
Novel Mechanisms of ALK Activation Revealed by Analysis of the Y1278S Neuroblastoma Mutation

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
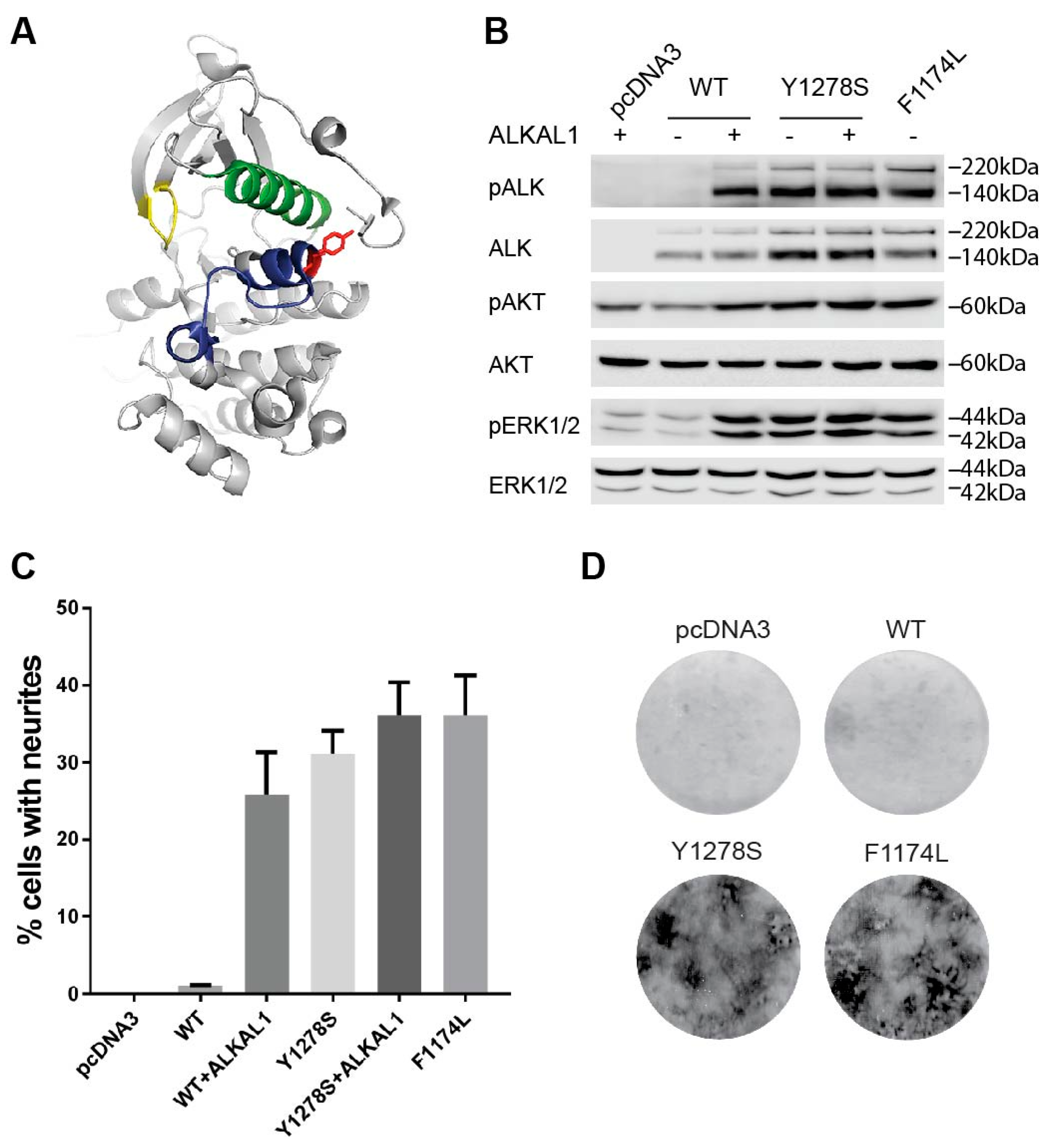
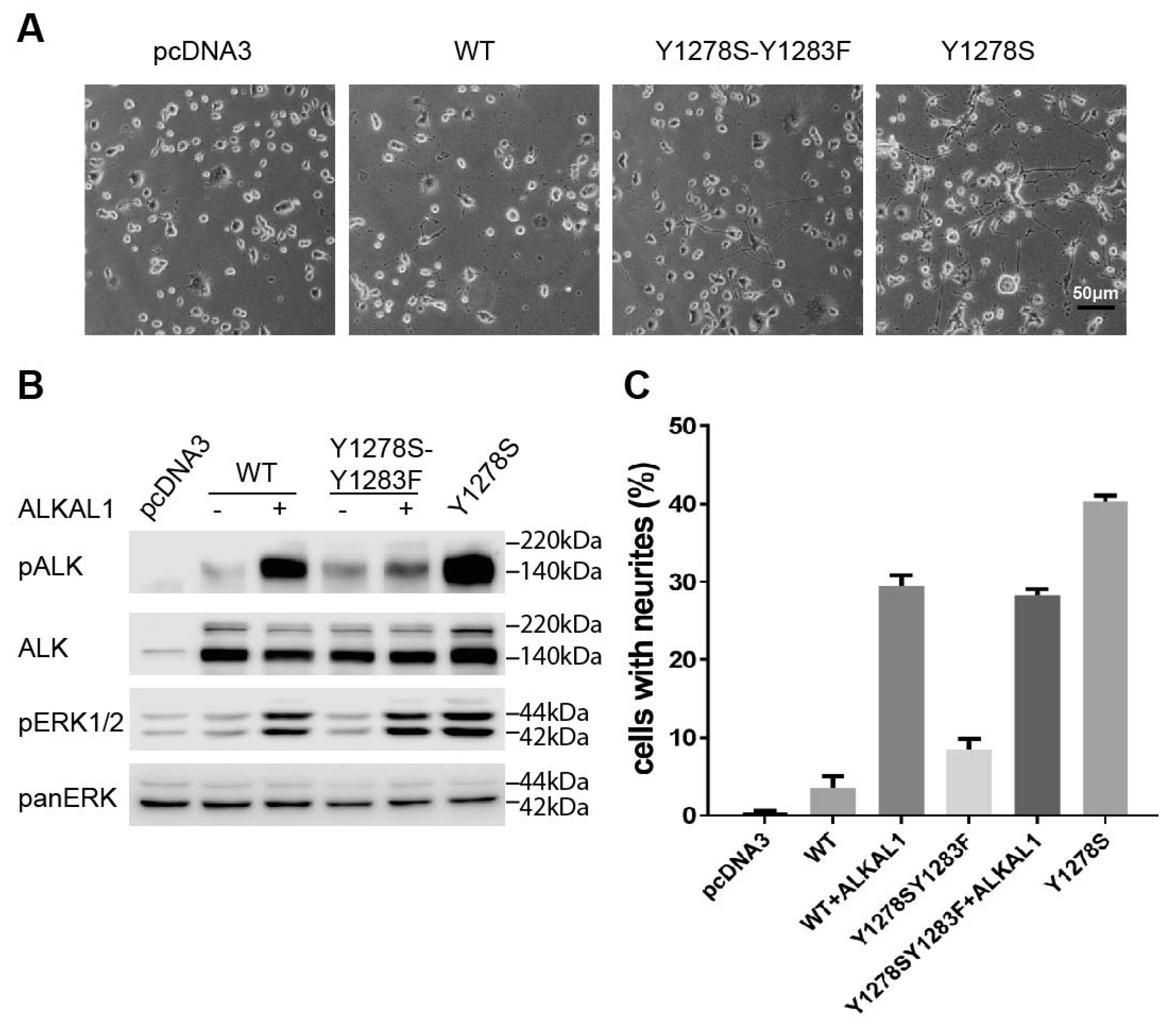
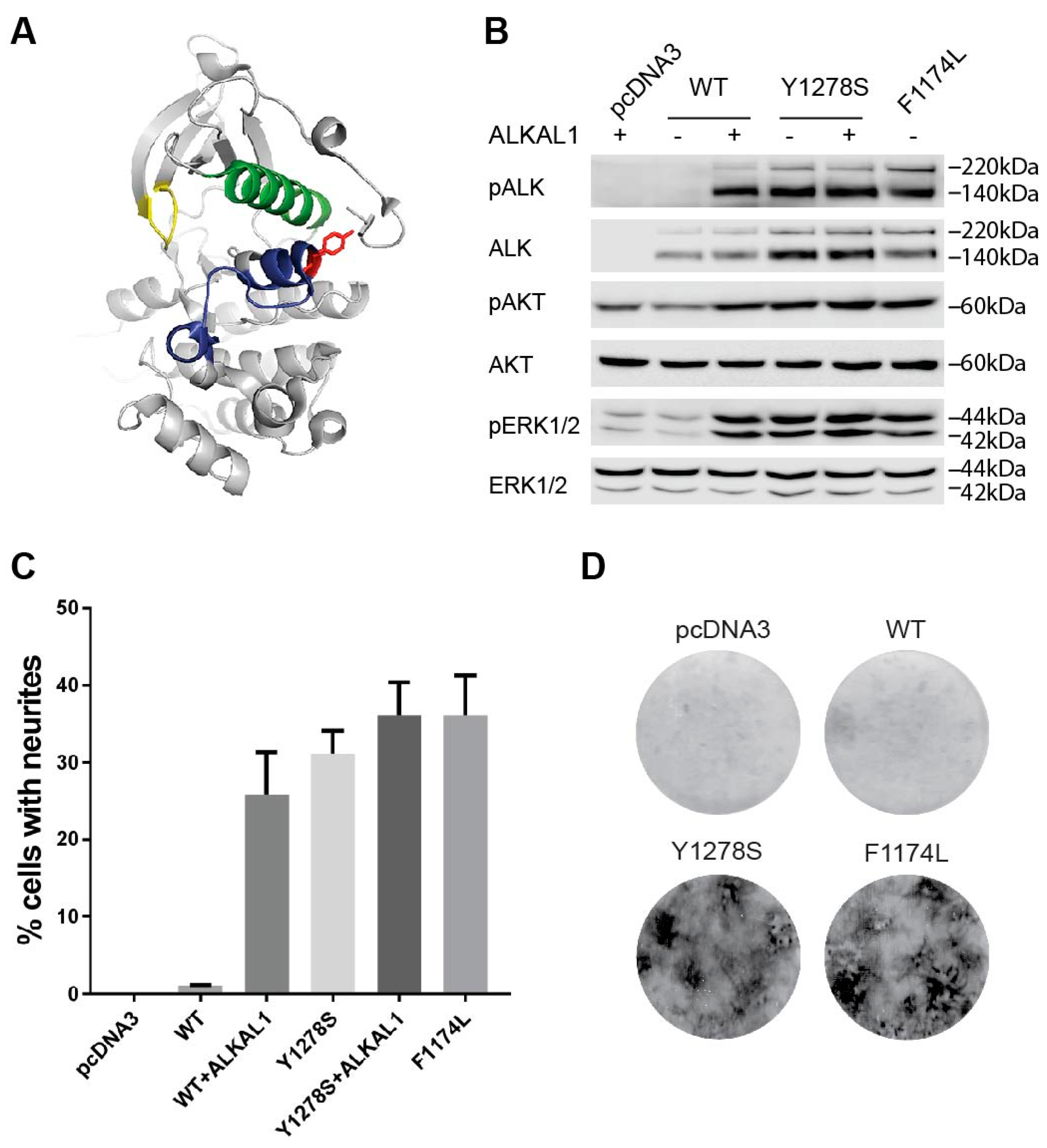
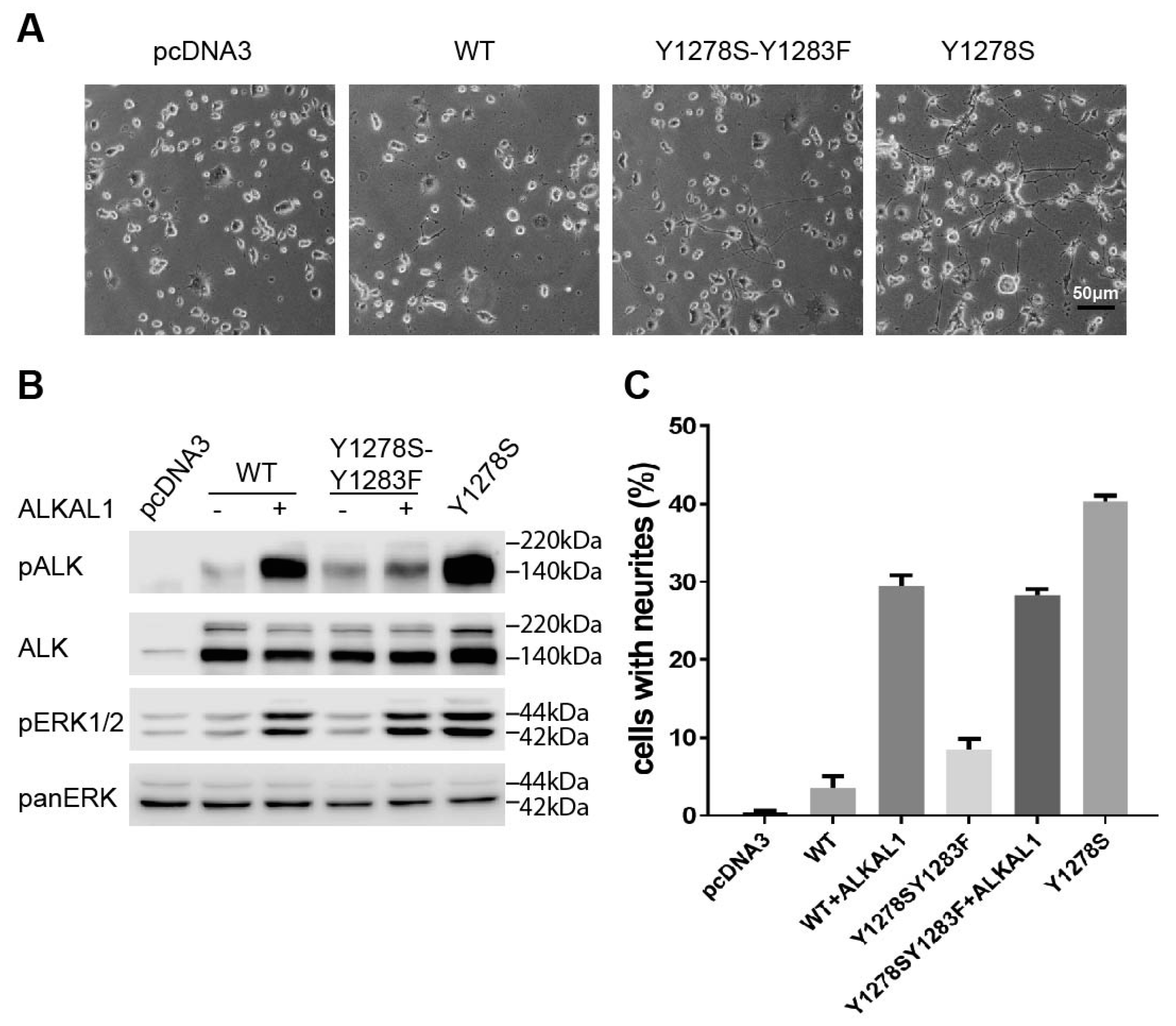
2.1. The Y1278S Neuroblastoma ALK Mutation Results in Ligand Independent Activation
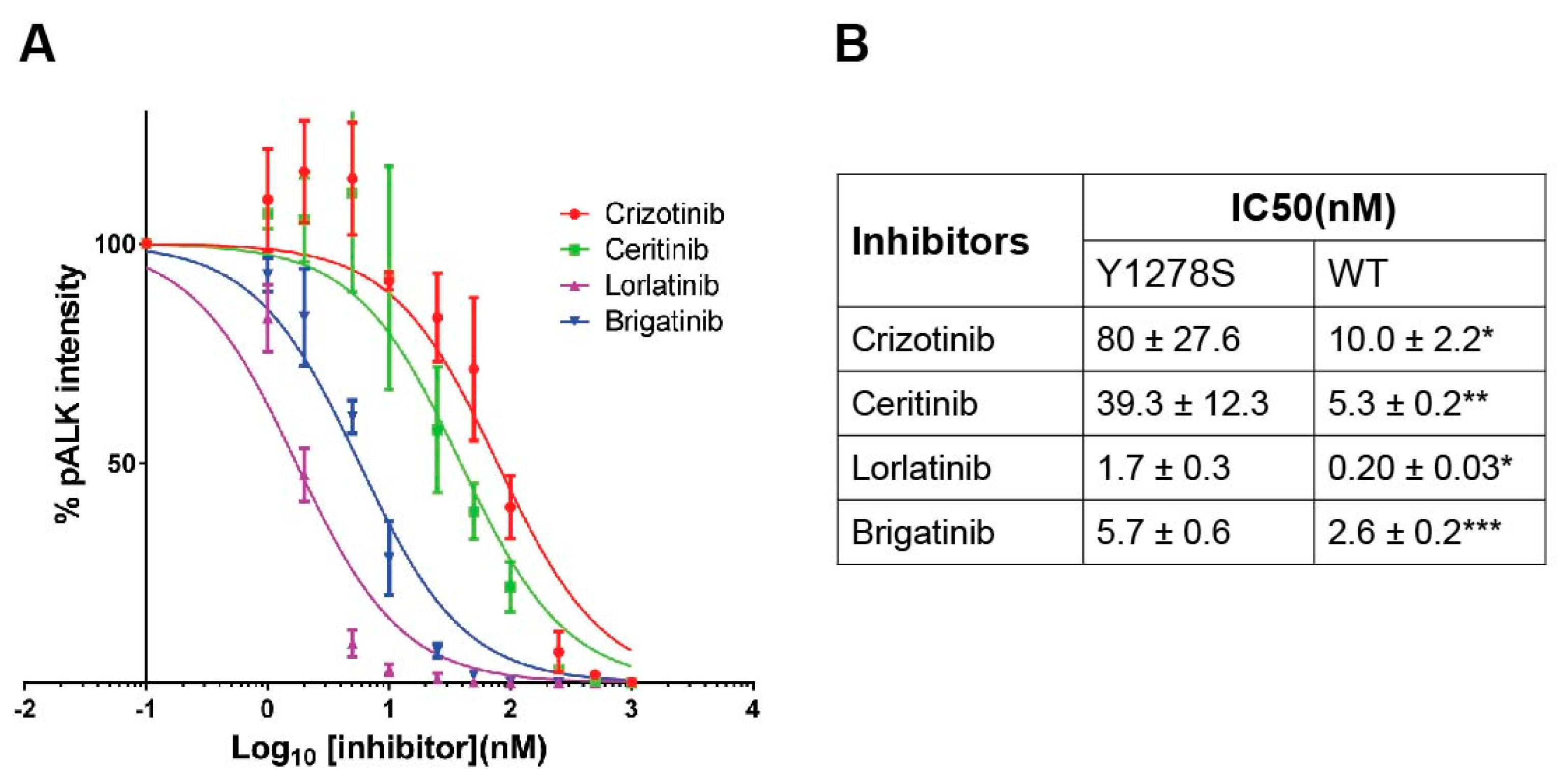
2.2. Sensitivity of ALK-Y1278S to ALK Inhibitors
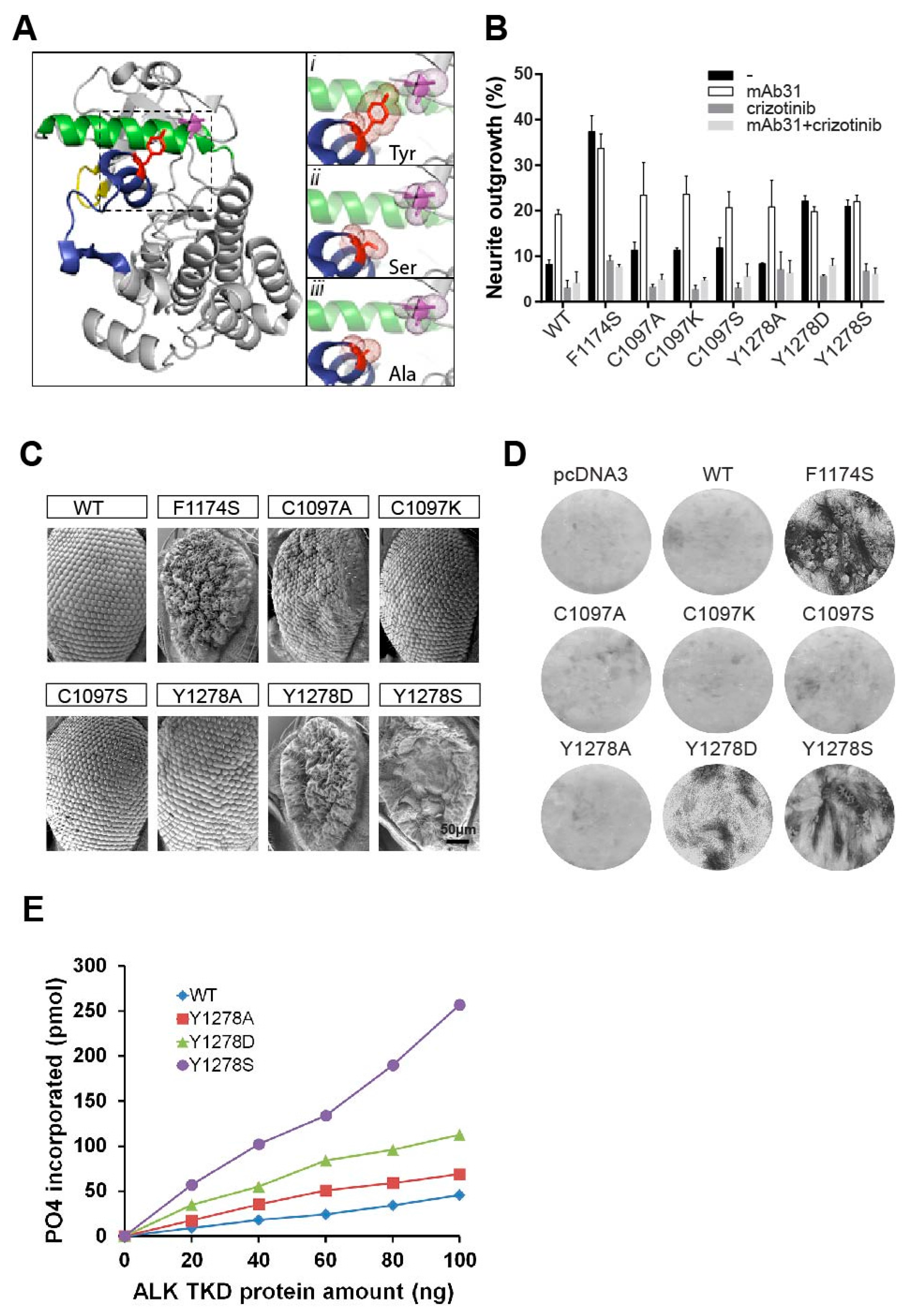
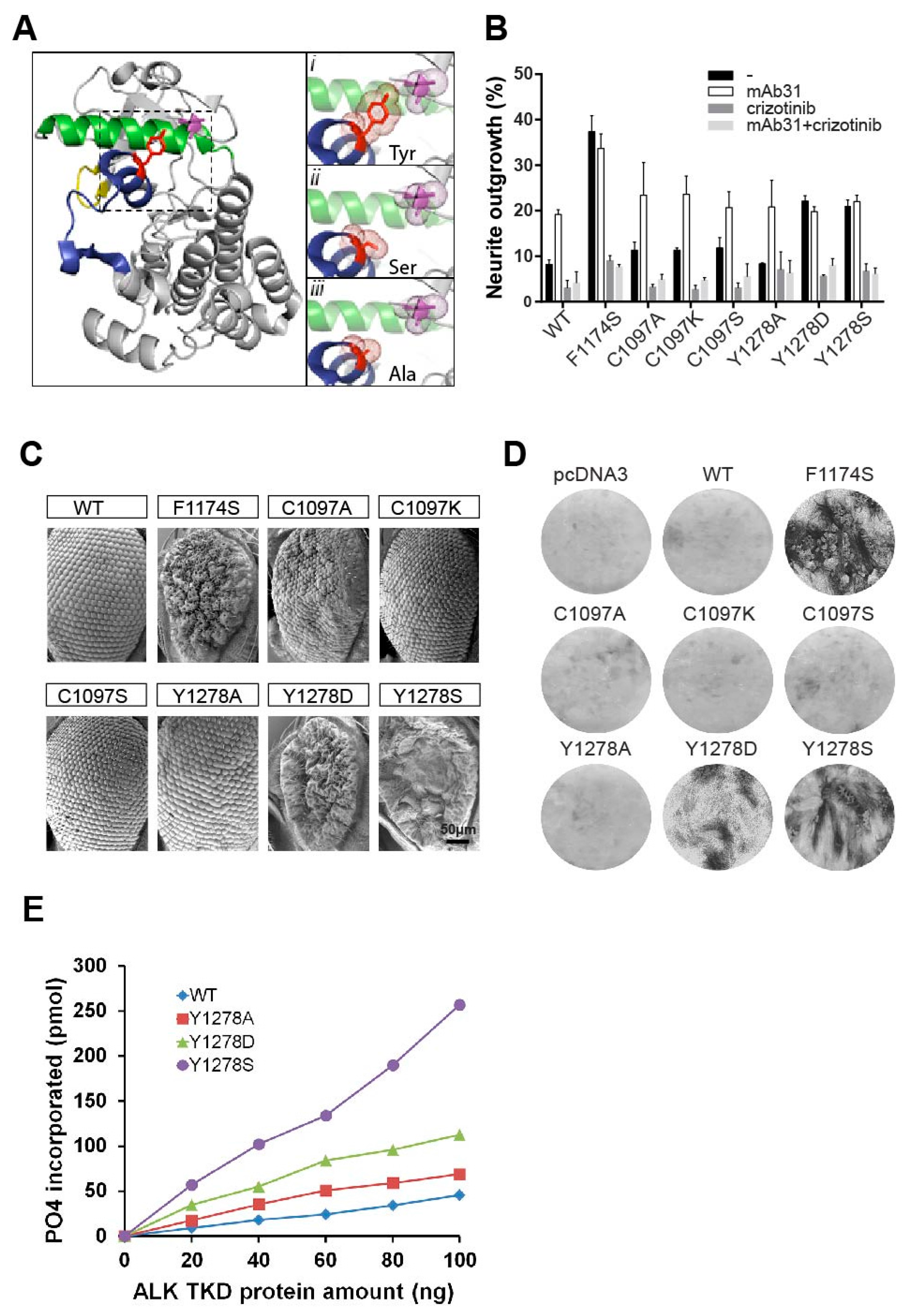
2.3. The Importance of the Hydrogen Bond between Y1278 and C1097
2.4. In Vitro Analysis of the ALK-Y1278S/A/D Activation Loop Mutations
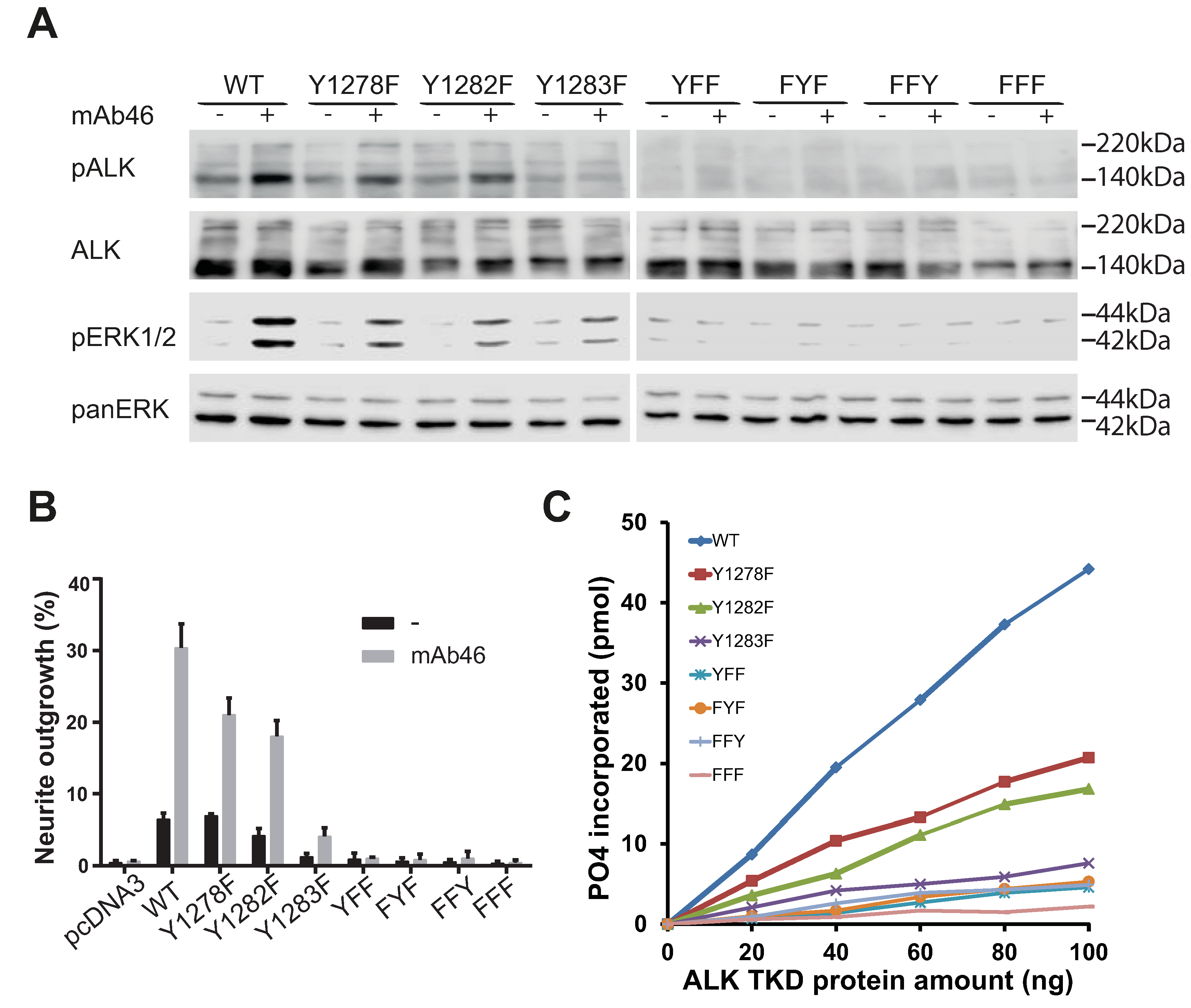
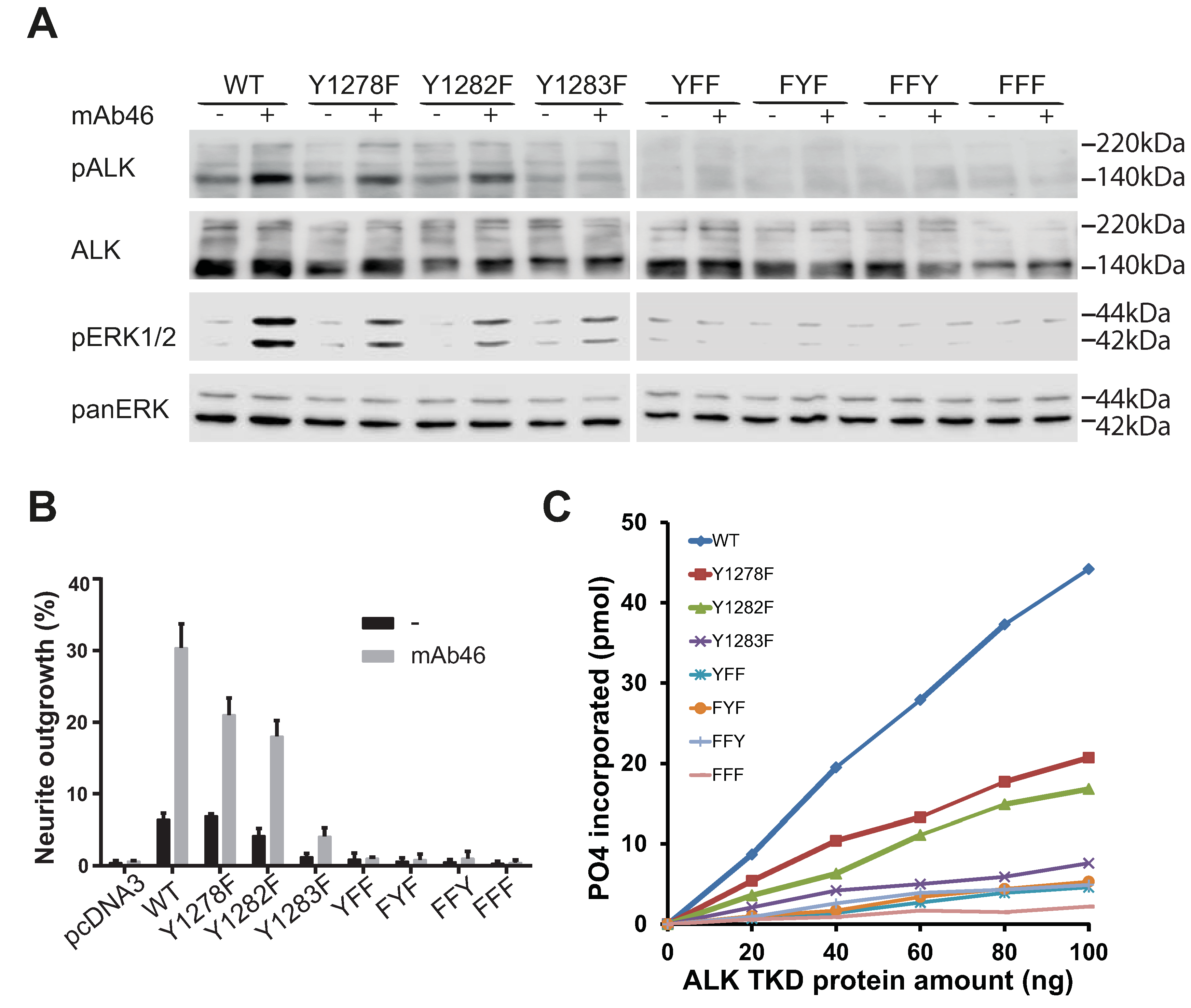
2.5. Analysis of the Tyrosines in the Activation Loop Motif 1278-YRASYY-1283 of ALK
2.6. ALK-Y1283 Is Critical for Wild Type ALK Kinase Activity
3. Discussion
4. Material and Methods
4.1. Antibodies and Inhibitors
4.2. Generation of ALK Mutant Constructs
4.3. Neurite Outgrowth Assay
4.4. Cell Lysis and Immunoblotting
4.5. Transformation Assay
4.6. ALK Inhibitor Profiles on ALK-Y1278S Mutant
4.7. Generation of Recombinant ALK TKD Proteins
4.8. In Vitro Kinase Assays
4.9. Expression of Human ALK Mutants in Drosophila Eye
4.10. Expression of Human ALK Mutants in Drosophila Eye
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s Lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Nakamura, S.; Ichinohasama, R.; Abe, M.; Akagi, T.; Takeshita, M.; Mori, N.; Fujimoto, J.; Miyauchi, J.; Mikata, A.; et al. Anaplastic large cell lymphomas expressing the novel chimeric protein p80NPM/ALK: A distinct clinicopathologic entity. Blood 1995, 86, 1954–1960. [Google Scholar] [PubMed]
- Hallberg, B.; Palmer, R.H. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat. Rev. Cancer 2013, 13, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.M. Neuroblastoma: Biological insights into a clinical enigma. Nat. Rev. Cancer 2003, 3, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, B.; Palmer, R.H. The role of the ALK receptor in cancer biology. Ann. Oncol. 2016, 27, iii4–iii15. [Google Scholar] [CrossRef] [PubMed]
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Primers 2016, 2, 16078. [Google Scholar] [CrossRef] [PubMed]
- De Brouwer, S.; De Preter, K.; Kumps, C.; Zabrocki, P.; Porcu, M.; Westerhout, E.M.; Lakeman, A.; Vandesompele, J.; Hoebeeck, J.; Van Maerken, T.; et al. Meta-analysis of neuroblastomas reveals a skewed ALK mutation spectrum in tumors with MYCN amplification. Clin. Cancer Res. 2010, 16, 4353–4362. [Google Scholar] [CrossRef] [PubMed]
- Michels, E.; Vandesompele, J.; De Preter, K.; Hoebeeck, J.; Vermeulen, J.; Schramm, A.; Molenaar, J.J.; Menten, B.; Marques, B.; Stallings, R.L.; et al. Arraycgh-based classification of neuroblastoma into genomic subgroups. Genes Chromosomes Cancer 2007, 46, 1098–1108. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; Baudis, M.; De Preter, K.; Van Roy, N.; Ambros, P.; Bown, N.; Brinkschmidt, C.; Christiansen, H.; Combaret, V.; Lastowska, M.; et al. Unequivocal delineation of clinicogenetic subgroups and development of a new model for improved outcome prediction in neuroblastoma. J. Clin. Oncol. 2005, 23, 2280–2299. [Google Scholar] [CrossRef] [PubMed]
- Chand, D.; Yamazaki, Y.; Ruuth, K.; Schonherr, C.; Martinsson, T.; Kogner, P.; Attiyeh, E.F.; Maris, J.; Morozova, O.; Marra, M.A.; et al. Cell culture and drosophila model systems define three classes of anaplastic lymphoma kinase mutations in neuroblastoma. Dis. Models Mech. 2013, 6, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Umapathy, G.; Yamazaki, Y.; Wolfstetter, G.; Mendoza, P.; Pfeifer, K.; Mohammed, A.; Hugosson, F.; Zhang, H.; Hsu, A.W.; et al. FAM150A and FAM150B are activating ligands for anaplastic lymphoma kinase. Elife 2015, 4, e09811. [Google Scholar] [CrossRef] [PubMed]
- Reshetnyak, A.V.; Murray, P.B.; Shi, X.; Mo, E.S.; Mohanty, J.; Tome, F.; Bai, H.; Gunel, M.; Lax, I.; Schlessinger, J. Augmentor alpha and beta (FAM150) are ligands of the receptor tyrosine kinases ALK and LTK: Hierarchy and specificity of ligand-receptor interactions. Proc. Natl. Acad. Sci. USA 2015, 112, 15862–15867. [Google Scholar] [CrossRef] [PubMed]
- Bossi, R.T.; Saccardo, M.B.; Ardini, E.; Menichincheri, M.; Rusconi, L.; Magnaghi, P.; Orsini, P.; Avanzi, N.; Borgia, A.L.; Nesi, M.; et al. Crystal structures of anaplastic lymphoma kinase in complex with ATP competitive inhibitors. Biochemistry 2010, 49, 6813–6825. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Jia, Y.; Li, N.; Sun, X.; Ng, K.; Ambing, E.; Gao, M.Y.; Hua, S.; Chen, C.; Kim, S.; et al. Crystal structure of the anaplastic lymphoma kinase (ALK) catalytic domain. Biochem. J. 2010, 430, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, S.R. The Insulin receptor: Both a prototypical and atypical receptor tyrosine kinase. Cold Spring Harb. Perspect. Biol. 2013, 5, a008946. [Google Scholar] [CrossRef] [PubMed]
- Donella-Deana, A.; Marin, O.; Cesaro, L.; Gunby, R.H.; Ferrarese, A.; Coluccia, A.M.; Tartari, C.J.; Mologni, L.; Scapozza, L.; Gambacorti-Passerini, C.; et al. Unique substrate specificity of anaplastic lymphoma kinase (ALK): Development of phosphoacceptor peptides for the assay of ALK activity. Biochemistry 2005, 44, 8533–8542. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Hubbard, S.R.; Hendrickson, W.A.; Ellis, L. Expression, characterization, and crystallization of the catalytic core of the human Insulin receptor protein-tyrosine kinase domain. J. Biol. Chem. 1995, 270, 8122–8130. [Google Scholar] [CrossRef] [PubMed]
- Tartari, C.J.; Gunby, R.H.; Coluccia, A.M.; Sottocornola, R.; Cimbro, B.; Scapozza, L.; Donella-Deana, A.; Pinna, L.A.; Gambacorti-Passerini, C. Characterization of some molecular mechanisms governing autoactivation of the catalytic domain of the anaplastic lymphoma kinase. J. Biol. Chem. 2008, 283, 3743–3750. [Google Scholar] [CrossRef] [PubMed]
- Bresler, S.C.; Weiser, D.A.; Huwe, P.J.; Park, J.H.; Krytska, K.; Ryles, H.; Laudenslager, M.; Rappaport, E.F.; Wood, A.C.; McGrady, P.W.; et al. ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell 2014, 26, 682–694. [Google Scholar] [CrossRef] [PubMed]
- Janoueix-Lerosey, I.; Lequin, D.; Brugieres, L.; Ribeiro, A.; de Pontual, L.; Combaret, V.; Raynal, V.; Puisieux, A.; Schleiermacher, G.; Pierron, G.; et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 2008, 455, 967–970. [Google Scholar] [CrossRef] [PubMed]
- Schleiermacher, G.; Javanmardi, N.; Bernard, V.; Leroy, Q.; Cappo, J.; Rio Frio, T.; Pierron, G.; Lapouble, E.; Combaret, V.; Speleman, F.; et al. Emergence of new ALK mutations at relapse of neuroblastoma. J. Clin. Oncol. 2014, 32, 2727–2734. [Google Scholar] [CrossRef] [PubMed]
- Cowley, S.; Paterson, H.; Kemp, P.; Marshall, C.J. Activation of map kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell 1994, 77, 841–852. [Google Scholar] [CrossRef]
- Schonherr, C.; Ruuth, K.; Yamazaki, Y.; Eriksson, T.; Christensen, J.; Palmer, R.H.; Hallberg, B. Activating ALK mutations found in neuroblastoma are inhibited by crizotinib and NVP-TAE684. Biochem. J. 2011, 440, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Siaw, J.T.; Wan, H.; Pfeifer, K.; Rivera, V.M.; Guan, J.; Palmer, R.H.; Hallberg, B. Brigatinib, an anaplastic lymphoma kinase inhibitor, abrogates activity and growth in ALK-positive neuroblastoma cells, drosophila and mice. Oncotarget 2016, 7, 29011–29022. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Wolfstetter, G.; Siaw, J.; Chand, D.; Hugosson, F.; Palmer, R.H.; Hallberg, B. Anaplastic lymphoma kinase L1198F AND G1201E mutations identified in anaplastic thyroid cancer patients are not ligand-independent. Oncotarget 2017, 8, 11566–11578. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Tucker, E.R.; Wan, H.; Chand, D.; Danielson, L.S.; Ruuth, K.; El Wakil, A.; Witek, B.; Jamin, Y.; Umapathy, G.; et al. The ALK inhibitor PF-06463922 is effective as a single agent in neuroblastoma driven by expression of ALK and mycn. Dis. Models Mech. 2016, 9, 941–952. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wu, F.; Ma, Y.P.; Li, L.; Lai, R.; Young, L.C. Functional characterization of the kinase activation loop in nucleophosmin (NPM)-anaplastic lymphoma kinase (ALK) using tandem affinity purification and liquid chromatography-mass spectrometry. J. Biol. Chem. 2010, 285, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Martinsson, T.; Eriksson, T.; Abrahamsson, J.; Caren, H.; Hansson, M.; Kogner, P.; Kamaraj, S.; Schonherr, C.; Weinmar, J.; Ruuth, K.; et al. Appearance of the novel activating F1174S ALK mutation in neuroblastoma correlates with aggressive tumour progression and unresponsiveness to therapy. Cancer Res. 2011, 71, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Englund, C.; Loren, C.E.; Grabbe, C.; Varshney, G.K.; Deleuil, F.; Hallberg, B.; Palmer, R.H. Jeb signals through the ALK receptor tyrosine kinase to drive visceral muscle fusion. Nature 2003, 425, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.L.; Eriksson, T.; Vernersson, E.; Vigny, M.; Hallberg, B.; Palmer, R.H. The ligand Jelly Belly (Jeb) activates the drosophila ALK RTK to drive PC12 cell differentiation, but is unable to activate the mouse ALK RTK. J. Exp. Zool. B 2007, 308, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Moog-Lutz, C.; Degoutin, J.; Gouzi, J.Y.; Frobert, Y.; Brunet-de Carvalho, N.; Bureau, J.; Creminon, C.; Vigny, M. Activation and inhibition of anaplastic lymphoma kinase receptor tyrosine kinase by monoclonal antibodies and absence of agonist activity of pleiotrophin. J. Biol. Chem. 2005, 280, 26039–26048. [Google Scholar] [CrossRef] [PubMed]
- Schonherr, C.; Yang, H.L.; Vigny, M.; Palmer, R.H.; Hallberg, B. Anaplastic lymphoma kinase activates the small GTPase RAP1 via the RAP1-specific GEF C3G in both neuroblastoma and PC12 cells. Oncogene 2010, 29, 2817–2830. [Google Scholar] [CrossRef] [PubMed]
- Hatzi, E.; Breit, S.; Zoephel, A.; Ashman, K.; Tontsch, U.; Ahorn, H.; Murphy, C.; Schweigerer, L.; Fotsis, T. Mycn oncogene and angiogenesis: Down-regulation of endothelial growth inhibitors in human neuroblastoma cells. Purification, structural, and functional characterization. Adv. Exp. Med. Biol. 2000, 476, 239–248. [Google Scholar] [PubMed]
- Brand, A.H.; Perrimon, N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 1993, 118, 401–415. [Google Scholar] [PubMed]
- Hubbard, S.R. Crystal structure of the activated Insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J. 1997, 16, 5572–5581. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guan, J.; Yamazaki, Y.; Chand, D.; Van Dijk, J.R.; Ruuth, K.; Palmer, R.H.; Hallberg, B. Novel Mechanisms of ALK Activation Revealed by Analysis of the Y1278S Neuroblastoma Mutation. Cancers 2017, 9, 149. https://doi.org/10.3390/cancers9110149
Guan J, Yamazaki Y, Chand D, Van Dijk JR, Ruuth K, Palmer RH, Hallberg B. Novel Mechanisms of ALK Activation Revealed by Analysis of the Y1278S Neuroblastoma Mutation. Cancers. 2017; 9(11):149. https://doi.org/10.3390/cancers9110149
Chicago/Turabian StyleGuan, Jikui, Yasuo Yamazaki, Damini Chand, Jesper R. Van Dijk, Kristina Ruuth, Ruth H. Palmer, and Bengt Hallberg. 2017. "Novel Mechanisms of ALK Activation Revealed by Analysis of the Y1278S Neuroblastoma Mutation" Cancers 9, no. 11: 149. https://doi.org/10.3390/cancers9110149