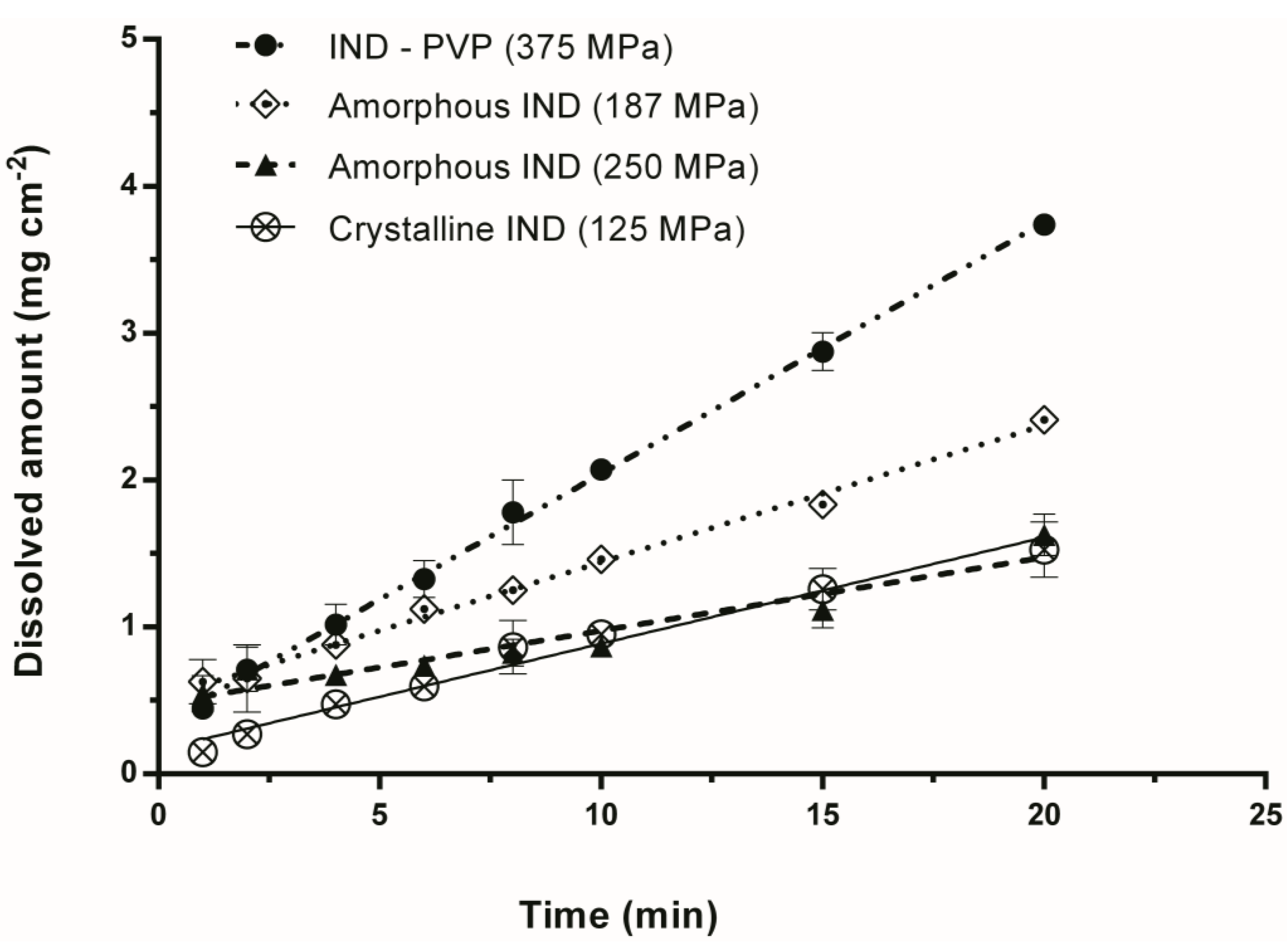
3.2. Intrinsic Dissolution Studies




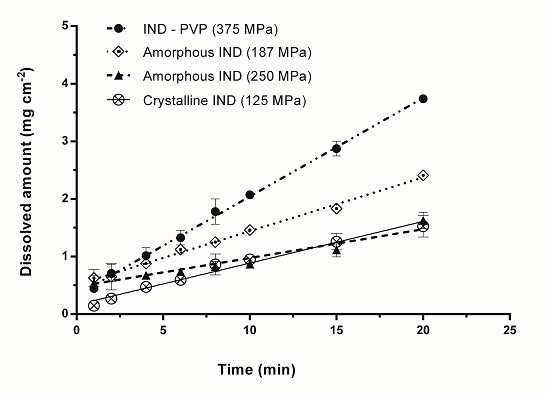{kind=link}
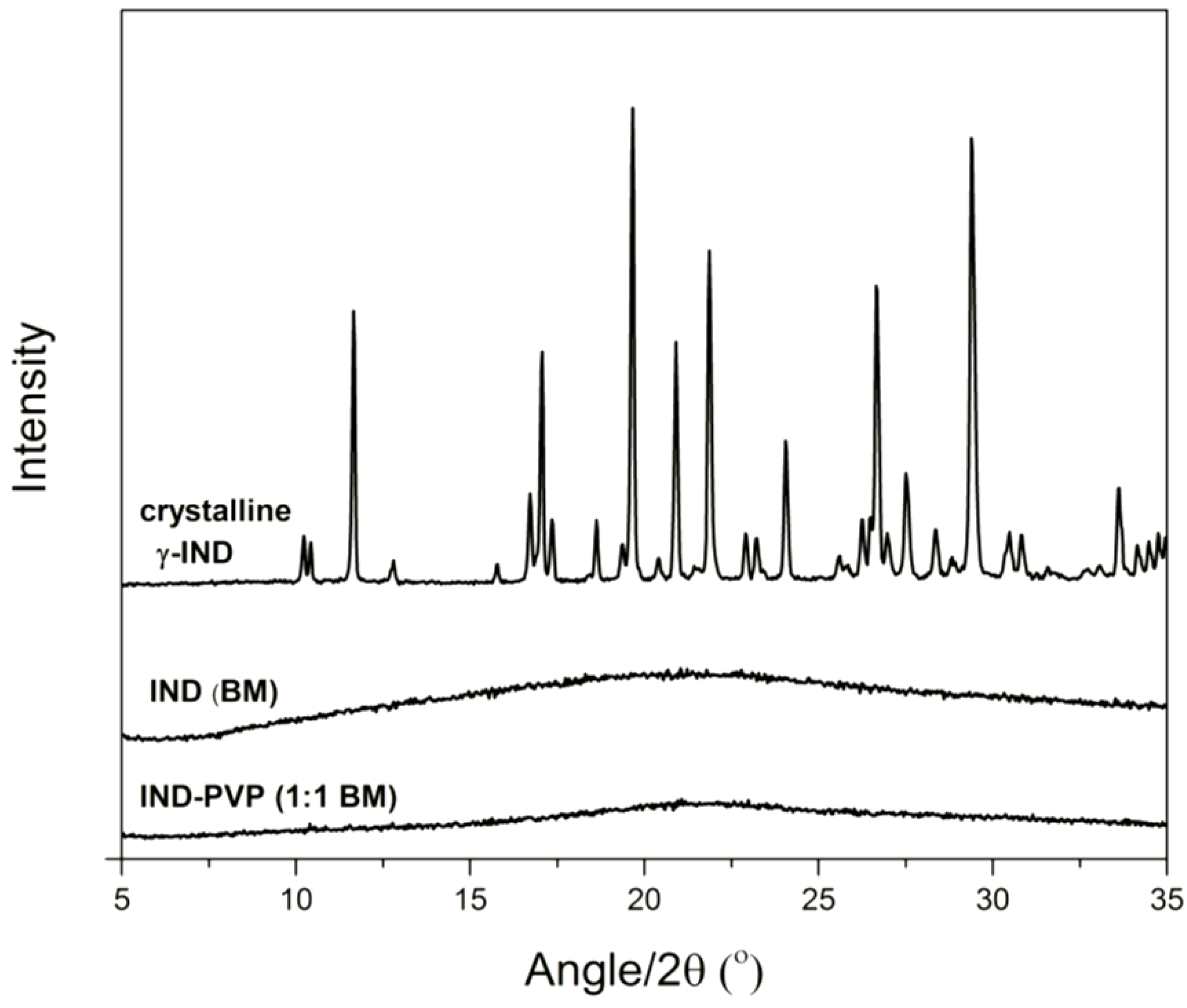{kind=link}
{kind=link}
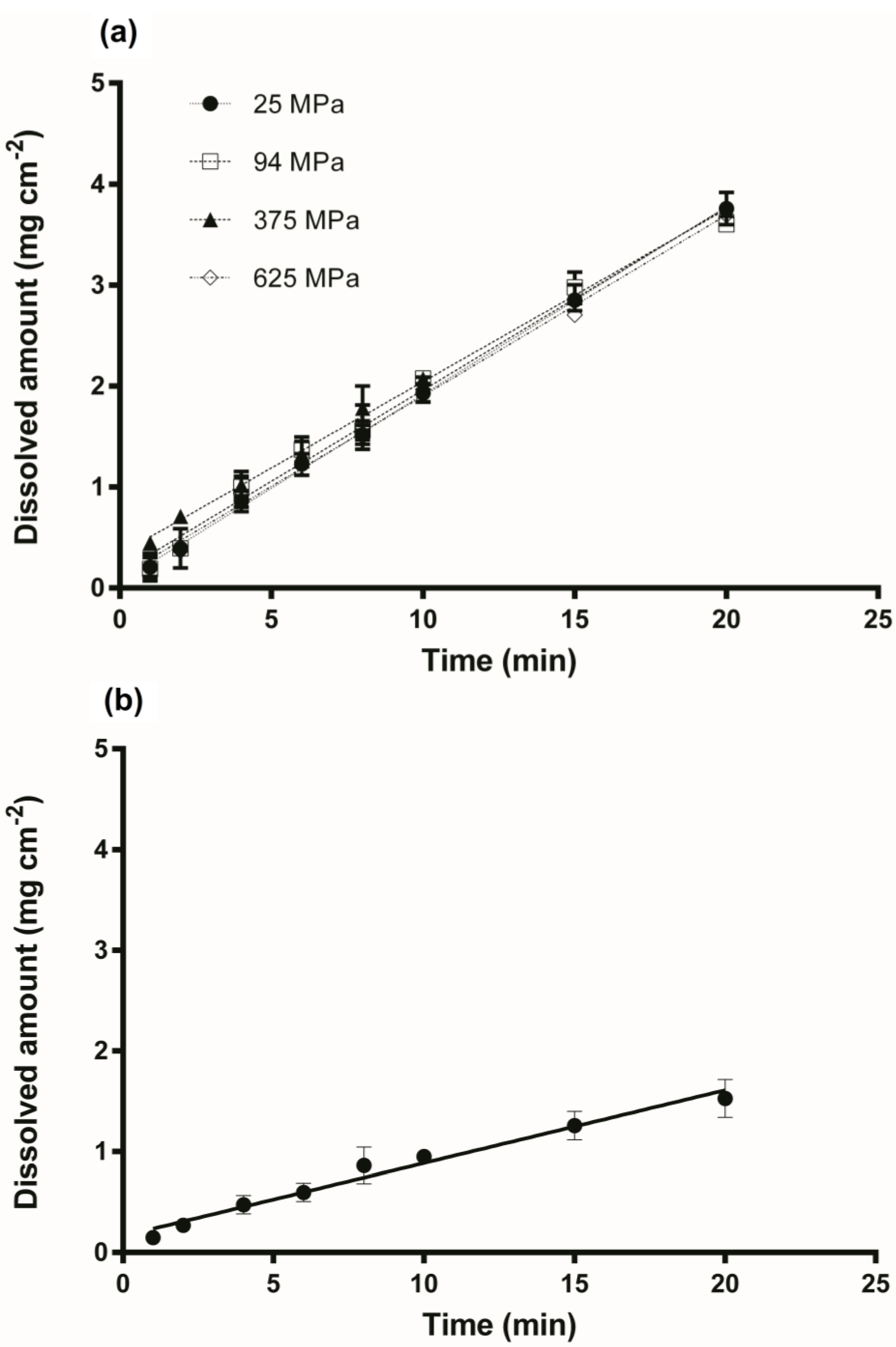{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Compression Pressure (MPa) | IDR (mg min−1 cm−2) |
|---|---|---|
| IND–PVP | 25 | 0.186 ± 0.003 |
| 94 | 0.181 ± 0.005 | |
| 375 | 0.171 ± 0.004 | |
| 625 | 0.179 ± 0.004 | |
| Amorphous IND (milled) | 37 | 0.099 ± 0.004 |
| 125 | 0.107 ± 0.004 | |
| 187 | 0.093 ± 0.003 | |
| 219 | 0.111 ± 0.005 | |
| 250 | 0.049 ± 0.006 | |
| 312 | 0.062 ± 0.007 | |
| Crystalline IND | 125 | 0.072 ± 0.004 |
| Quenched-in IND | – | 0.043 ± 0.004 |


4. Conclusions
Author Contributions
Conflicts of Interest
References
- Khan, K.A.; Rhodes, C. Effect of variation in compaction force on properties of six direct compression tablet formulations. J. Pharm. Sci. 1976, 65, 1835–1837. [Google Scholar] [CrossRef] [PubMed]
- Iranloye, T.A.; Parrott, E.L. Effects of compression force, particle size, and lubricants on dissolution rate. J. Pharm. Sci. 1978, 67, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Velasco, M.; Ford, J.L.; Rowe, P.; Rajabi-Siahboomi, A.R. Influence of drug: Hydroxypropylmethylcellulose ratio, drug and polymer particle size and compression force on the release of diclofenac sodium from hpmc tablets. J. Control. Release 1999, 57, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Tenho, M.; Heinänen, P.; Tanninen, V.P.; Lehto, V.-P. Does the preferred orientation of crystallites in tablets affect the intrinsic dissolution? J. Pharm. Biomed. Anal. 2007, 43, 1315–1323. [Google Scholar] [CrossRef]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Hulse, W.L.; Gray, J.; Forbes, R.T. A discriminatory intrinsic dissolution study using UV area imaging analysis to gain additional insights into the dissolution behaviour of active pharmaceutical ingredients. Int. J. Pharm. 2012, 434, 133–139. [Google Scholar] [PubMed]
- Kawabata, Y.; Wada, K.; Nakatani, M.; Yamada, S.; Onoue, S. Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: Basic approaches and practical applications. Int. J. Pharm. 2011, 420, 1–10. [Google Scholar] [PubMed]
- Aaltonen, J.; Rades, T. Commentary: Towards physicorelevant dissolution. Dissolut. Technol. 2009, 16, 47–54. [Google Scholar]
- Anjana, M.; Joseph, J.; Nair, S.C. Solubility enhancement methods—A promising technology for poorly water soluble drugs. Int. J. Pharm. Sci. Rev. Res. 2013, 20, 127–134. [Google Scholar]
- Morris, K.R.; Griesser, U.J.; Eckhardt, C.J.; Stowell, J.G. Theoretical approaches to physical transformations of active pharmaceutical ingredients during manufacturing processes. Adv. Drug Deliv. Rev. 2001, 48, 91–114. [Google Scholar] [CrossRef] [PubMed]
- Karmwar, P.; Graeser, K.; Gordon, K.C.; Strachan, C.J.; Rades, T. Effect of different preparation methods on the dissolution behaviour of amorphous indomethacin. Eur. J. Pharm. Biopharm. 2012, 80, 459–464. [Google Scholar] [PubMed]
- Nagapudi, K.; Jona, J. Amorphous active pharmaceutical ingredients in preclinical studies: Preparation, characterization, and formulation. Curr. Bioact. Comp. 2008, 4, 213–224. [Google Scholar] [CrossRef]
- Hilden, L.R.; Morris, K.R. Physics of amorphous solids. J. Pharm. Sci. 2004, 93, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Van den Mooter, G. The use of amorphous solid dispersions: A formulation strategy to overcome poor solubility and dissolution rate. Drug Discov. Today Technol. 2012, 9, e79–e85. [Google Scholar]
- Aso, Y.; Yoshioka, S.; Kojima, S. Molecular mobility-based estimation of the crystallization rates of amorphous nifedipine and phenobarbital in poly(vinylpyrrolidone) solid dispersions. J. Pharm. Sci. 2004, 93, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Yu, L. Amorphous pharmaceutical solids: Preparation, characterization and stabilization. Adv. Drug Deliv. Rev. 2001, 48, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Hancock, B.C.; Parks, M. What is the true solubility advantage for amorphous pharmaceuticals? Pharm. Res. 2000, 17, 397–404. [Google Scholar]
- Worku, Z.A.; Aarts, J.; van den Mooter, G. Influence of compression forces on the structural stability of naproxen/PVP–VA 64 solid dispersions. Mol. Pharm. 2014, 11, 1102–1108. [Google Scholar] [CrossRef] [PubMed]
- Ayenew, Z.; Paudel, A.; Rombaut, P.; van den Mooter, G. Effect of compression on non-isothermal crystallization behaviour of amorphous indomethacin. Pharm. Res. 2012, 29, 2489–2498. [Google Scholar] [CrossRef] [PubMed]
- Greco, K.; Bogner, R. Crystallization of amorphous indomethacin during dissolution: Effect of processing and annealing. Mol. Pharm. 2010, 7, 1406–1418. [Google Scholar] [CrossRef] [PubMed]
- Nanjwade, B.K. Effect of compression pressure on dissolution and solid state characterization of cefuroxime axetil. J. Anal. Bioanal. Tech. 2010, 1, 112. [Google Scholar]
- Wlodarski, K.; Sawicki, W.; Paluch, K.; Tajber, L.; Grembecka, M.; Hawelek, L.; Wojnarowska, Z.; Grzybowska, K.; Talik, E.; Paluch, M. The influence of amorphization methods on the apparent solubility and dissolution rate of tadalafil. Eur. J. Pharm. Sci. 2014, 62, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Hendriksen, B.; Williams, J. Characterization of calcium fenoprofen 2. Dissolution from formulated tablets and compressed rotating discs. Int. J. Pharm. 1991, 69, 175–180. [Google Scholar]
- Lim, R.T.Y.; Ng, W.K.; Widjaja, E.; Tan, R.B. Comparison of the physical stability and physicochemical properties of amorphous indomethacin prepared by co-milling and supercritical anti-solvent co-precipitation. J. Supercrit. Fluids 2013, 79, 186–201. [Google Scholar] [CrossRef]
- Okumura, T.; Ishida, M.; Takayama, K.; Otsuka, M. Polymorphic transformation of indomethacin under high pressures. J. Pharm. Sci. 2006, 95, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Savolainen, M.; Heinz, A.; Strachan, C.; Gordon, K.C.; Yliruusi, J.; Rades, T.; Sandler, N. Screening for differences in the amorphous state of indomethacin using multivariate visualization. Eur. J. Pharm. Sci. 2007, 30, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Perez-Marcos, B.; Iglesias, R.; Gomez-Amoza, J.; Martinez-Pacheco, R.; Souto, C.; Concheiro, A. Mechanical and drug-release properties of atenolol-carbomer hydrophilic matrix tablets. J. Control. Release 1991, 17, 267–276. [Google Scholar]
- Yadav, V.; Yadav, A. Enhancement of solubility and dissolution rate of indomethacin with different polymers by compaction process. Int. J. ChemTech Res. 2009, 1, 1072–1078. [Google Scholar]
- Guilbaud, J.B.; Cummings, L.; Khimyak, Y.Z. Encapsulation of indomethacin in PVP: Solid-state NMR studies. Macromol. Symp. 2007, 251, 41–46. [Google Scholar] [CrossRef]
- Matsumoto, T.; Zografi, G. Physical properties of solid molecular dispersions of indomethacin with poly(vinylpyrrolidone) and poly(vinylpyrrolidone-co-vinyl-acetate) in relation to indomethacin crystallization. Pharm. Res. 1999, 16, 1722–1728. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Löbmann, K.; Flouda, K.; Qiu, D.; Tsolakou, T.; Wang, W.; Rades, T. The Influence of Pressure on the Intrinsic Dissolution Rate of Amorphous Indomethacin. Pharmaceutics 2014, 6, 481-493. https://doi.org/10.3390/pharmaceutics6030481
Löbmann K, Flouda K, Qiu D, Tsolakou T, Wang W, Rades T. The Influence of Pressure on the Intrinsic Dissolution Rate of Amorphous Indomethacin. Pharmaceutics. 2014; 6(3):481-493. https://doi.org/10.3390/pharmaceutics6030481
Chicago/Turabian StyleLöbmann, Korbinian, Konstantina Flouda, Danwen Qiu, Theodosia Tsolakou, Wenbo Wang, and Thomas Rades. 2014. "The Influence of Pressure on the Intrinsic Dissolution Rate of Amorphous Indomethacin" Pharmaceutics 6, no. 3: 481-493. https://doi.org/10.3390/pharmaceutics6030481