STAT3: An Anti-Invasive Factor in Colorectal Cancer?

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Colorectal Cancer (CRC)
2. STAT3 Signaling and Its Role in Cancers
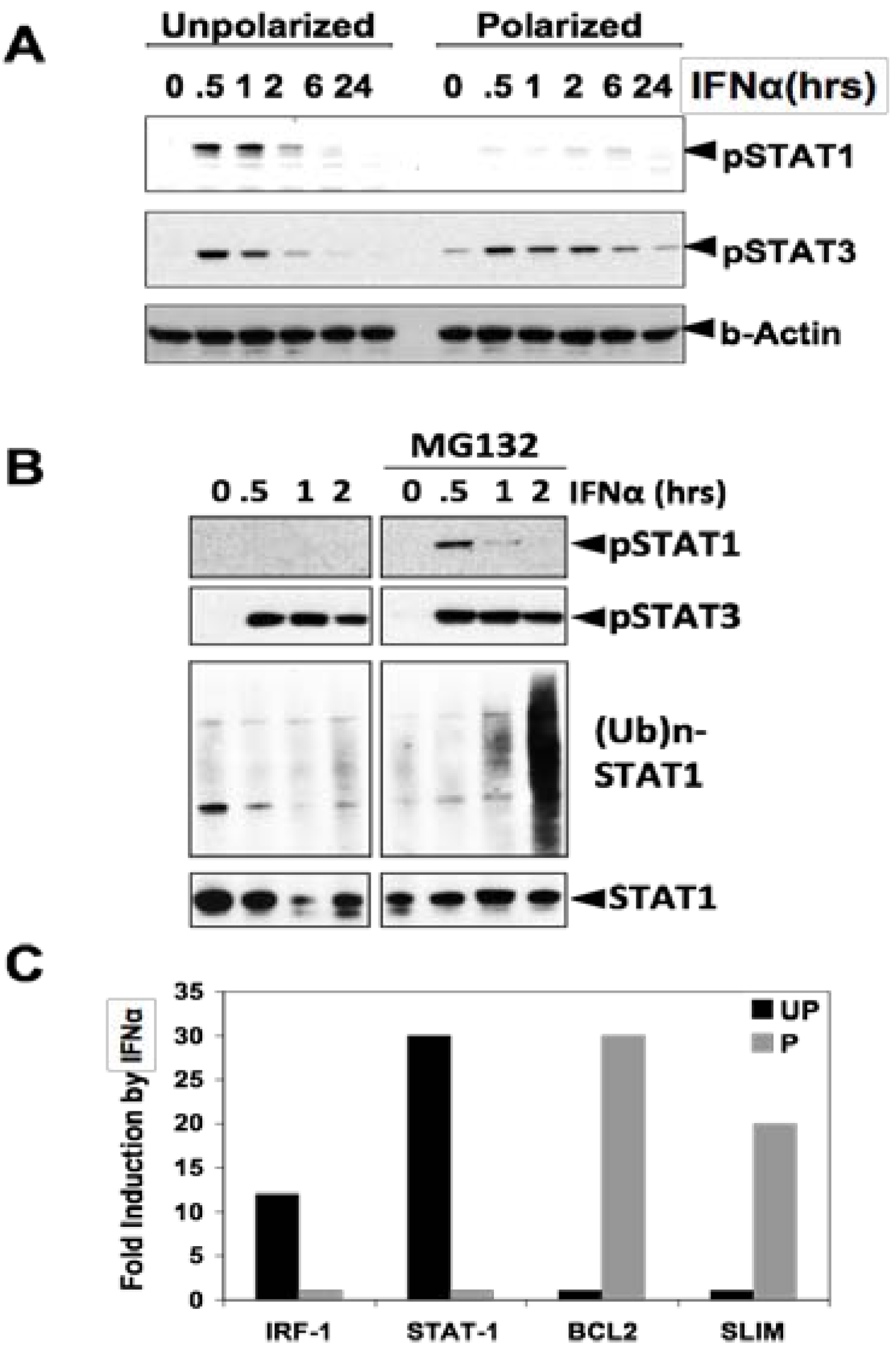
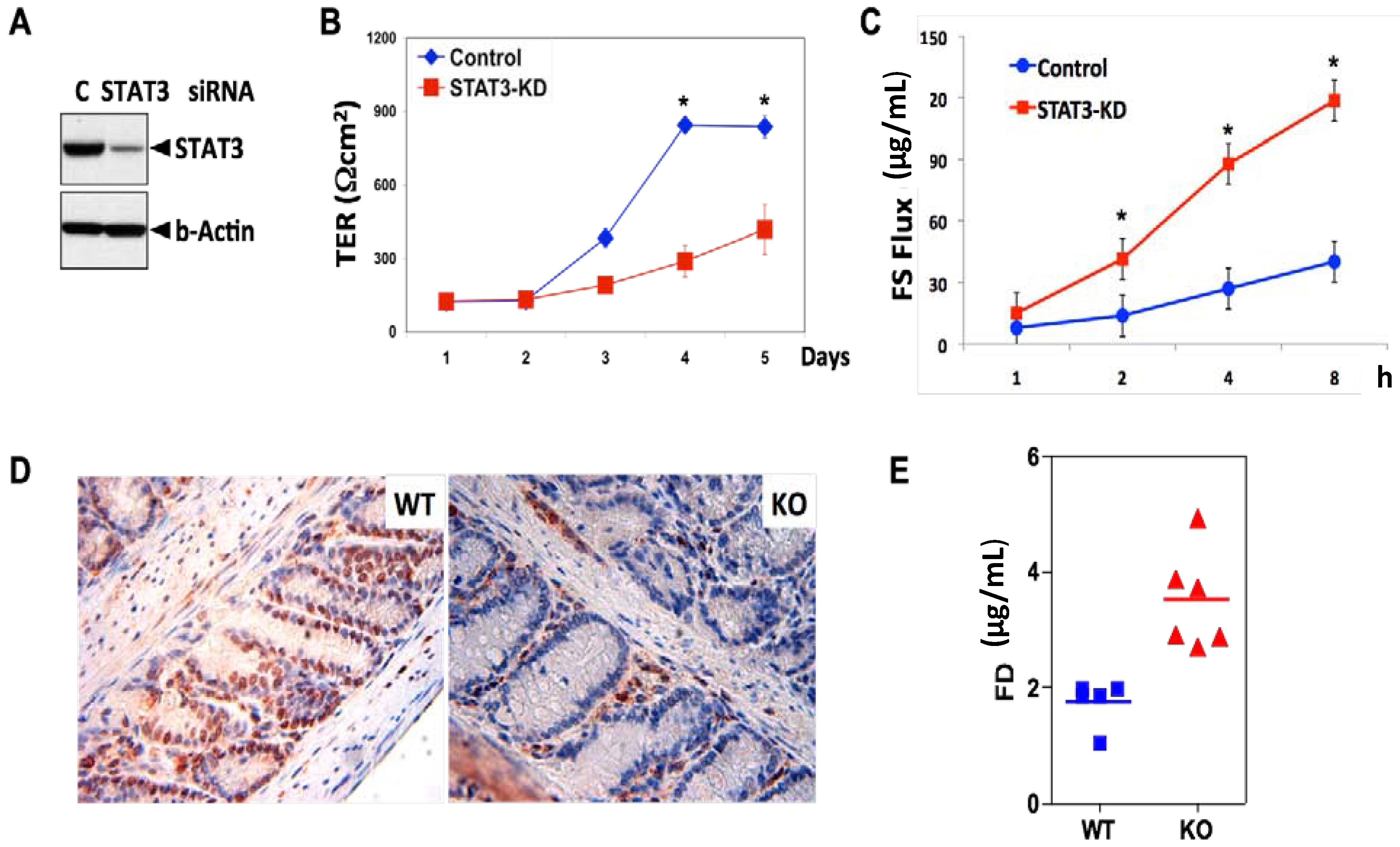
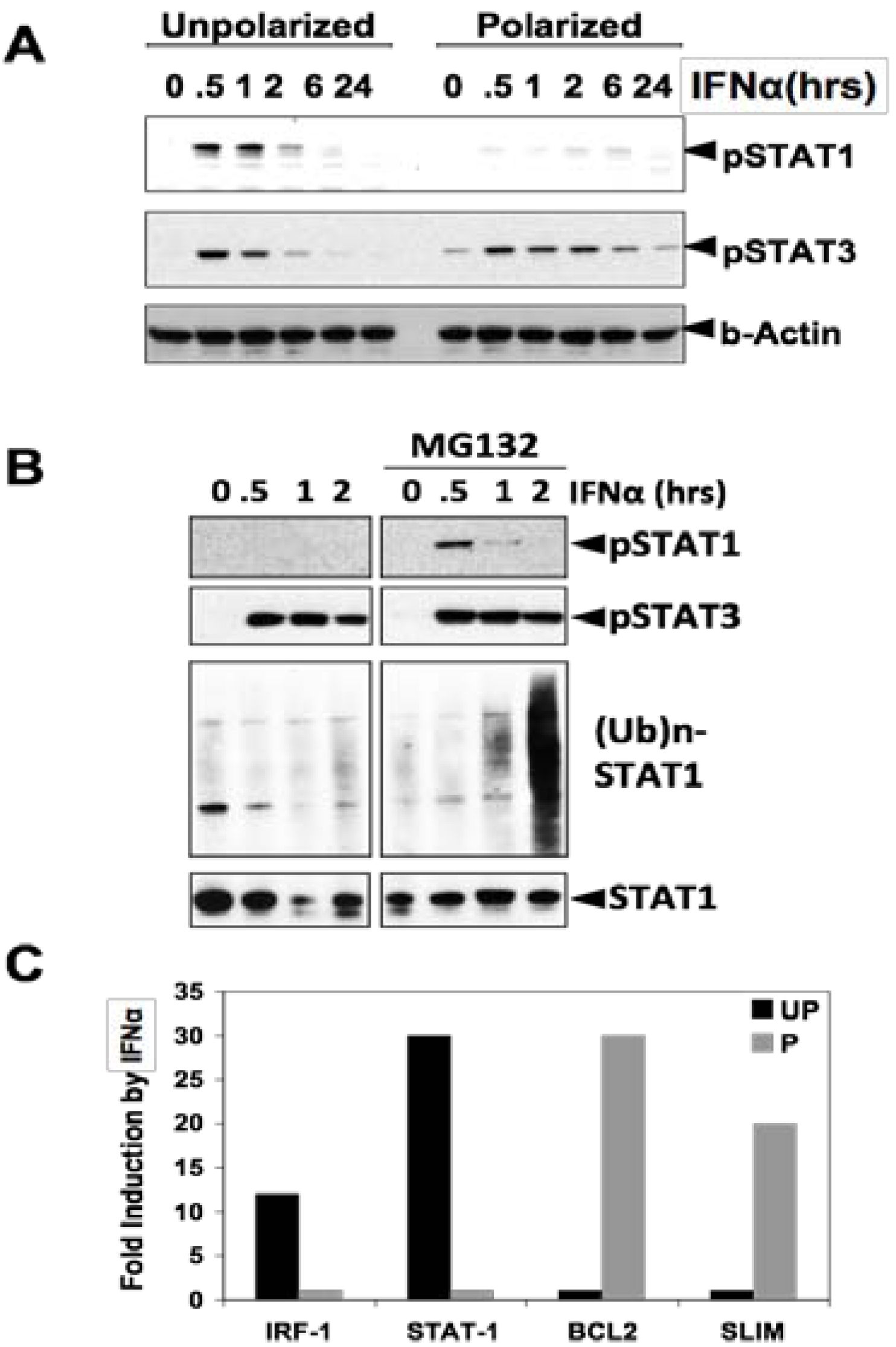
3. STAT3 in Intestinal Epithelial Cells (IEC) Promotes Cell Survival and Epithelial Barrier Function
4. STAT3 Does Not Affect Intestinal Tumor Initiation
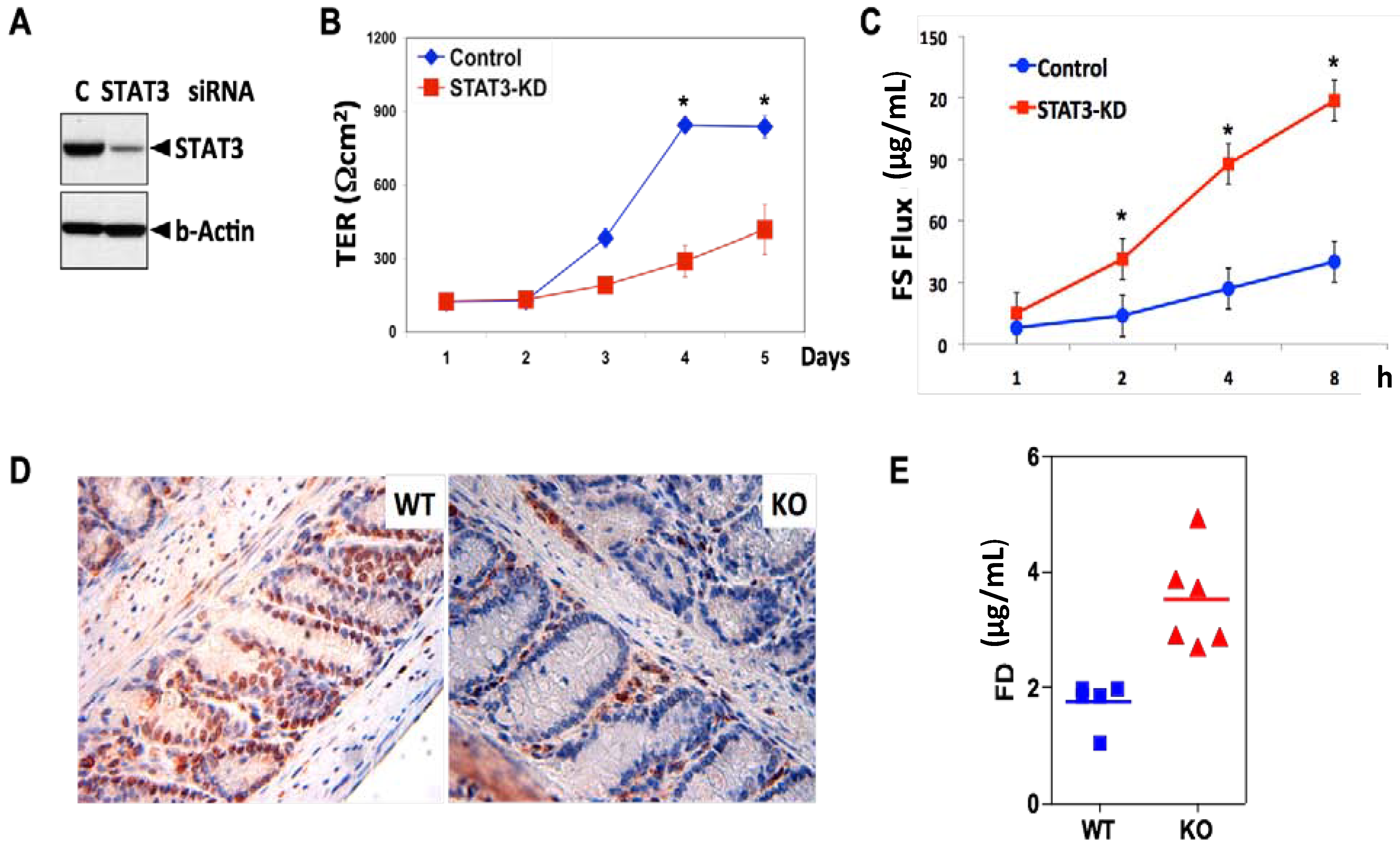
5. STAT3 Suppresses Adenoma to Adenocarcinoma Transition
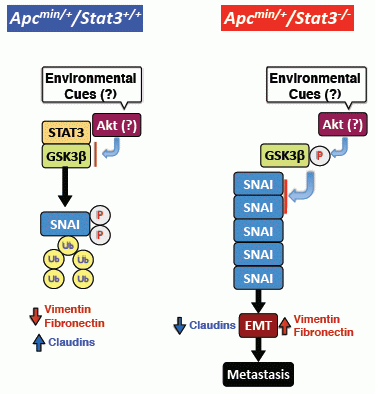
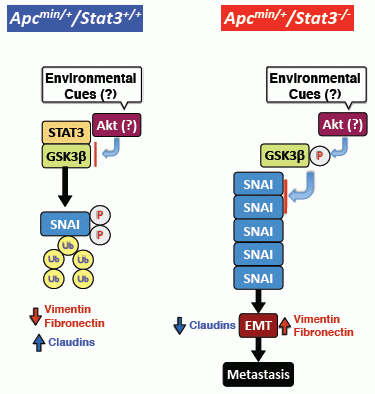
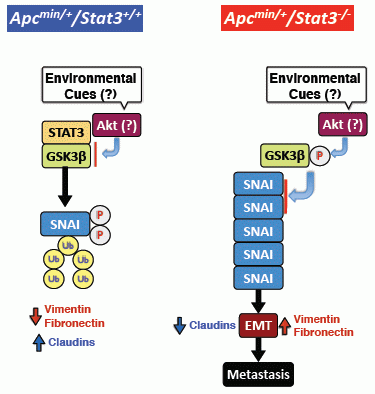
6. STAT3 Inhibits Tumor Invasion via Regulation of an EMT Inducer SNAI-1
7. STAT3 Coordinates SNAI-1 Stability as a Molecular Adaptor
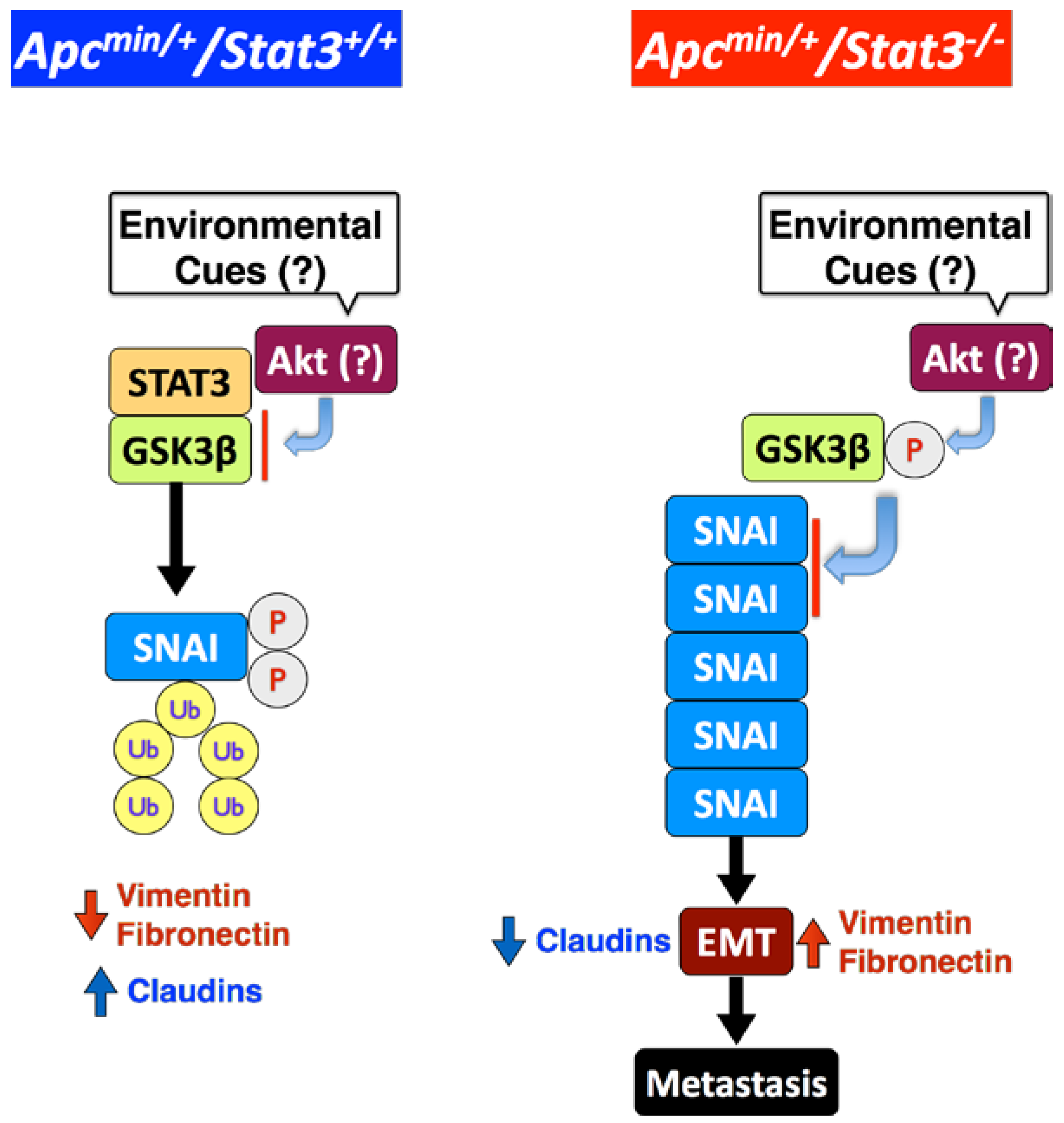
8. Clinical Implications
9. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Jemal, A.; Center, M.M.; DeSantis, C.; Ward, E.M. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol. Biomark. Prev. 2010, 19, 1893–1907. [Google Scholar] [CrossRef]
- Grady, W.M.; Carethers, J.M. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology 2008, 135, 1079–1099. [Google Scholar] [CrossRef]
- Rajagopalan, H.; Nowak, M.A.; Vogelstein, B.; Lengauer, C. The significance of unstable chromosomes in colorectal cancer. Nat. Rev. Cancer 2003, 3, 695–701. [Google Scholar] [CrossRef]
- Pino, M.S.; Chung, D.C. The chromosomal instability pathway in colon cancer. Gastroenterology 2010, 138, 2059–2072. [Google Scholar] [CrossRef]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instability in colorectal cancers. Nature 1997, 386, 623–627. [Google Scholar] [CrossRef]
- Fensterer, H.; Radlwimmer, B.; Strater, J.; Buchholz, M.; Aust, D.E.; Julie, C.; Radvanyi, F.; Nordlinger, B.; Belluco, C.; van Cutsem, E.; et al. Matrix-comparative genomic hybridization from multicenter formalin-fixed paraffin-embedded colorectal cancer tissue blocks. BMC Cancer 2007, 7. [Google Scholar] [CrossRef]
- Knutsen, T.; Padilla-Nash, H.M.; Wangsa, D.; Barenboim-Stapleton, L.; Camps, J.; McNeil, N.; Difilippantonio, M.J.; Ried, T. Definitive molecular cytogenetic characterization of 15 colorectal cancer cell lines. Genes Chromosom. Cancer 2010, 49, 204–223. [Google Scholar]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular origins of cancer: Molecular basis of colorectal cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef]
- Walther, A.; Johnstone, E.; Swanton, C.; Midgley, R.; Tomlinson, I.; Kerr, D. Genetic prognostic and predictive markers in colorectal cancer. Nat. Rev. Cancer 2009, 9, 489–499. [Google Scholar] [CrossRef]
- De Sousa, E.M.F.; Wang, X.; Jansen, M.; Fessler, E.; Trinh, A.; de Rooij, L.P.; de Jong, J.H.; de Boer, O.J.; van Leersum, R.; Bijlsma, M.F.; et al. Poor-prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nat. Med. 2013, 19, 614–618. [Google Scholar] [CrossRef]
- Sadanandam, A.; Lyssiotis, C.A.; Homicsko, K.; Collisson, E.A.; Gibb, W.J.; Wullschleger, S.; Ostos, L.C.; Lannon, W.A.; Grotzinger, C.; Del Rio, M.; et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat. Med. 2013, 19, 619–625. [Google Scholar] [CrossRef]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Nordlinger, B.; Cervantes, A.; Group, E.G.W. Advanced colorectal cancer: ESMO Clinical Practice Guidelines for treatment. Ann. Oncol. 2010, 21, v93–v97. [Google Scholar] [CrossRef]
- Lee, J.; Kim, J.C.; Lee, S.E.; Quinley, C.; Kim, H.; Herdman, S.; Corr, M.; Raz, E. Signal transducer and activator of transcription 3 (STAT3) protein suppresses adenoma-to-carcinoma transition in Apcmin/+ mice via regulation of Snail-1 (SNAI) protein stability. J. Biol. Chem. 2012, 287, 18182–18189. [Google Scholar]
- Musteanu, M.; Blaas, L.; Mair, M.; Schlederer, M.; Bilban, M.; Tauber, S.; Esterbauer, H.; Mueller, M.; Casanova, E.; Kenner, L.; et al. Stat3 is a negative regulator of intestinal tumor progression in Apc(Min) mice. Gastroenterology 2010, 138. [Google Scholar] [CrossRef]
- Akira, S. Roles of STAT3 defined by tissue-specific gene targeting. Oncogene 2000, 19, 2607–2611. [Google Scholar] [CrossRef]
- Kane, A.; Deenick, E.K.; Ma, C.S.; Cook, M.C.; Uzel, G.; Tangye, S.G. STAT3 is a central regulator of lymphocyte differentiation and function. Curr. Opin. Immunol. 2014, 28C, 49–57. [Google Scholar]
- Kamran, M.Z.; Patil, P.; Gude, R.P. Role of STAT3 in cancer metastasis and translational advances. BioMed Res. Int. 2013, 2013. [Google Scholar] [CrossRef]
- Rebe, C.; Vegran, F.; Berger, H.; Ghiringhelli, F. STAT3 activation: A key factor in tumor immunoescape. Jak-stat 2013, 2, e23010. [Google Scholar] [CrossRef]
- Yuan, Z.L.; Guan, Y.J.; Chatterjee, D.; Chin, Y.E. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 2005, 307, 269–273. [Google Scholar] [CrossRef]
- Yang, J.; Chatterjee-Kishore, M.; Staugaitis, S.M.; Nguyen, H.; Schlessinger, K.; Levy, D.E.; Stark, G.R. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res. 2005, 65, 939–947. [Google Scholar]
- Yang, J.; Liao, X.; Agarwal, M.K.; Barnes, L.; Auron, P.E.; Stark, G.R. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NF-κb. Genes Dev. 2007, 21, 1396–1408. [Google Scholar] [CrossRef]
- Pfeffer, L.M.; Mullersman, J.E.; Pfeffer, S.R.; Murti, A.; Shi, W.; Yang, C.H. STAT3 as an adapter to couple phosphatidylinositol 3-kinase to the IFNAR1 chain of the type I interferon receptor. Science 1997, 276, 1418–1420. [Google Scholar]
- Ng, D.C.; Lin, B.H.; Lim, C.P.; Huang, G.; Zhang, T.; Poli, V.; Cao, X. Stat3 regulates microtubules by antagonizing the depolymerization activity of stathmin. J. Cell Biol. 2006, 172, 245–257. [Google Scholar] [CrossRef]
- Silver, D.L.; Naora, H.; Liu, J.; Cheng, W.; Montell, D.J. Activated signal transducer and activator of transcription (STAT) 3: Localization in focal adhesions and function in ovarian cancer cell motility. Cancer Res. 2004, 64, 3550–3558. [Google Scholar]
- Frank, D.A. STAT3 as a central mediator of neoplastic cellular transformation. Cancer Lett. 2007, 251, 199–210. [Google Scholar] [CrossRef]
- Bar-Natan, M.; Nelson, E.A.; Xiang, M.; Frank, D.A. STAT signaling in the pathogenesis and treatment of myeloid malignancies. JAK-STAT 2012, 1, 55–64. [Google Scholar] [CrossRef]
- Kusaba, T.; Nakayama, T.; Yamazumi, K.; Yakata, Y.; Yoshizaki, A.; Nagayasu, T.; Sekine, I. Expression of p-STAT3 in human colorectal adenocarcinoma and adenoma; correlation with clinicopathological factors. J. Clin. Pathol. 2005, 58, 833–838. [Google Scholar] [CrossRef]
- Morikawa, T.; Baba, Y.; Yamauchi, M.; Kuchiba, A.; Nosho, K.; Shima, K.; Tanaka, N.; Huttenhower, C.; Frank, D.A.; Fuchs, C.S.; et al. STAT3 expression, molecular features, inflammation patterns, and prognosis in a database of 724 colorectal cancers. Clin. Cancer Res. 2011, 17, 1452–1462. [Google Scholar] [CrossRef]
- Koskela, H.L.; Eldfors, S.; Ellonen, P.; van Adrichem, A.J.; Kuusanmaki, H.; Andersson, E.I.; Lagstrom, S.; Clemente, M.J.; Olson, T.; Jalkanen, S.E.; et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N. Engl. J. Med. 2012, 366, 1905–1913. [Google Scholar] [CrossRef]
- Lee, J.; Mo, J.H.; Katakura, K.; Alkalay, I.; Rucker, A.N.; Liu, Y.T.; Lee, H.K.; Shen, C.; Cojocaru, G.; Shenouda, S.; et al. Maintenance of colonic homeostasis by distinctive apical TLR9 signalling in intestinal epithelial cells. Nat. Cell Biol. 2006, 8, 1327–1336. [Google Scholar] [CrossRef]
- Mellman, I.; Nelson, W.J. Coordinated protein sorting, targeting and distribution in polarized cells. Nat. Rev. Mol. Cell Biol. 2008, 9, 833–845. [Google Scholar] [CrossRef]
- Tanaka, T.; Soriano, M.A.; Grusby, M.J. SLIM is a nuclear ubiquitin E3 ligase that negatively regulates STAT signaling. Immunity 2005, 22, 729–736. [Google Scholar] [CrossRef]
- Jarnicki, A.; Putoczki, T.; Ernst, M. Stat3: Linking inflammation to epithelial cancer—More than a “gut” feeling? Cell Div. 2010, 5. [Google Scholar] [CrossRef]
- Kawada, M.; Seno, H.; Uenoyama, Y.; Sawabu, T.; Kanda, N.; Fukui, H.; Shimahara, Y.; Chiba, T. Signal transducers and activators of transcription 3 activation is involved in nuclear accumulation of beta-catenin in colorectal cancer. Cancer Res. 2006, 66, 2913–2917. [Google Scholar] [CrossRef]
- Lee, S.H.; Hu, L.L.; Gonzalez-Navajas, J.; Seo, G.S.; Shen, C.; Brick, J.; Herdman, S.; Varki, N.; Corr, M.; Lee, J.; et al. ERK activation drives intestinal tumorigenesis in Apc(min/+) mice. Nat. Med. 2010, 16, 665–670. [Google Scholar] [CrossRef]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef]
- Liang, J.; Nagahashi, M.; Kim, E.Y.; Harikumar, K.B.; Yamada, A.; Huang, W.C.; Hait, N.C.; Allegood, J.C.; Price, M.M.; Avni, D.; et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell 2013, 23, 107–120. [Google Scholar] [CrossRef]
- Clapper, M.L.; Cooper, H.S.; Chang, W.C. Dextran sulfate sodium-induced colitis-associated neoplasia: A promising model for the development of chemopreventive interventions. Acta Pharmacol. Sin. 2007, 28, 1450–1459. [Google Scholar] [CrossRef]
- Takahashi, M.; Nakatsugi, S.; Sugimura, T.; Wakabayashi, K. Frequent mutations of the beta-catenin gene in mouse colon tumors induced by azoxymethane. Carcinogenesis 2000, 21, 1117–1120. [Google Scholar] [CrossRef]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef]
- Guo, F.; Parker Kerrigan, B.C.; Yang, D.; Hu, L.; Shmulevich, I.; Sood, A.K.; Xue, F.; Zhang, W. Post-transcriptional regulatory network of epithelial-to-mesenchymal and mesenchymal-to-epithelial transitions. J. Hematol. Oncol. 2014, 7. [Google Scholar] [CrossRef]
- Tam, W.L.; Weinberg, R.A. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 2013, 19, 1438–1449. [Google Scholar] [CrossRef]
- Nieto, M.A. Epithelial plasticity: A common theme in embryonic and cancer cells. Science 2013, 342. [Google Scholar] [CrossRef]
- Oka, H.; Shiozaki, H.; Kobayashi, K.; Inoue, M.; Tahara, H.; Kobayashi, T.; Takatsuka, Y.; Matsuyoshi, N.; Hirano, S.; Takeichi, M.; et al. Expression of E-cadherin cell adhesion molecules in human breast cancer tissues and its relationship to metastasis. Cancer Res. 1993, 53, 1696–1701. [Google Scholar]
- Schipper, J.H.; Frixen, U.H.; Behrens, J.; Unger, A.; Jahnke, K.; Birchmeier, W. E-cadherin expression in squamous cell carcinomas of head and neck: Inverse correlation with tumor dedifferentiation and lymph node metastasis. Cancer Res. 1991, 51, 6328–6337. [Google Scholar]
- Umbas, R.; Isaacs, W.B.; Bringuier, P.P.; Schaafsma, H.E.; Karthaus, H.F.; Oosterhof, G.O.; Debruyne, F.M.; Schalken, J.A. Decreased E-cadherin expression is associated with poor prognosis in patients with prostate cancer. Cancer Res. 1994, 54, 3929–3933. [Google Scholar]
- Yadav, A.; Kumar, B.; Datta, J.; Teknos, T.N.; Kumar, P. IL-6 promotes head and neck tumor metastasis by inducing epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling pathway. Mol. Cancer Res. 2011, 9, 1658–1667. [Google Scholar] [CrossRef]
- Xie, T.X.; Huang, F.J.; Aldape, K.D.; Kang, S.H.; Liu, M.; Gershenwald, J.E.; Xie, K.; Sawaya, R.; Huang, S. Activation of Stat3 in human melanoma promotes brain metastasis. Cancer Res. 2006, 66, 3188–3196. [Google Scholar] [CrossRef]
- Lee, J.; Mo, J.H.; Raz, E.; Department of Medicine, University of California, San Diego, CA, USA. Unpublished data. 2014.
- Chen, X.; Halberg, R.B.; Burch, R.P.; Dove, W.F. Intestinal adenomagenesis involves core molecular signatures of the epithelial-mesenchymal transition. J. Mol. Histol. 2008, 39, 283–294. [Google Scholar] [CrossRef]
- Tsareva, S.A.; Moriggl, R.; Corvinus, F.M.; Wiederanders, B.; Schutz, A.; Kovacic, B.; Friedrich, K. Signal transducer and activator of transcription 3 activation promotes invasive growth of colon carcinomas through matrix metalloproteinase induction. Neoplasia 2007, 9, 279–291. [Google Scholar] [CrossRef]
- Ota, I.; Li, X.Y.; Hu, Y.; Weiss, S.J. Induction of a MT1-MMP and MT2-MMP-dependent basement membrane transmigration program in cancer cells by Snail1. Proc. Natl. Acad. Sci. USA 2009, 106, 20318–20323. [Google Scholar] [CrossRef]
- Hotary, K.B.; Allen, E.D.; Brooks, P.C.; Datta, N.S.; Long, M.W.; Weiss, S.J. Membrane type I matrix metalloproteinase usurps tumor growth control imposed by the three-dimensional extracellular matrix. Cell 2003, 114, 33–45. [Google Scholar] [CrossRef]
- Kanazawa, A.; Oshima, T.; Yoshihara, K.; Tamura, S.; Yamada, T.; Inagaki, D.; Sato, T.; Yamamoto, N.; Shiozawa, M.; Morinaga, S.; et al. Relation of MT1-MMP gene expression to outcomes in colorectal cancer. J. Surg. Oncol. 2010, 102, 571–575. [Google Scholar] [CrossRef]
- Zucker, S.; Cao, J. Selective matrix metalloproteinase (MMP) inhibitors in cancer therapy: Ready for prime time? Cancer Biol. Ther. 2009, 8, 2371–2373. [Google Scholar] [CrossRef]
- Xiong, H.; Hong, J.; Du, W.; Lin, Y.W.; Ren, L.L.; Wang, Y.C.; Su, W.Y.; Wang, J.L.; Cui, Y.; Wang, Z.H.; et al. Roles of STAT3 and ZEB1 proteins in E-cadherin down-regulation and human colorectal cancer epithelial-mesenchymal transition. J. Biol. Chem. 2012, 287, 5819–5832. [Google Scholar] [CrossRef]
- Zhou, C.; Tong, Y.; Wawrowsky, K.; Melmed, S. PTTG acts as a STAT3 target gene for colorectal cancer cell growth and motility. Oncogene 2014, 33, 851–861. [Google Scholar] [CrossRef]
- Calon, A.; Espinet, E.; Palomo-Ponce, S.; Tauriello, D.V.; Iglesias, M.; Cespedes, M.V.; Sevillano, M.; Nadal, C.; Jung, P.; Zhang, X.H.; et al. Dependency of colorectal cancer on a TGF-beta-driven program in stromal cells for metastasis initiation. Cancer Cell 2012, 22, 571–584. [Google Scholar]
- Zhou, B.P.; Deng, J.; Xia, W.; Xu, J.; Li, Y.M.; Gunduz, M.; Hung, M.C. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat. Cell Biol. 2004, 6, 931–940. [Google Scholar] [CrossRef]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef]
- Doble, B.W.; Patel, S.; Wood, G.A.; Kockeritz, L.K.; Woodgett, J.R. Functional redundancy of GSK-3α and GSK-3β in Wnt/beta-catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev. Cell 2007, 12, 957–971. [Google Scholar] [CrossRef]
- Waldner, M.J.; Foersch, S.; Neurath, M.F. Interleukin-6—A key regulator of colorectal cancer development. Int. J. Biol. Sci. 2012, 8, 1248–1253. [Google Scholar]
- Senft, C.; Priester, M.; Polacin, M.; Schroder, K.; Seifert, V.; Kogel, D.; Weissenberger, J. Inhibition of the JAK-2/STAT3 signaling pathway impedes the migratory and invasive potential of human glioblastoma cells. J. Neuro-Oncol. 2011, 101, 393–403. [Google Scholar] [CrossRef]
- Sai, K.; Wang, S.; Balasubramaniyan, V.; Conrad, C.; Lang, F.F.; Aldape, K.; Szymanski, S.; Fokt, I.; Dasgupta, A.; Madden, T.; et al. Induction of cell-cycle arrest and apoptosis in glioblastoma stem-like cells by WP1193, a novel small molecule inhibitor of the JAK2/STAT3 pathway. J. Neuro-Oncol. 2012, 107, 487–501. [Google Scholar] [CrossRef]
- Sen, M.; Thomas, S.M.; Kim, S.; Yeh, J.I.; Ferris, R.L.; Johnson, J.T.; Duvvuri, U.; Lee, J.; Sahu, N.; Joyce, S.; et al. First-in-human trial of a STAT3 decoy oligonucleotide in head and neck tumors: Implications for cancer therapy. Cancer Discov. 2012, 2, 694–705. [Google Scholar] [CrossRef]
- Li, G.H.; Wei, H.; Chen, Z.T.; Lv, S.Q.; Yin, C.L.; Wang, D.L. STAT3 silencing with lentivirus inhibits growth and induces apoptosis and differentiation of U251 cells. J. Neuro-Oncol. 2009, 91, 165–174. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
De Jong, P.R.; Mo, J.-H.; Harris, A.R.; Lee, J.; Raz, E. STAT3: An Anti-Invasive Factor in Colorectal Cancer? Cancers 2014, 6, 1394-1407. https://doi.org/10.3390/cancers6031394
De Jong PR, Mo J-H, Harris AR, Lee J, Raz E. STAT3: An Anti-Invasive Factor in Colorectal Cancer? Cancers. 2014; 6(3):1394-1407. https://doi.org/10.3390/cancers6031394
Chicago/Turabian StyleDe Jong, Petrus Rudolf, Ji-Hun Mo, Alexandra R. Harris, Jongdae Lee, and Eyal Raz. 2014. "STAT3: An Anti-Invasive Factor in Colorectal Cancer?" Cancers 6, no. 3: 1394-1407. https://doi.org/10.3390/cancers6031394