Nutrigenetics and Nutrigenomics Insights into Diabetes Etiopathogenesis



Abstract
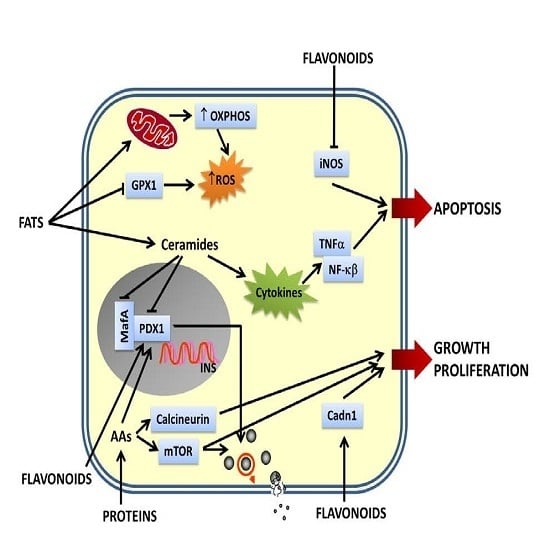
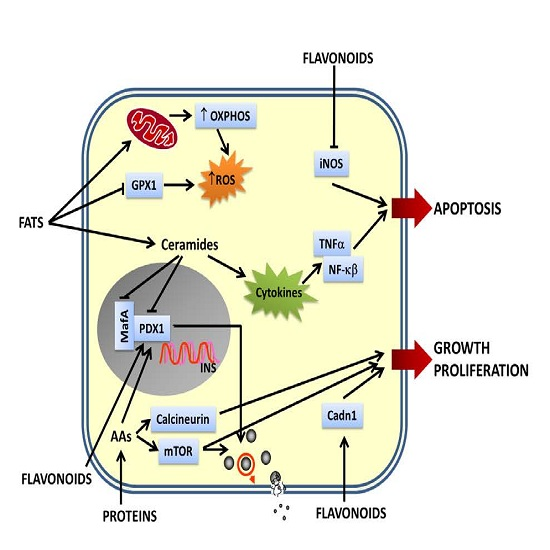
:
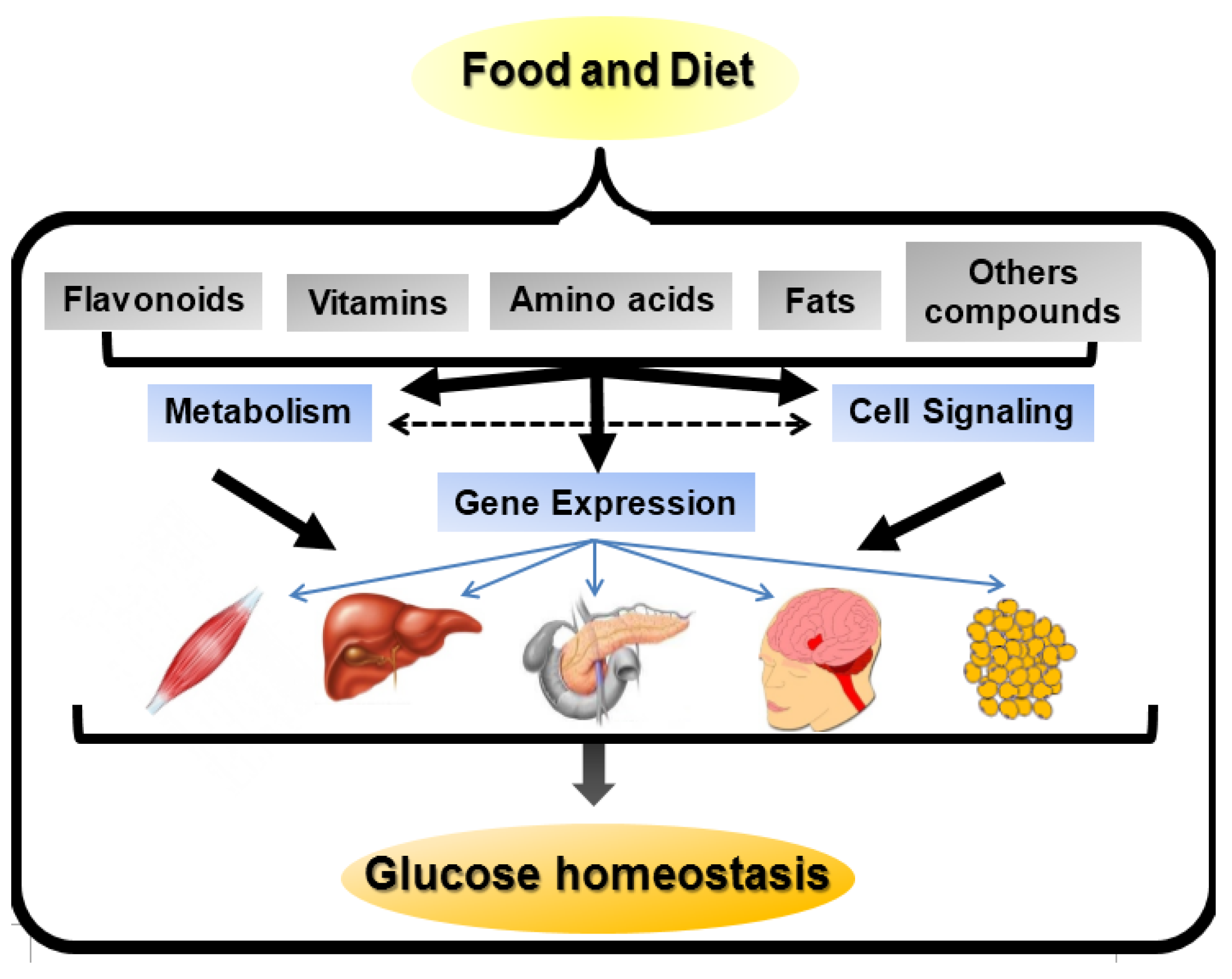
1. Introduction
2. Nutrigenetics Approximation to DM

{kind=link}
{kind=link}
{kind=link}
| Loci | |||||
|---|---|---|---|---|---|
| NOTCH2 | PSMD6 | VGEFA | CHCHD9 | DCD | CMIP |
| ADAM30 | CACNA1D | CDKAL1 | GAS1 | HMGA2 | WWOX |
| SLC44A3 | PPARG | C6orf57 | CAMK1D | TMEM19 | SGSM2 |
| SNX7 | SYN2 | TP53INP1 | CDC123 | LGR5 | SRR |
| PROX1 | ZPLD1 | GCK | VPS26A | TSPAN8 | HNF1B |
| CR2 | PLS1 | CPVL | KIF11 | IGF1 | LPIN2 |
| PCNXL2 | SLC2A2 | JAZF1 | HHEK | HNF1A | PAPL |
| BCL11A | PEX5L | DGKB | ADRA2A | TRIAP1 | PEPD |
| THADA | IGF2BP2 | ACHE | TCF7L2 | SPRY2 | GIPR |
| GCKR | ST6GAL1 | GCC1 | TCERG1L | C14orf70 | HNF4A |
| ITGB2 | PPP2R2C | PAX4 | CRY2 | ATP10A | HUNK |
| RBM43 | WFS1 | KLF14 | MADD | C2CD4A | PCBP3 |
| RND3 | MAEA | ZMAT4 | KCNJ11 | C2CD4B | SEZ6L |
| ITGB6 | ZBED3 | KCNU1 | GALNTL4 | VPS13C | DUSP9 |
| RBMS1 | AP3B1 | CSMD1 | LOC72903 | LARP6 | |
| GRB14 | CETN3 | SLC30A8 | KCNQ1 | HMG20A | |
| G6PC2 | LOC72901 | CDKN2A | ARAP1 | ZFAND6 | |
| TMEFF2 | PCSK1 | CDKN2B | MTNR1B | AP3S2 | |
| IRS1 | KCNK16 | PTRD | BARX2 | PRC1 | |
| ADAMTS9 | ZFAND3 | GLIS3 | TMEM45B | FTO | |
3. Gene-Nutrient or Dietary Pattern Interactions in The Development of T2DM
| Gene | Region | SNP | Allele Change | T2DM-Related Traits | Dietary Factors | References |
|---|---|---|---|---|---|---|
| PPARG | 3p25.2 | rs1801282 | C > G | HOMA-IR index | PUFA intake | [51,52,53] |
| TCF7L2 | 10q25.3 | rs12573128 | A > G | HOMA-IR index Oral glucose tolerance test | Fat intake | [54] |
| rs12255372 | G > T | T2DM risk | Carbohydrate intake | [55] | ||
| FTO | 16q12.2 | rs9939609 | A > T | T2DM risk | Adherence to Mediterranean Diet | [56,57] |
| SLC30A8 | 8q24.11 | rs11558471 | A > G | Fasting glucose levels | Zinc intake Magnesium intake | [58,59] |
| rs13266634 | C > T | T2DM risk | Trans- and cis-beta-carotene and gamma-tocoferol intake | [60] | ||
| TRPM6 | 9q21.13 | rs2274924 | C > T | Fasting glucose levels | Magnesium intake | [59] |
| AS3MT | 10q14.32 | rs3740393 | G > C | Fasting glucose levels | Magnesium intake | [59] |
| IRS1 | 2q36.3 | rs2943641 | C > T | HOMA-IR index | Vitamin D | [61] |
| GCKR | 2p23 | rs780094 | C > T | Fasting insulin levels | Whole-grain intake | [62] |
| ADIPOQ * | 3q27 | SNP276 G > T | G > T | Fasting glucose levels | Carbohydrate intake | [63,64,65,66] |
| SNP45 G > T | G > T | T2DM lower risk | Omega-3 intake | [67] | ||
| FABP2 | 4q28.31 | Ala54Thr polymorphism | G > A | HOMA-IR index | SFA intake | [68] |
| CAV2 | 7q31.1 | rs2270188 | C > T | T2DM risk | SFA intake | [69] |
| PLIN | 15q26.1 | 11482 G > A | G > A | HOMA-IR index | SFA fat and carbohydrates intake | [70] |
| 14995 A > T | A > T | HOMA-IR index | SFA fat and carbohydrates intake | [70] | ||
| CEBPA | 19q13.1 | rs12691 | C > T | Oral glucose tolerance test HOMA-IR index | Fat intake | [71] |
| CLOCK | 4q12 | rs1801260 | T > C | Fasting insulin levels HOMA-IR index QUICKI index | Fat and MUFA intake | [72] |
| CRY1 | 12q24.1 | Rs2287161 | G > C | Fasting insulin levels HOMA-IR index QUICKI index | Carbohydrate intake | [73] |
| SIRT1 | 10q21.3 | rs7895833 | A > G | Oral glucose tolerance test | Famine in prenatal life | [74] |
| rs1467568 | A > G | Oral glucose tolerance test | Famine in prenatal life | [74] |
3.1. Most Relevant T2DM Susceptibility Genes
3.2. Other T2DM Susceptibility Genes
3.3. CLOCK Gene Variants Linked to Diabetes
3.4. Importance of Genotype by Macronutrient Interactions for T2DM-Related Traits
4. The Effects of Nutrients on Gene Expression: Their Importance in DM

4.1. Flavonoids-Gene Interactions in DM Pathogenesis
| Nutrient | Gene Interaction | Function | Experimental Model | References | |
|---|---|---|---|---|---|
| Flavonoids | |||||
| Epigallocatechingallate (EGCG) | ↑Irs2, ↑Akt, ↑Foxo1, ↑Pdx1 | ↑Viability of β-cell, ↑insulin secretion | RIN-m5F cells | [87] | |
| ↑Bcl-2 | ↓Apoptosis, ↑glucose uptake | RINm5F cells | [88] | ||
| ↓L-Cpt-1, ↓Ddit3, ↓Ppp1r15a, ↓Cdkn1a | ↑Insulin secretion, preserve islet structure | db/db mice | [89] | ||
| Naringin or hesperidin | ↑Gk (liver), ↑Glut4 (WAT), ↑Pparγ | ↓Hyperglycemia | db/db mice | [90] | |
| Naringin | ↑Pparγ, ↑Hsp | ↓Hyperglycemia, ↓hyperinsulinemia, ↓insulin resistance, ↑β cell function | HFD-STZ-induced T2DM rats | [91] | |
| Anthocyanins | ↑Glut4 (WAT, muscle), ↑Pparα, ↑Aco, ↑L-Cpt-1 | ↓Hyperglycemia, ↑insulin sensitivity | T2DM mice | [92] | |
| ↓Lipogenic genes | ↓Hyperglycemia, ↓hyperinsulinemia | HFD-DM mice | [93] | ||
| Quercetin | ↓Cdkn1a | ↓Hyperglycemia, ↑insulin plasma levels, ↑pancreatic cell proliferation | STZ-induced DM mice | [94] | |
| Luteolin, apigenin | ↓iNos | ↓Apoptosis | RINm5F cells | [95] | |
| Genistein | ↑Ccnd1, ↓iNos | ↓Hyperglycemia, ↑glucose tolerance, ↑insulin plasma levels | STZ-induced DM mice | [96] | |
| Another bioactive compounds | |||||
| Oleanolic acid | ↑Antioxidant enzymes genes, ↑phase II detoxification enzymes genes, ↓NF-κB | ↑β-cell survival | Pancreatic islets | [97] | |
| Berberine | ↑Cyp7a1, ↑Igfbp1, ↑cell cycle genes, ↑NADPH metabolism genes | ↓Fasting glucose, ↓insulin resistance | Diabetic Zucker rats | [98] | |
| ↑CuZn-superoxide dismutase | ↓Hyperglycemia | STZ-nicotinamide diabetic mice | [99] | ||
| Vitamins | |||||
| Vitamin D | ↓Islet cytokine and chemokine genes | ↓Insulitis | NOD mice | [100,101] | |
| Biotin | ↑Foxa2, ↑Pdx-1, ↑Hnf-4α, ↑Ins, ↑Gk, ↑Cacna1d, ↑Acac | ↑Insulin secretion, ↑islet function | Mice | [102] | |
| Riboflavin | ↑IL-6 | ↓Cytokines-induced inflammation | NIT-1 cells | [103] | |
| Nicotinamide | ↑MafA | ↑Insulin synthesis | INS-1 cells, pancreatic islets | [104] | |
| Amino acids | |||||
| Leucine | ↑mTor | ↑Growth and proliferation | Pancreatic islets | [105] | |
| Taurine | ↑Pdx1, ↑Sur-1, ↑Gk, ↑Glut-2, ↑Ins | ↑Insulin secretion, ↑insulin synthesis | OF1 mice, pancreatic islets culture | [106] | |
| l-glutamine | ↑Pdx1, ↑Calcineurin, ↑Acac | ↑Insulin secretion, ↑proliferation | BRIN-BD11 beta-cells | [107] | |
| Dietary fats | |||||
| Palmitate | ↓Ins | ↓Insulin secretion | Pancreatic islets | [108] | |
| ↓Pdx-1, ↓MafA | ↓Insulin secretion | [109] | |||
| HFD | ↓Gpx1 | ↓Antioxidant defenses of β-cells | C57BL/6J mice | [110] | |
| ↑Growth and development, ↑oxidative metabolism, ↑insulin processing and secretion, ↑signaling, ↑redox status | T2DM | NZO-mice | [111] | ||
| CHFD plus HFD | ↓Pdx-1, ↓MafA, ↓Nkx6.1 | ↓Insulin secretion, ↓insulin synthesis, ↓β-cell survival | NZO-mice | [112] | |
| Lipoic acid | ↑Frk, ↑Gk, ↑G6pc2, ↑Phox | Protection against T2DM | High fructose-fed Wistar rats | [113] | |
| Oleic acid | ↓NPY, ↓AgRP | ↓Food intake, ↓glucose production, ↓plasma glucose levels, ↓insulin plasma levels | Sprague-Dawley rats Sprague-Dawley or Zucker fatty rats | [114,115] | |
4.2. Bioactive Compounds-Gene Interactions in DM Pathogenesis
4.3. Vitamins-Gene Interactions in DM Pathogenesis
4.4. Amino Acids-Gene Interaction in DM Pathogenesis
4.5. Dietary Fat-Gene Interactions and Their Role in T2DM
4.6. Food-Gene Interactions in DM Pathogenesis: Human Studies
4.7. Nutrient-Gene Interactions in Hypothalamus Are Also Involved in DM Pathogenesis

5. Epigenetics, Micro RNAs (miRNAs) and Diet: Are They Involved in DM?
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Georgoulis, M.; Kontogianni, M.D.; Yiannakouris, N. Mediterranean diet and diabetes: Prevention and treatment. Nutrients 2014, 6, 1406–1423. [Google Scholar] [CrossRef] [PubMed]
- International Diabetes Federation. IDF Diabetes Atlas, 6th ed.; International Diabetes Federation: Brussels, Belgium, 2013; p. 11. [Google Scholar]
- Dib, S.A.; Gomes, M.B. Etiopathogenesis of type 1 diabetes mellitus: Prognostic factors for the evolution of residual beta cell function. Diabetol. Metab. Syndr. 2009, 1, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, L.; Pedersen, O. Genetics of type 2 diabetes mellitus: Status and perspectives. Diabetes Obes. Metab. 2005, 7, 122–135. [Google Scholar] [CrossRef] [PubMed]
- Schulze, M.B.; Hu, F.B. Primary prevention of diabetes: What can be done and how much can be prevented? Annu. Rev. Public Health 2005, 26, 445–467. [Google Scholar] [CrossRef]
- Corella, D.; Ordovas, J.M. Nutrigenomics in cardiovascular medicine. Circ. Cardiovasc. Genet. 2009, 2, 637–651. [Google Scholar] [CrossRef] [PubMed]
- Brennan, R.O. Nutrigenetics: New Concepts for Relieving Hypoglycemia; M. Evans and Company Inc.: New York, NY, USA, 1976. [Google Scholar]
- Corella, D.; Carrasco, P.; Sorlí, J.V.; Estruch, R.; Rico-Sanz, J.; Martínez-González, M.Á.; Salas-Salvadó, J.; Covas, M.I.; Coltell, O.; Arós, F.; et al. Mediterranean diet reduces the adverse effect of the TCF7L2-rs7903146 polymorphism on cardiovascular risk factors and stroke incidence: A randomized controlled trial in a high-cardiovascular-risk population. Diabetes Care 2013, 36, 3803–3811. [Google Scholar] [CrossRef]
- Daar, A.S.; Singer, P.A.; Persad, D.L.; Pramming, S.K.; Matthews, D.R.; Beaglehole, R.; Bernstein, A.; Borysiewicz, L.K.; Colagiuri, S.; Ganguly, N.; et al. Grand challenges in chronic non-communicable diseases. Nature 2007, 450, 494–496. [Google Scholar] [CrossRef]
- Scott, J. Molecular genetics of common diseases. Br. Med. J. 1987, 295, 769–771. [Google Scholar] [CrossRef]
- Sladek, R.; Rocheleau, G.; Rung, J.; Dina, C.; Shen, L.; Serre, D.; Boutin, P.; Vincent, D.; Belisle, A.; Hadjadj, S.; et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 2007, 445, 881–885. [Google Scholar] [CrossRef]
- Stratigopoulos, G.; Padilla, S.L.; LeDuc, C.A.; Watson, E.; Hattersley, A.T.; McCarthy, M.I.; Zeltser, L.M.; Chung, W.K.; Leibel, R.L. Regulation of FTO/FTM gene expression in mice and humans. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R1185–R1196. [Google Scholar] [CrossRef] [PubMed]
- Thorisson, G.A.; Stein, L.D. The SNP Consortium website: Past, present and future. Nucleic Acid Res. 2003, 31, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, A.A.; Chakravarti, A.; Cutler, D.J. On the probability that a novel variant is a disease-causing mutation. Genome Res. 2005, 15, 960–966. [Google Scholar] [CrossRef] [PubMed]
- Bailey-Wilson, J.E.; Wilson, A.F. Linkage analysis in the next generation sequencing era. Hum. Hered. 2011, 72, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Zanda, M.; Onengut-Gumuscu, S.; Walker, N.; Shtir, C.; Gallo, D.; Wallace, C.; Smyth, D.; Todd, J.A.; Hurles, M.E.; Plagnol, V.; et al. A genome-wide assessment of the role of untagged copy number variants in type 1 diabetes. PLoS Genet. 2014, 29, e1004367. [Google Scholar] [CrossRef]
- Ziegler, A.G.; Nepom, G.T. Prediction and pathogenesis in type 1 diabetes. Immunity 2010, 32, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, S.M.; Knip, M. Nutritional risk predictors of beta-cell autoimmunity and type 1 diabetes at a young age. Am. J. Clin. Nutr. 2003, 78, 1053–1067. [Google Scholar] [PubMed]
- Knip, M.; Virtanen, S.M.; Becker, D.; Dupré, J.; Krischer, J.P.; Åkerblom, H.K.; TRIGR Study Group. Early feeding risk of type 1 diabetes: Experiences from the Trial to Reduce Insulin-dependent diabetes mellitus in the Genetically at Risk (TRIGR). Am. J. Clin. Nutr. 2011, 94, 1814S–1820S. [Google Scholar] [CrossRef]
- Norris, J.M.; Barriga, K.; Klingensmith, G.; Hoffman, M.; Eisenbarth, G.S.; Erlich, H.A.; Rewers, M. Timing of initial cereal exposure in infancy and risk of islet autoimmunity. JAMA 2003, 290, 1713–1720. [Google Scholar] [CrossRef] [PubMed]
- Hyppönen, E.; Läärä, E.; Reunanen, A.; Järvelin, M.R.; Virtanen, S.M. Intake of vitamin D and risk of type 1 diabetes: A birth-cohort study. Lancet 2001, 358, 1500–1503. [Google Scholar] [CrossRef]
- Sildorf, S.M.; Fredheim, S.; Svensson, J.; Buschard, K. Remission without insulin therapy on gluten-free diet in a 6-year old boy with type 1 diabetes mellitus. BMJ Case Rep. 2012, 2012, bcr0220125878. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterocytes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef]
- Patrick, C.; Wang, G.S.; Lefebvre, D.E.; Crookshank, J.A.; Sonier, B.; Eberhard, C.; Mojibian, M.; Kennedy, C.R.; Brooks, S.P.; Kalmokoff, M.L.; et al. Promotion of autoimmune diabetes by cereal diet in the presence or absence of microbes associated with gut immune activation, regulatory imbalance, and altered cathelicidin antimicrobial peptide. Diabetes 2013, 62, 2036–2047. [Google Scholar]
- Bobbala, D.; Chen, X.L.; Leblanc, C.; Mayhue, M.; Stankova, J.; Tanaka, T.; Chen, Y.G.; Ilangumaran, S.; Ramanathan, S. Interleukin-15 plays an essential role in the pathogenesis of autoimmune diabetes in the NOD mouse. Diabetologia 2012, 55, 3010–3020. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Ono, M.; Setoguchi, R.; Yagi, H.; Hori, S.; Fehervari, Z.; Shimizu, J.; Takahashi, T.; Nomura, T. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol. Rev. 2006, 212, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Salmond, R.J.; Filby, A.; Qureshi, I.; Caserta, S.; Zamoyska, R. T-cell receptor proximal signaling via the Src-family kinases, Lck and Fyn, influences T-cell activation, differentiation, and tolerance. Immunol. Rev. 2009, 228, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Nijnik, A.; Pistolic, J.; Wyatt, A.; Tam, S.; Hancock, R.E. Human cathelicidin peptide LL-37 modulates the effects of IFN-gamma on APCs. J. Immunol. 2009, 183, 5788–5798. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C.M. Nutrigenetics and metabolic disease: Current status and implications for personalized nutrition. Nutrients 2013, 5, 32–57. [Google Scholar] [CrossRef] [PubMed]
- Vimaleswaran, K.S.; Loos, R.J. Progress in the genetics of common obesity and type 2 diabetes. Expert Rev. Mol. Med. 2010, 12, e7. [Google Scholar] [CrossRef] [PubMed]
- Temelkova-Kurktschiev, T.; Stefanov, T.S. Lifestyle and genetics in obesity and type 2 diabetes. Exp. Clin. Endocrinol. Diabetes 2012, 120, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kaput, J.; Noble, J.; Hatipoglu, B.; Kohrs, K.; Dawson, K.; Bartholomew, A. Application of nutrigenomic concepts to type 2 diabetes mellitus. Nutr. Metab. Cardiovasc. Dis. 2007, 17, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Flint, J.; Valdar, W.; Shifman, S.; Mott, R. Strategies for mapping and cloning quantitative trait genes in rodents. Nat. Rev. Genet. 2005, 6, 271–286. [Google Scholar] [CrossRef] [PubMed]
- Kaput, J.; Dawson, K. Complexity of type 2 diabetes mellitus data sets emerging from nutrigenomic research: A case for dimensionality reduction? Mutat. Res. 2007, 622, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Hindorff, L.A.; MacArthur, J.; Morales, J.; Junkins, H.A.; Hall, P.N.; Klemm, A.K.; Manolio, T.A. A Catalog of Published Genome-Wide Association Studies. Available online: http://www.genome.gov/gwastudies (accessed on 23 June 2014).
- Dupuis, J.; Langenberg, C.; Prokopenko, I.; Saxena, R.; Soranzo, N.; Jackson, A.U.; Wheeler, E.; Glazer, N.L.; Bouatia-Naji, N.; Gloyn, A.L.; et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat. Genet. 2010, 42, 105–116. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, M.I. Genomics, type 2 diabetes, and obesity. N. Engl. J. Med. 2010, 363, 2339–2350. [Google Scholar] [CrossRef] [PubMed]
- Saxena, R.; Hivert, M.F.; Langenberg, C.; Tanaka, T.; Pankow, J.S.; Vollenweider, P.; Lyssenko, V.; Bouatia-Naji, N.; Dupuis, J.; Jackson, A.U.; et al. Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nat. Genet. 2010, 42, 142–148. [Google Scholar] [CrossRef]
- Strawbridge, R.J.; Dupuis, J.; Prokopenko, I.; Barker, A.; Ahlqvist, E.; Rybin, D.; Petrie, J.R.; Travers, M.E.; Bouatia-Naji, N.; Dimas, A.S.; et al. Genome-wide association identifies nine common variants associated with fasting proinsulin levels and provides new insights into the pathophysiology of type 2 diabetes. Diabetes 2011, 60, 2624–2634. [Google Scholar] [CrossRef] [PubMed]
- Florez, J.C. Newly identified loci highlight beta cell dysfunction as a key cause of type 2 diabetes: Where are the insulin resistance genes? Diabetologia 2008, 51, 1100–1110. [Google Scholar] [CrossRef] [PubMed]
- Grant, S.F.; Thorleifsson, G.; Reynisdottir, I.; Benediktsson, R.; Manolescu, A.; Sainz, J.; Helgason, A.; Stefansson, H.; Emilsson, V.; Helgadottir, A.; et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat. Genet. 2006, 38, 320–323. [Google Scholar] [CrossRef] [PubMed]
- Zeggini, E.; McCarthy, M.I. TCF7L2: The biggest story in diabetes genetics since HLA? Diabetologia 2007, 50, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Chandak, G.R.; Janipalli, C.S.; Bhaskar, S.; Kulkarni, S.R.; Mohankrishna, P.; Hattersley, A.T.; Frayling, T.M.; Yajnik, C.S. Common variants in the TCF7L2 gene are strongly associated with type 2 diabetes mellitus in the Indian population. Diabetologia 2007, 50, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Horikoshi, M.; Hara, K.; Ito, C.; Nagai, R.; Froguel, P.; Kadowaki, T. A genetic variation of the transcription factor 7-like 2 gene is associated with risk of type 2 diabetes in the Japanese population. Diabetologia 2007, 50, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Lehman, D.M.; Hunt, K.J.; Leach, R.J.; Hamlington, J.; Arya, R.; Abboud, H.E.; Duggirala, R.; Blangero, J.; Göring, H.H.; Stern, M.P.; et al. Haplotypes of transcription factor 7-like 2 (TCF7L2) gene and its upstream region are associated with type 2 diabetes and age of onset in Mexican Americans. Diabetes 2007, 56, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Helgason, A.; Pálsson, S.; Thorleifsson, G.; Grant, S.F.; Emilsson, V.; Gunnarsdottir, S.; Adeyemo, A.; Chen, Y.; Chen, G.; Reynisdottir, I.; et al. Refining the impact of TCF7L2 gene variants on type 2 diabetes and adaptive evolution. Nat. Genet. 2007, 39, 218–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyssenko, V.; Lupi, R.; Marchetti, P.; Del Guerra, S.; Orho-Melander, M.; Almgren, P.; Sjögren, M.; Ling, C.; Eriksson, K.F.; Lethagen, A.L.; et al. Mechanisms by which common variants in the TCF7L2 gene increase the risk of type 2 diabetes. J. Clin. Investig. 2007, 117, 2155–2163. [Google Scholar]
- Cornelis, M.C.; Hu, F.B. Gene-enviroment interactions in the development of type 2 diabetes: Recent progress and continuing challenges. Annu. Rev. Nutr. 2012, 32, 245–259. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.; Lai, C.Q.; Ordovas, J.M.; Parnell, L.D. A database of gene-enviroment interactions pertaining to blood lipid traits, cardiovascular disease and type 2 diabetes. J. Data Mining Genomics Proteomics 2011, 2, 106. [Google Scholar] [CrossRef] [PubMed]
- Harrington, J.M.; Phillips, C.M. Nutrigenetics: Bridging two worlds to understand type 2 diabetes. Curr. Diabetes Rep. 2014, 14, 477. [Google Scholar] [CrossRef]
- Gouda, H.N.; Sagoo, G.S.; Harding, A.H.; Yates, J.; Sandhu, M.S.; Higgins, J.P. The association between the peroxisome proliferator-activated-receptor gamma 2 (PPARG2) Pro12Ala gene variant and type 2 diabetes mellitus: HuGe review and meta-analysis. Am. J. Epidemiol. 2010, 171, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Lamri, A.; Abi-Khalil, C.; Jaziri, R.; Velho, G.; Lantieri, O.; Vol, S.; Froguel, P.; Balkau, B.; Marre, M.; Fumeron, F.; et al. Dietary fat intake and polymorphisms at the PPARG locus modulate BMI and type 2 diabetes risk in the D.E.S.I.R. prospective study. Int. J. Obes. 2012, 36, 218–224. [Google Scholar] [CrossRef]
- Luan, J.; Browne, P.O.; Harding, A.H.; Halsall, D.J.; O’Rahilly, S.; Chatterjee, V.K.; Wareham, N.J. Evidence for gene-nutrient interaction at the PPARgamma locus. Diabetes 2001, 50, 686–689. [Google Scholar] [CrossRef] [PubMed]
- Ruchat, S.M.; Elks, C.E.; Loos, R.J.; Vohl, M.C.; Weisnagel, S.J.; Rankinen, T.; Bouchard, C.; Pérusse, L. Evidence of interaction between type 2 diabetes susceptibility genes and dietary fat intake for adiposity and glucose homeostasis-related phenotypes. J. Nutrigenet. Nutrigenomics 2009, 2, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Cornelis, M.C.; Qi, L.; Kraft, P.; Hu, F.B. TCF7L2, dietary carbohydrate, and risk of type 2 diabetes in US women. Am. J. Clin. Nutr. 2009, 89, 1256–1262. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Azorín, C.; Sorlí, J.V.; Asensio, E.M.; Coltell, O.; Martínez-González, M.Á.; Salas-Salvadó, J.; Covas, M.I.; Arós, F.; Lapetra, J.; Serra-Majem, L.; et al. Associations of the FTO rs9939609 and the MC4R rs17782313 polymorphisms with type 2 diabetes are modulated by diet, being higher when adherence to the Mediterranean diet pattern is low. Cardiovasc. Diabetol. 2012, 11, 137. [Google Scholar] [CrossRef] [PubMed]
- Steemburgo, T.; Azevedo, M.J.; Gross, J.L.; Milagro, F.I.; Campión, J.; Martínez, J.A. The rs9939609 polymorphism in the FTO gene is associated with fat and fiber intakes in patients with type 2 diabetes. J. Nutrigenet. Nutrigenomics 2013, 6, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Kanoni, S.; Nettleton, J.A.; Hivert, M.F.; Ye, Z.; van Rooij, F.J.; Shungin, D.; Sonestedt, E.; Ngwa, J.S.; Wojczynski, M.K.; Lemaitre, R.N.; et al. Total zinc intake may modify the glucose-raising effect of a zinc transporter (SLC30A8) variant: A 14-cohort meta-analysis. Diabetes 2011, 60, 2407–2416. [Google Scholar] [CrossRef] [PubMed]
- Hruby, A.; Ngwa, J.S.; Renström, F.; Wojczynski, M.K.; Ganna, A.; Hallmans, G.; Houston, D.K.; Jacques, P.F.; Kanoni, S.; Lehtimäki, T.; et al. Higher magnesium intake is associated with lower fasting glucose and insulin, with no evidence of interaction with select genetic loci, in a meta-analysis of 15 CHARGE Consortium Studies. J. Nutr. 2013, 143, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Patel, C.J.; Chen, R.; Kodama, K.; Ioannidis, J.P.A.; Butte, A.J. Systematic identification of interactions effects between genome- and environment-wide associations in type 2 diabetes mellitus. Hum. Genet. 2013, 132, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.S.; Parnell, L.D.; Smith, C.E.; Lee, Y.C.; Jamal-Allial, A.; Ma, Y.; Li, D.; Tucker, K.L.; Ordovas, J.M.; Lai, C.Q.; et al. Circulating 25-hydroxyvitamin D, IRS1 variant rs2943641, and insulin resistance: Replication of a gene-nutrient interaction in 4 populations of different ancestries. Clin. Chem. 2014, 60, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Nettleton, J.A.; McKeown, N.M.; Kanoni, S.; Lemaitre, R.N.; Hivert, M.F.; Ngwa, J.; van Rooij, F.J.; Sonestedt, E.; Wojczynski, M.K.; Ye, Z.; et al. Interactions of dietary whole-grain intake with fasting glucose- and insulin-related genetic loci in individuals of European descent: A meta-analysis of 14 cohort studies. Diabetes Care 2010, 33, 2684–2691. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.Y.; Park, J.E.; Choi, Y.J.; Huh, K.B.; Chang, N.; Kim, W.Y. Carbohydrate intake interacts with SNP276G > T polymorphism in the adiponectin gene to affect fasting blood glucose, HbA1C, and HDL cholesterol in Korean patients with type 2 diabetes. J. Am. Coll. Nutr. 2013, 32, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Boutin, P.; Mori, Y.; Tobe, K.; Dina, C.; Yasuda, K.; Yamauchi, T.; Otabe, S.; Okada, T.; Eto, K.; et al. Genetic variation in the gene encoding adiponectin is associated with an increased risk of type 2 diabetes in the Japanese population. Diabetes 2002, 51, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Yang, Y.C.; Chen, C.L.; Wu, I.L.; Lu, J.Y.; Lu, F.H.; Tai, T.Y. Adiponectin SNP276 is associated with obesity, metabolic syndrome, and diabetes in the elderly. Am. J. Clin. Nutr. 2007, 86, 509–513. [Google Scholar] [PubMed]
- Jang, Y.; Lee, J.H.; Kim, O.Y.; Koh, S.J.; Chae, J.S.; Woo, J.H.; Cho, H.; Lee, J.E.; Ordovas, J.M. The SNP276 G > T polymorphism in the adiponectin (ACDC) gene is more strongly associated with insulin resistance and cardiovascular disease risk than SNP45 T > G in nonobese/nondiabetic Korean men independent of abdominal adiposity and circulating plasma adiponectin. Metabolism 2006, 55, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Alsaleh, A.; Crepostnaia, D.; Maniou, Z.; Lewis, F.J.; Hall, W.L.; Sanders, T.A.; O’Dell, S.D.; MARINA Study Team. Adiponectin gene variant interacts with fish oil supplementation to influence serum adiponectin in older individuals. J. Nutr. 2013, 143, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Marín, C.; Perez-Jimenez, F.; Gomez, P.; Delgado, J.; Paniagua, J.A.; Lozano, A.; Cortes, B.; Jimenez-Gomez, Y.; Gomez, M.J.; Lopez-Miranda, J.; et al. The Ala54Thr polymorphism of the fatty acid-binding protein 2 gene is associated with a change in insulin sensitivity after a change in the type of dietary fat. Am. J. Clin. Nutr. 2005, 82, 196–200. [Google Scholar] [PubMed]
- Fisher, E.; Schreiber, S.; Joost, H.G.; Boeing, H.; Döring, F. A two-step association study identifies CAV2 rs2270188 single nucleotide polymorphism interaction with fat intake in type 2 diabetes risk. J. Nutr. 2011, 141, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Corella, D.; Qi, L.; Tai, E.S.; Deurenberg-Yap, M.; Tan, C.E.; Chew, S.K.; Ordovas, J.M. Perilipin gene variation determines higher susceptibility to insulin resistance in Asian women when consuming a high-saturated fat, low-carbohydrate diet. Diabetes Care 2006, 29, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Lista, J.; Perez-Martinez, P.; García-Ríos, A.; Philipis, C.M.; Hall, W.; Gjelstad, I.M.F.; Lairon, D.; Saris, W.; Kiec-Wilk, B.; Karlström, B.; et al. A gene variation (rs12691) in the CCAT/enhancer binding protein α modulates glucose metabolism in metabolic syndrome. Nutr. Metab. Cardiovasc. 2013, 23, 417–423. [Google Scholar] [CrossRef]
- García-Ríos, A.; Gomez-Delgado, F.J.; Garaulet, M.; Alcala-Diaz, J.F.; Delgado-Lista, F.J.; Marin, C.; Rangel-Zúñiga, O.A.; Rodriguez-Cantalejo, F.; Gómez-Luna, P.; Ordovas, J.M.; et al. Beneficial effect of CLOCK gene polymorphism rs1801260 in combination with low-fat diet on insulin metabolism in patients with metabolic syndrome. Chronobiol. Int. 2014, 31, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Dashti, H.S.; Smith, C.E.; Lee, Y.C.; Parnell, L.D.; Lai, C.Q.; Arnett, D.K.; Ordovas, J.M.; Garaulet, M. CRY1 circadian gene variants interacts with carbohydrate intake for insulin resistance in two independent populations: Mediterranean and North American. Chronobiol. Int. 2014, 31, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Botden, I.P.; Zillikens, M.C.; de Rooij, S.R.; Langendonk, J.G.; Danser, A.H.; Sijbrands, E.J.; Roseboom, T.J. Variants in the SIRT1 gene may affect diabetes risk in interaction with prenatal exposure to famine. Diabetes Care 2012, 35, 424–426. [Google Scholar] [CrossRef] [PubMed]
- Lazar, M.A. PPAR gamma, 10 years later. Biochimie 2005, 87, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Fisher, E.; Boeing, H.; Fritsche, A.; Doering, F.; Joost, H.G.; Schulze, M.B. Whole-grain consumption and transcription factor-7-like 2 (TCF7L2) rs7903146: Gene-diet interaction in modulating type 2 diabetes risk. Br. J. Nutr. 2009, 101, 478–481. [Google Scholar] [CrossRef] [PubMed]
- Florez, J.C.; Jablonski, K.A.; Bayley, N.; Pollin, T.I.; de Bakker, P.I.; Shuldiner, A.R.; Knowler, W.C.; Nathan, D.M.; Altshuler, D.; Diabetes Prevention Program Research Group. TCF7L2 polymorphisms and progression to diabetes in the Diabetes Prevention Program. N. Engl. J. Med. 2006, 355, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Haupt, A.; Thamer, C.; Heni, M.; Ketterer, C.; Machann, J.; Schick, F.; Machicao, F.; Stefan, N.; Claussen, C.D.; Häring, H.U.; et al. Gene variants of TCF7L2 influence weight loss and body composition during lifestyle intervention in a population at risk for type 2 diabetes. GDiabetes 2010, 59, 747–750. [Google Scholar]
- Reinehr, T.; Friedel, S.; Mueller, T.D.; Toschke, A.M.; Hebebrand, J.; Hinney, A. Evidence for an influence of TCF7L2 polymorphism rs7903146 on insulin resistance and sensitivity indices in overweight children and adolescents during a lifestyle intervention. Int. J. Obes. 2008, 32, 1521–1524. [Google Scholar] [CrossRef]
- Chimienti, F. Zinc, pancreatic islet function and diabetes: New insights into an old story. Nutr. Res. Rev. 2013, 26, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Shan, Z.; Bao, W.; Zhang, Y.; Rong, Y.; Wang, X.; Jin, Y.; Song, Y.; Yao, P.; Sun, C.; Hu, F.B.; et al. Interactions between zinc transporter-8 gene (SLC30A8) and plasma zinc concentrations for impaired glucose regulation and type 2 diabetes. Diabetes 2014, 63, 1796–1803. [Google Scholar] [CrossRef] [PubMed]
- Marcheva, B.; Ramsey, K.M.; Buhr, E.D.; Kobayashi, Y.; Su, H.; Ko, C.H.; Ivanova, G.; Omura, C.; Mo, S.; Vitaterna, M.H.; et al. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature 2010, 466, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.S.; Arnett, D.K.; Lee, Y.C.; Shen, J.; Parnell, L.D.; Smith, C.E.; Richardson, K.; Li, D.; Borecki, I.B.; Ordovás, J.M.; et al. Genome-wide contribution of genotype by environment interaction to variation of diabetes-related traits. PLoS One 2013, 8, e77442. [Google Scholar] [CrossRef] [PubMed]
- Babu, P.V.; Liu, D.; Gilbert, E.R. Recent advances in understanding the anti-diabetic actions of dietary flavonoids. J. Nutr. Biochem. 2013, 11, 1777–1789. [Google Scholar] [CrossRef]
- Wedick, N.M.; Pan, A.; Cassidy, A.; Rimm, E.B.; Sampson, L.; Rosner, B.; Willett, W.; Hu, F.B.; Sun, Q.; van Dam, R.M.; et al. Dietary flavonoid intakes and risk of type 2 diabetes in US men and women. Am. J. Clin. Nutr. 2012, 95, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Hanhineva, K.; Torronen, R.; Bondia-Pons, I.; Pekkinen, J.; Kolehmainen, M.; Mykkanen, H.; Poutanen, K. Impact of dietary polyphenols on carbohydrate metabolism. Int. J. Mol. Sci. 2010, 11, 1365–1402. [Google Scholar] [CrossRef] [PubMed]
- Cai, E.P.; Lin, J.K. Epigallocatechingallate (EGCG) and rutin suppress the glucotoxicity through activating IRS2 and AMPK signaling in rat pancreatic beta cells. J. Agric. Food Chem. 2009, 57, 9817–9827. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ding, Y.; Dai, X.; Wang, J.; Li, Y. Epigallocatechin-3-gallate protects pro-inflammatory cytokine induced injuries in insulin-producing cells through the mitochondrial pathway. Eur. J. Pharmacol. 2011, 670, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Ortsater, H.; Grankvist, N.; Wolfram, S.; Kuehn, N.; Sjoholm, A. Diet supplementation with green tea extract epigallocatechingallate prevents progression to glucose intolerance in db/db mice. Nutr. Metab. 2012, 9, 11–13. [Google Scholar] [CrossRef]
- Jung, U.J.; Lee, M.K.; Park, Y.B.; Kang, M.A.; Choi, M.S. Effect of citrus flavonoids on lipid metabolism and glucose-regulating enzyme mRNA levels in type-2 diabetic mice. Int. J. Biochem. Cell Biol. 2006, 38, 1134–1145. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.K.; Bharti, S.; Ojha, S.; Bhatia, J.; Kumar, N.; Ray, R.; Kumari, S.; Arya, D.S. Up-regulation of PPARgamma, heat shock protein-27 and -72 by naringin attenuates insulin resistance, beta-cell dysfunction, hepatic steatosis and kidney damage in a rat model of type 2 diabetes. Br. J. Nutr. 2011, 106, 1713–1723. [Google Scholar] [CrossRef] [PubMed]
- Takikawa, M.; Inoue, S.; Horio, F.; Tsuda, T. Dietary anthocyanin-rich bilberry extract ameliorates hyperglycemia and insulin sensitivity via activation of AMP-activated protein kinase in diabetic mice. J. Nutr. 2010, 140, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, T.; Horio, F.; Uchida, K.; Aoki, H.; Osawa, T. Dietary cyanidin 3-O-beta-d-glucoside-rich purple corn color prevents obesity and ameliorates hyperglycemia in mice. J. Nutr. 2003, 133, 2125–2130. [Google Scholar] [PubMed]
- Kobori, M.; Masumoto, S.; Akimoto, Y.; Takahashi, Y. Dietary quercetin alleviates diabetic symptoms and reduces streptozotocin-induced disturbance of hepatic gene expression in mice. Mol. Nutr. Food Res. 2009, 53, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Kwon, K.B.; Song, M.Y.; Han, M.J.; Lee, J.H.; Lee, Y.R.; Ryu, D.G.; Park, B.H.; Park, J.W. Flavonoids protect against cytokine-induced pancreatic beta-cell damage through suppression of nuclear factor kappa B activation. Pancreas 2007, 35, e1–e9. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Zhang, W.; Zhen, W.; Lum, H.; Nadler, J.; Bassaganya-Riera, J.; Jia, Z.; Wang, Y.; Misra, H.; Liu, D.; et al. Genistein induces pancreatic beta-cell proliferation through activation of multiple signaling pathways and prevents insulin-deficient diabetes in mice. Endocrinology 2010, 151, 3026–3037. [Google Scholar] [CrossRef] [PubMed]
- Castellano, J.M.; Guinda, A.; Delgado, T.; Rada, M.; Cayuela, J.A. Biochemical basis of the antidiabetic activity of oleanolic acid and related pentacyclic triterpenes. Diabetes 2013, 62, 1791–1799. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.L.; Sui, Y.; Qiao, C.F.; Yip, K.Y.; Leung, R.K.; Tsui, S.K.; Lee, H.M.; Wong, H.K.; Zhu, X.; Siu, J.J.; et al. Sustained antidiabetic effects of a berberine-containing Chinese herbal medicine through regulation of hepatic gene expression. Diabetes 2012, 61, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Chatuphonprasert, W.; Lao-Ong, T.; Jarukamjorn, K. Improvement of superoxide dismutase and catalase in streprozotocin-nicotinamide-induced type 2 diabetes in mice by berberine and glibenclamide. Pharm. Biol. 2013, in press. [Google Scholar]
- Gysemans, C.A.; Cardozo, A.K.; Callewaert, H.; Giulietti, A.; Hulshagen, L.; Bouillon, R.; Eizirik, D.L.; Mathieu, C. 1,25-Dihydroxyvitamin D3 modulates expression of chemokines and cytokines in pancreatic islets: Implications for prevention of diabetes in nonobese diabetic mice. Endocrinology 2005, 146, 1956–1964. [Google Scholar] [CrossRef] [PubMed]
- Wolden-Kirk, H.; Rondas, D.; Bugliani, M.; Korf, H.; van lommel, L.; Brusgaard, K.; Christesen, H.T.; Schuit, F.; Proost, P.; Masini, M.; et al. Discovery of molecular pathways mediating 1,25-dihydroxyvitamin D3 protection against cytokine-induced inflammation and damage of human and male mouse islets of Langerhans. Endocrinology 2014, 155, 736–747. [Google Scholar] [CrossRef] [PubMed]
- Lazo de la Vega-Monroy, M.L.; Larrieta, E.; German, M.S.; Baez-Saldana, A.; Fernandez-Mejia, C. Effects of biotin supplementation in the diet on insulin secretion, islet gene expression, glucose homeostasis and beta-cell proportion. J. Nutr. Biochem. 2013, 24, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Cobianchi, L.; Fornoni, A.; Pileggi, A.; Molano, R.D.; Sanabria, N.Y.; Gonzalez-Quintana, J.; Bocca, N.; Marzorati, S.; Zahr, E.; Hogan, A.R.; et al. Riboflavin inhibits IL-6 expression and p38 activation in islet cells. Cell Transplant. 2008, 17, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.Z.; Tai, M.H.; Linning, K.D.; Szabo, C.; Olson, L.K. MafA expression and insulin promoter activity are induced by nicotinamide and related compounds in INS-1 pancreatic beta-cells. Diabetes 2006, 55, 742–750. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Kwon, G.; Cruz, W.S.; Marshall, C.A.; McDaniel, M.L. Metabolic regulation by leucine of translation initiation through the mTOR signaling pathway by pancreatic beta-cells. Diabetes 2001, 50, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, E.M.; Latorraca, M.Q.; Araujo, E.; Beltrá, M.; Oliveras, M.J.; Navarro, M.; Berná, G.; Bedoya, F.J.; Velloso, L.A.; Soria, B.; et al. Taurine supplementation modulates glucose homeostasis and islet function. J. Nutr. Biochem. 2009, 20, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Corless, M.; Kiely, A.; McClenaghan, N.H.; Flatt, P.R.; Newsholme, P. Glutamine regulates expression of key transcription factor, signal transduction, metabolic gene, and protein expression in a clonal pancreatic beta-cell line. J. Endocrinol. 2006, 190, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Ritz-Laser, B.; Meda, P.; Constant, I.; Klages, N.; Charollais, A.; Morales, A.; Magnan, C.; Ktorza, A.; Philippe, J. Glucose-induced preproinsulin gene expression is inhibited by the free fatty acid palmitate. Endocrinology 1999, 140, 4005–4014. [Google Scholar] [PubMed]
- Hagman, D.K.; Hays, L.B.; Parazzoli, S.D.; Poitout, V. Palmitate inhibits insulin gene expression by altering PDX-1 nuclear localization and reducing MafA expression in isolated rat islets of Langerhans. J. Biol. Chem. 2005, 16, 32413–32418. [Google Scholar] [CrossRef]
- Qiu, L.; List, E.O.; Kopchick, J.J. Differentially expressed proteins in the pancreas of diet-induced diabetic mice. Mol. Cell Proteomics 2005, 4, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Dreja, T.; Jovanovic, Z.; Rasche, A.; Kluge, R.; Herwig, R.; Tung, Y.C.; Joost, H.G.; Yeo, G.S.; Al-Hasani, H. Diet-induced gene expression of isolated pancreatic islets from a polygenic mouse model of the metabolic syndrome. Diabetologia 2010, 53, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Kluth, O.; Mirhashemi, F.; Scherneck, S.; Kaiser, D.; Kluge, R.; Neschen, S.; Joost, H.G.; Schürmann, A. Dissociation of lipotoxicity and glucotoxicity in a mouse model of obesity associated diabetes: Role of forkhead box O1 (FOXO1) in glucose-induced beta cell failure. Diabetologia 2011, 54, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.C.; Francini, F.; Gagliardino, J.J.; Massa, M.L. Lipoic acid prevents fructose-induced changes in liver carbohydrate metabolism: Role of oxidative stress. Biochim. Biophys. Acta 2014, 1840, 1145–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obici, S.; Feng, Z.; Morgan, K.; Stein, D.; Karkanias, G.; Rossetti, L. Central administration of oleic acid inhibits glucose production and food intake. Diabetes 2002, 51, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Morgan, K.; Obici, S.; Rossetti, L. Hypothalamic responses to long-chain fatty acids are nutritionally regulated. J. Biol. Chem. 2004, 279, 31139–31148. [Google Scholar] [CrossRef] [PubMed]
- Jäger, S.; Trojan, H.; Kopp, T.; Laszczky, M.N.; Scheffler, A. Pentacyclictriterpene distribution in various plant-rich sources for a new group of multi-potent plant extracts. Molecules 2009, 14, 2016–2031. [Google Scholar] [CrossRef] [PubMed]
- Guinda, A.; Rada, M.; Delgado, T.; Gutierrez-Adane, P.; Castellano, J.M. Pentacyclictriterpenoids from olive fruit and leaf. J. Agric. Food Chem. 2010, 58, 9685–9691. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Wang, N.; Zhao, L.; Lu, F. Berberine in the treatment of type 2 diabetes mellitus: A systemic review and meta-analysis. Evid. Based Complement. Altern. Med. 2012, 2012, 591654. [Google Scholar] [CrossRef]
- De Oliveira, B.F.; Costa, D.C.; Nogueira-Machado, J.A.; Chaves, M.M. β-Carotene, α-tocopherol and ascorbic acid: Differential profile of antioxidant, inflammatory status and regulation of gene expression in human mononuclear cells of diabetic donors. Diabetes Metab. Rev. Res. 2013, 29, 636–645. [Google Scholar] [CrossRef]
- De Oliveira, C.A.; Latorraca, M.Q.; de Mello, M.A.; Carneiro, E.M. Mechanisms of insulin secretion in malnutrition: Modulation by amino acids in rodent models. Amino Acids 2011, 40, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Dickson, L.M.; Rhodes, C.J. Pancreatic beta-cell growth and survival in the onset of type 2 diabetes: A role for protein kinase B in the Akt? Am. J. Physiol. Endocrinol. Metab. 2004, 287, E192–E198. [Google Scholar] [CrossRef] [PubMed]
- Kwon, G.; Marshall, C.A.; Pappan, K.L.; Remedi, M.S.; McDaniel, M.L. Signaling elements involved in the metabolic regulation of mTOR by nutrients, incretins, and growth factors in islets. Diabetes 2004, 53, S225–S232. [Google Scholar] [CrossRef] [PubMed]
- Newsholme, P.; Brennan, L.; Rubi, B.; Maechler, P. New insights into amino acid metabolism, beta-cell function and diabetes. Clin. Sci. 2005, 108, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Newsholme, P.; Bender, K.; Kiely, A.; Brennan, L. Amino acid metabolism, insulin secretion and diabetes. Biochem. Soc. Trans. 2007, 35, 1180–1186. [Google Scholar] [CrossRef] [PubMed]
- Lindström, J.; Ilanne-Parikka, P.; Peltonen, M.; Aunola, S.; Eriksson, J.G.; Hemiö, K.; Hämäläinen, H.; Härkönen, P.; Keinänen-Kiukaanniemi, S.; Laakso, M.; et al. Sustained reduction in the incidence of type 2 diabetes by lifestyle intervention: Follow-up of the Finnish Diabetes Prevention Study. Lancet 2006, 11, 1673–1679. [Google Scholar] [CrossRef]
- Imai, Y.; Patel, H.R.; Doliba, N.M.; Matschinsky, F.M.; Tobias, J.W.; Ahima, R.S. Analysis of gene expression in pancreatic islets from diet-induced obese mice. Physiol. Genomics 2008, 12, 43–51. [Google Scholar] [CrossRef]
- Marchetti, P.; Bugliani, M.; Boggi, U.; Masini, M.; Marselli, L. The pancreatic beta cells in human type 2 diabetes. Adv. Exp. Med. Biol. 2012, 771, 288–309. [Google Scholar] [PubMed]
- Udupa, A.; Nahar, P.; Shah, S.; Kshirsagar, M.; Ghongane, B. A comparative study of effects of omega-3 fatty acids, alpha lipoic acid and vitamin E in type 2 diabetes mellitus. Ann. Med. Health Sci. Res. 2013, 3, 442–446. [Google Scholar] [CrossRef] [PubMed]
- Igoillo-Esteve, M.; Marselli, L.; Cunha, D.A.; Ladrière, L.; Ortis, F.; Grieco, F.A.; Dotta, F.; Weir, G.C.; Marchetti, P.; Eizirik, D.L.; et al. Palmitate induces a pro-inflammatory response in human pancreatic islets that mimics CCL2 expression by beta cells in type 2 diabetes. Diabetologia 2010, 53, 1395–1405. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Hwang, S.; Lee, S.H.; Lee, Y.R.; Shin, J.; Park, K.S.; Cho, Y.M. Genome-wide identification of palmitate-regulated immediate early genes and target genes in pancreatic beta-cells reveals a central role of NF-κB. Mol. Biol. Rep. 2012, 39, 6781–6789. [Google Scholar] [CrossRef] [PubMed]
- Kharroubi, I.; Ladrière, L.; Cardozo, A.K.; Dogusan, Z.; Cnop, M.; Eizirik, D.L. Free fatty acids and cytokines induce pancreatic beta-cell apoptosis by different mechanisms: Role of nuclear factor-kappaB and endoplasmic reticulum stress. Endocrinology 2004, 145, 5087–5096. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidou, V.; Khymenets, O.; Covas, M.I.; de la Torre, R.; Muñoz-Aguayo, D.; Anglada, R.; Farré, M.; Fito, M. Time course of changes in the expression of insulin sensitivity-related genes after an acute load of virgin olive oil. OMICS 2009, 13, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Kallio, P.; Kolehmainen, M.; Laaksonen, D.E.; Kekäläinen, J.; Salopuro, T.; Sivenius, K.; Pulkkinen, L.; Mykkänen, H.M.; Niskanen, L.; Uusitupa, M.; et al. Dietary carbohydrate modification induces alterations in gene expression in abdominal subcutaneous adipose tissue in persons with the metabolic syndrome: The FUNGENUT Study. Am. J. Clin. Nutr. 2007, 85, 1417–1427. [Google Scholar] [PubMed]
- Crujeiras, A.B.; Parra, D.; Milagro, F.I.; Goyenechea, E.; Larrarte, E.; Margareto, J.; Martínez, J.A. Differential expression of oxidative stress and inflammation related genes in peripheral blood mononuclear cells in response to a low-calorie diet: A nutrigenomics study. OMICS 2008, 12, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, H.; Nakae, J.; Kitamura, T.; Park, B.C.; Dragatsis, I.; Accili, D. Transgenic rescue of insulin receptor-deficient mice. J. Clin. Investig. 2004, 114, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Bruning, J.C.; Gautam, D.; Burks, D.J.; Gillette, J.; Schubert, M.; Orban, P.C.; Klein, R.; Krone, W.; Müller-Wieland, D.; Kahn, C.R.; et al. Role of brain insulin receptor in control of body weight and reproduction. Science 2000, 289, 2122–2125. [Google Scholar] [CrossRef] [PubMed]
- Duca, F.A.; Yue, J.T. Fatty acid sensing in the gut and the hypothalamus: In vivo and in vitro perspectives. Mol. Cell Endocrinol. 2014, in press. [Google Scholar]
- Fick, L.J.; Belsham, D.D. Nutrient sensing and insulin signaling in neuropeptide-expressing immortalized, hypothalamic neurons. Cell Cycle 2010, 16, 3186–2193. [Google Scholar]
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Ambors, V. The function of animals micro RNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Saetrom, P.; Snove, O.; Rossi, J.J. MicroRNA-directed transcriptional gene silence in mammalian cells. Proc. Natl. Acad. Sci. USA 2008, 105, 16230–16235. [Google Scholar] [CrossRef] [PubMed]
- García-Segura, L.; Peréz-Andrade, M.; Miranda-Ríos, J. The emerging role of micro RNAs in the regulation of gene expression by nutrients. J. Nutrigenet. Nutrigenomics 2013, 6, 16–31. [Google Scholar] [CrossRef] [PubMed]
- Roseboom, T.; de Rooij, S.; Painter, R. The Dutch famine and its long-term consequences for adult health. Early Hum. Dev. 2006, 82, 485–491. [Google Scholar] [CrossRef] [PubMed]
- De Rooij, S.R.; Painter, R.C.; Roseboom, T.J.; Phillips, D.I.; Osmond, C.; Barker, D.J.; Tanck, M.W.; Michels, R.P.; Bossuyt, P.M.; Bleker, O.P.; et al. Glucose tolerance at age 58 and the decline of glucose tolerance in comparison with age 50 in people prenatally exposed to the Dutch famine. Diabetologia 2006, 49, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.F.; Lin, R.C.; Laybutt, D.R.; Barres, R.; Owens, J.A.; Morris, M.J. Chronic high-fat diet in fathers programs beta-cell dysfunction in female rat offspring. Nature 2010, 467, 963–966. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Kido, Y.; Ashara, S.; Matsuda, T.; Shibutani, Y.; Koyanagi, M.; Kasuga, M. Effect of intrauterine undernutrition during late gestation on pancreatic beta cell mass. BioMed. Res. 2009, 30, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Volkmar, M.; Dedeurwaerder, S.; Cunha, D.A.; Ndlovu, M.N.; Defrance, M.; Deplus, R.; Calonne, E.; Volkmar, U.; Igoillo-Esteve, M.; Naamane, N.; et al. DNA methylation profiling identifies epigenetic dysregulation in pancreatic islets from type 2 diabetic patients. EMBO J. 2012, 21, 1405–1426. [Google Scholar] [CrossRef]
- Fernández-Valverde, S.L.; Taft, R.J.; Mattick, J.S. Micro RNAs in β cell biology, insulin resistance, diabetes and its complications. Diabetes 2011, 60, 1825–1831. [Google Scholar] [CrossRef] [PubMed]
- Frost, R.J.A.; Olson, E.N. Control of glucose homeostasis and insulin sensitivity by the Let-7 family of micro RNAs. Proc. Natl. Acad. Sci. USA 2011, 108, 21075–21080. [Google Scholar] [CrossRef] [PubMed]
- Bladé, C.; Baselga-Escudero, L.; Salvadó, M.J.; Arola-Arnal, A. miRNAS, polyphenols and chronic disease. Mol. Nutr. Food Res. 2013, 57, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.D.; Soule, B.P.; Simone, B.A.; Zaorsky, N.G.; Jin, L.; Simone, N.L. Micro RNA expression altered by diet: Can food be medicinal? Ageing Res. Rev. 2014. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berná, G.; Oliveras-López, M.J.; Jurado-Ruíz, E.; Tejedo, J.; Bedoya, F.; Soria, B.; Martín, F. Nutrigenetics and Nutrigenomics Insights into Diabetes Etiopathogenesis. Nutrients 2014, 6, 5338-5369. https://doi.org/10.3390/nu6115338
Berná G, Oliveras-López MJ, Jurado-Ruíz E, Tejedo J, Bedoya F, Soria B, Martín F. Nutrigenetics and Nutrigenomics Insights into Diabetes Etiopathogenesis. Nutrients. 2014; 6(11):5338-5369. https://doi.org/10.3390/nu6115338
Chicago/Turabian StyleBerná, Genoveva, María Jesús Oliveras-López, Enrique Jurado-Ruíz, Juan Tejedo, Francisco Bedoya, Bernat Soria, and Franz Martín. 2014. "Nutrigenetics and Nutrigenomics Insights into Diabetes Etiopathogenesis" Nutrients 6, no. 11: 5338-5369. https://doi.org/10.3390/nu6115338