
A Tale of Two Signals: AR and WNT in Development and Tumorigenesis of Prostate and Mammary Gland


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
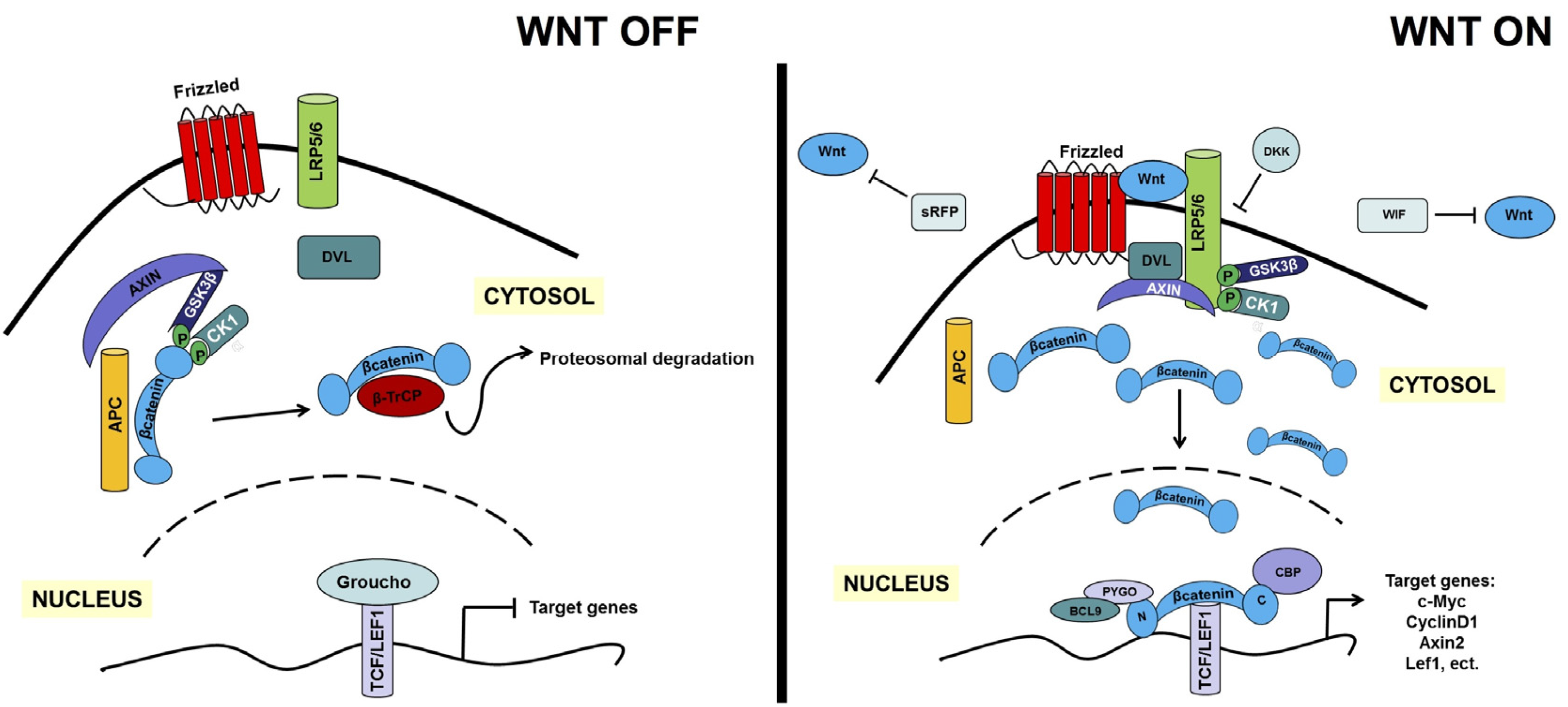
2. An Overview of the Canonical and Non-Canonical WNT Signaling Pathways
3. WNT Signaling in Prostate Development and Stem Cells
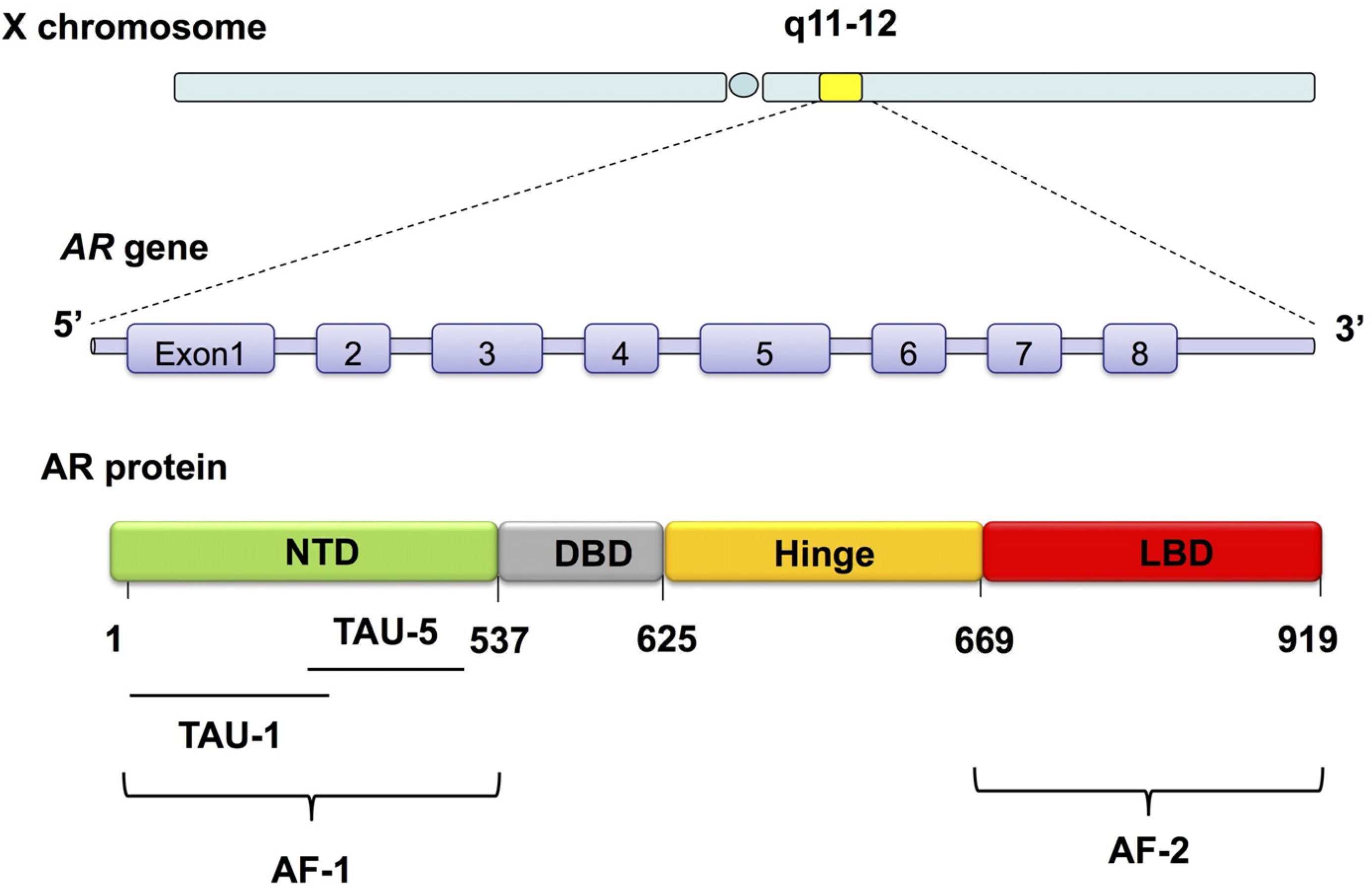
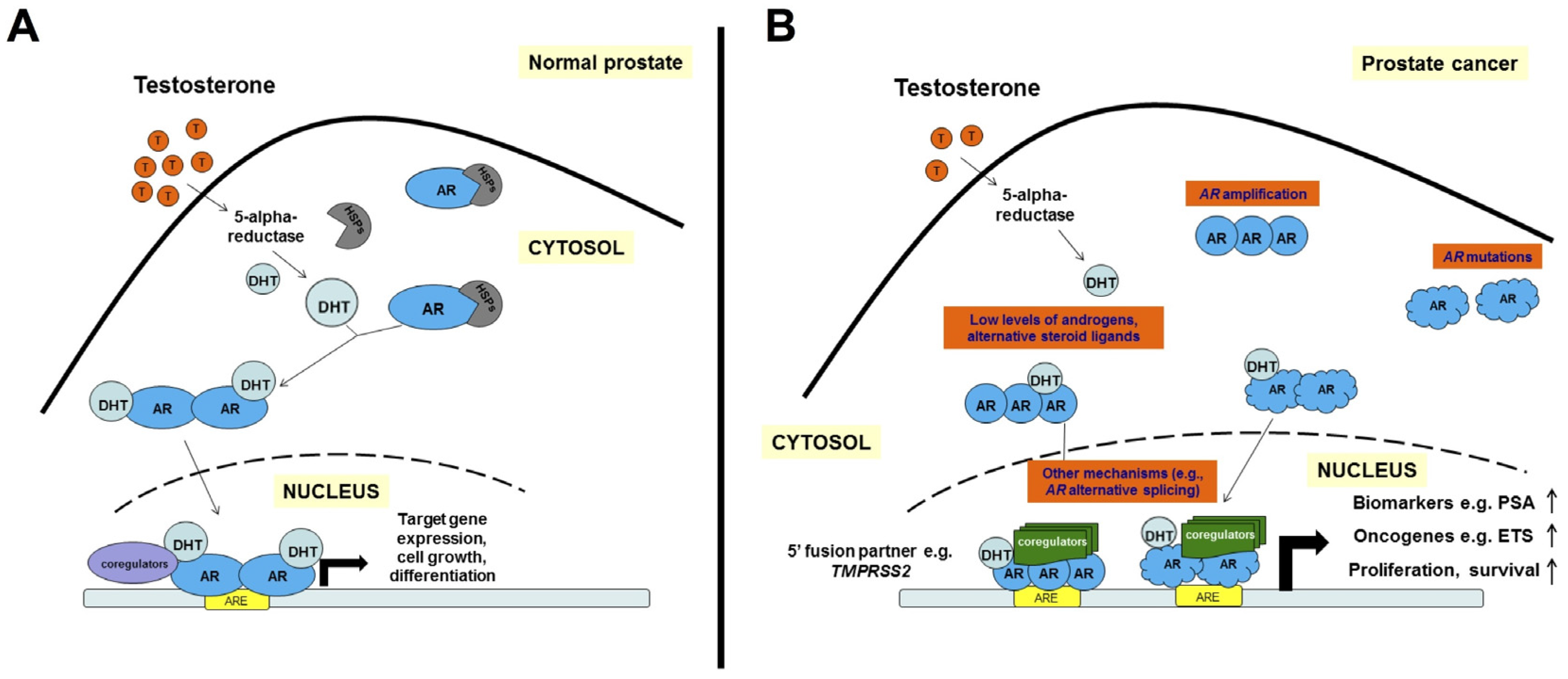
4. An Overview of AR and AR Signaling
5. The Emergence of Castration Resistance
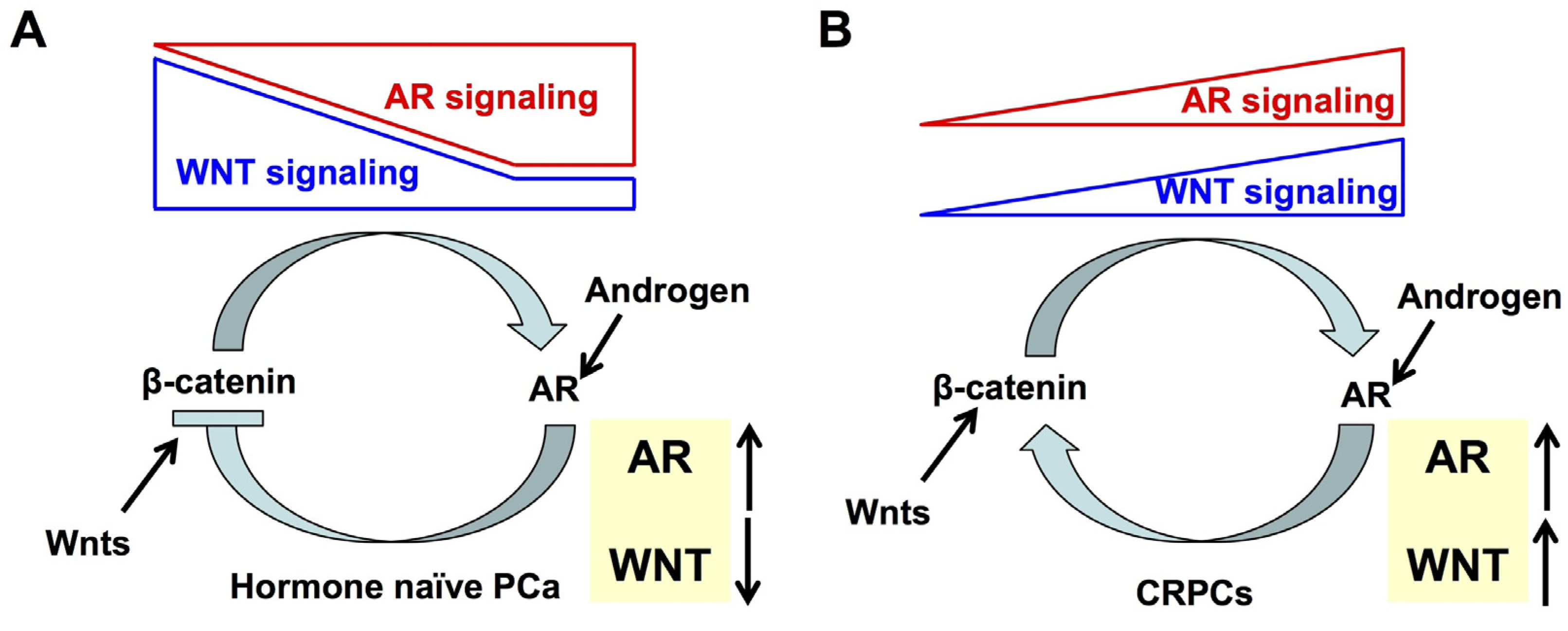
6. Interaction between AR and WNT Signaling in Prostate Cancer
7. Therapeutic Applications for Targeting WNT/β-catenin-AR Interactions in CRPC
8. AR and WNT Signaling in Mammary Gland Development and Breast Cancer
8.1. AR and WNT Signaling in Mammary Gland Development
8.2. AR Signaling in Breast Cancer
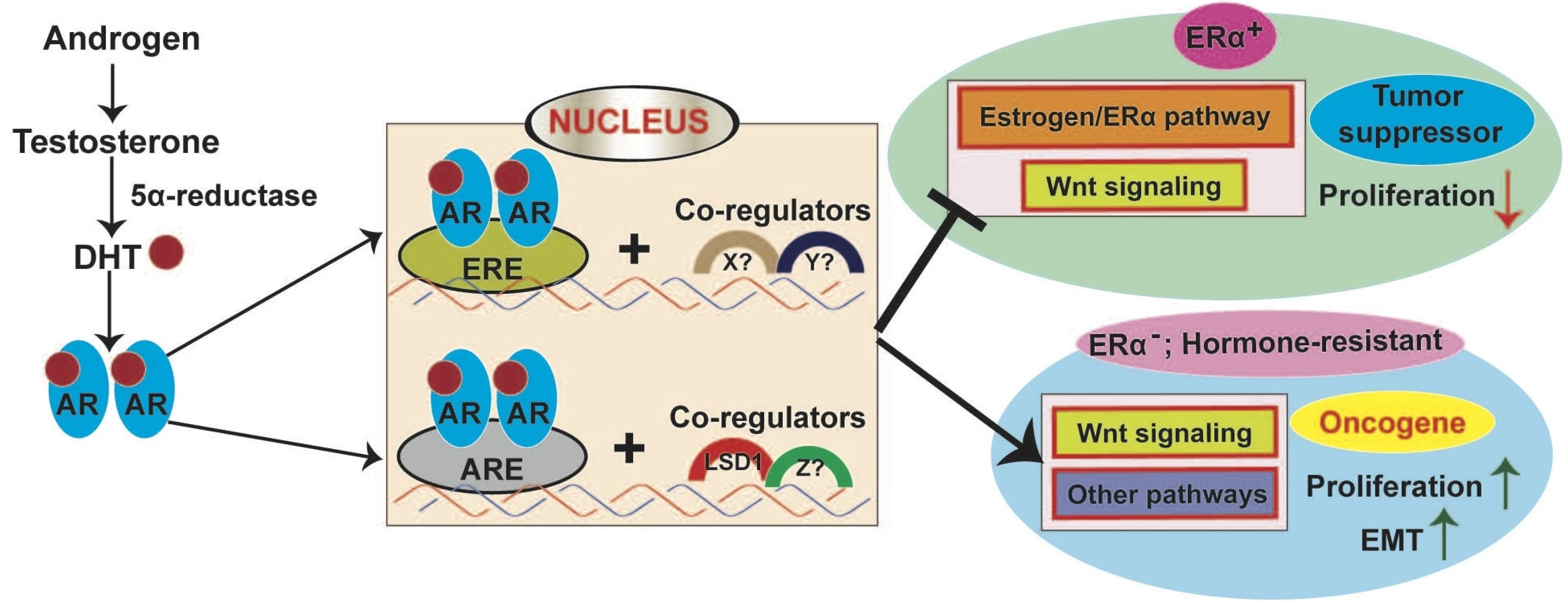
8.2.1. AR in ER+ Breast Cancer
8.2.2. AR Signaling in ER−/HER2+ Breast Cancer
8.2.3. AR Signaling in TNBC
8.3. Interaction between AR and WNT Signaling in Breast Cancer
9. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2013. CA: Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Ali, S.; Sarkar, F.H. Advances in androgen receptor targeted therapy for prostate cancer. J. Cell. Physiol. 2014, 229, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA: Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.R.; Lubahn, D.B.; Wilson, E.M.; Joseph, D.R.; French, F.S.; Migeon, C.J. Deletion of the steroid-binding domain of the human androgen receptor gene in one family with complete androgen insensitivity syndrome: Evidence for further genetic heterogeneity in this sy ndrome. Proc. Natl. Acad. Sci. USA 1988, 85, 8151–8155. [Google Scholar] [CrossRef] [PubMed]
- Lubahn, D.B.; Brown, T.R.; Simental, J.A.; Higgs, H.N.; Migeon, C.J.; Wilson, E.M.; French, F.S. Sequence of the intron/exon junctions of the coding region of the human androgen receptor gene and identification of a point mutation in a family with complete androgen insensitivity. Proc. Natl. Acad. Sci. USA 1989, 86, 9534–9538. [Google Scholar] [CrossRef] [PubMed]
- Huggins, C.; Hodges, C.V. Studies on prostatic cancer. I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. CA: Cancer J. Clin. 1972, 22, 232–240. [Google Scholar] [CrossRef]
- Heidenreich, A.; Bastian, P.J.; Bellmunt, J.; Bolla, M.; Joniau, S.; van der Kwast, T.; Mason, M.; Matveev, V.; Wiegel, T.; Zattoni, F.; et al. Eau guidelines on prostate cancer. Part ii: Treatment of advanced, relapsing, and castration-resistant prostate cancer. Eur. Urol. 2014, 65, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Prensner, J.R.; Rubin, M.A.; Wei, J.T.; Chinnaiyan, A.M. Beyond psa: The next generation of prostate cancer biomarkers. Sci. Transl. Med. 2012. [Google Scholar] [CrossRef] [PubMed]
- Holzbeierlein, J.; Lal, P.; LaTulippe, E.; Smith, A.; Satagopan, J.; Zhang, L.; Ryan, C.; Smith, S.; Scher, H.; Scardino, P.; et al. Gene expression analysis of human prostate carcinoma during hormonal therapy identifies androgen-responsive genes and mechanisms of therapy resistance. Am. J. Pathol. 2004, 164, 217–227. [Google Scholar] [CrossRef]
- Penney, K.L.; Schumacher, F.R.; Kraft, P.; Mucci, L.A.; Sesso, H.D.; Ma, J.; Niu, Y.; Cheong, J.K.; Hunter, D.J.; Stampfer, M.J.; et al. Association of KLK3 (PSA) genetic variants with prostate cancer risk and PSA levels. Carcinogenesis 2011, 32, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Parikh, H.; Wang, Z.; Pettigrew, K.A.; Jia, J.; Daugherty, S.; Yeager, M.; Jacobs, K.B.; Hutchinson, A.; Burdett, L.; Cullen, M.; et al. Fine mapping the KLK3 locus on chromosome 19q13.33 associated with prostate cancer susceptibility and psa levels. Hum. Genet. 2011, 129, 675–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldman, B.J.; Feldman, D. The development of androgen-independent prostate cancer. Nat. Rev. Cancer 2001, 1, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.; Finn, S.; Loda, M.; Hahn, W.C. Prostate cancer: Re-focusing on androgen receptor signaling. Int. J. Biochem. Cell Biol. 2007, 39, 1562–1568. [Google Scholar] [CrossRef] [PubMed]
- Snoek, R.; Cheng, H.; Margiotti, K.; Wafa, L.A.; Wong, C.A.; Wong, E.C.; Fazli, L.; Nelson, C.C.; Gleave, M.E.; Rennie, P.S. In vivo knockdown of the androgen receptor results in growth inhibition and regression of well-established, castration-resistant prostate tumors. Clin. Cancer Res. 2009, 15, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.L.; Massie, C.E.; Ramos-Montoya, A.; Zecchini, V.; Scott, H.E.; Lamb, A.D.; MacArthur, S.; Stark, R.; Warren, A.Y.; Mills, I.G.; et al. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell 2013, 23, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Pienta, K.J.; Bradley, D. Mechanisms underlying the development of androgen-independent prostate cancer. Clin. Cancer Res. 2006, 12, 1665–1671. [Google Scholar] [CrossRef] [PubMed]
- Kypta, R.M.; Waxman, J. Wnt/beta-Catenin signalling in prostate cancer. Nat. Rev. Urol. 2012, 9, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Watanabe, T.; Okada, M.; Inoue, K.; Ueda, T.; Takada, I.; Watabe, T.; Yamamoto, Y.; Fukuda, T.; Nakamura, T.; et al. Noncanonical wnt signaling mediates androgen-dependent tumor growth in a mouse model of prostate cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 4938–4943. [Google Scholar] [CrossRef] [PubMed]
- Uysal-Onganer, P.; Kawano, Y.; Caro, M.; Walker, M.M.; Diez, S.; Darrington, R.S.; Waxman, J.; Kypta, R.M. Wnt-11 promotes neuroendocrine-like differentiation, survival and migration of prostate cancer cells. Mol. Cancer 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verras, M.; Brown, J.; Li, X.; Nusse, R.; Sun, Z. Wnt3a growth factor induces androgen receptor-mediated transcription and enhances cell growth in human prostate cancer cells. Cancer Res. 2004, 64, 8860–8866. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Mazor, M.; Kawano, Y.; Walker, M.M.; Leung, H.Y.; Armstrong, K.; Waxman, J.; Kypta, R.M. Analysis of wnt gene expression in prostate cancer: Mutual inhibition by wnt11 and the androgen receptor. Cancer Res. 2004, 64, 7918–7926. [Google Scholar] [CrossRef] [PubMed]
- Nelson, W.J.; Nusse, R. Convergence of WNT, β-catenin, and cadherin pathways. Science 2004, 303, 1483–1487. [Google Scholar] [CrossRef] [PubMed]
- Logan, C.Y.; Nusse, R. The WNT signaling pathway in development and disease. Ann. Rev. Cell Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef] [PubMed]
- Grigoryan, T.; Wend, P.; Klaus, A.; Birchmeier, W. Deciphering the function of canonical wnt signals in development and disease: Conditional loss- and gain-of-function mutations of beta-catenin in mice. Genes Dev. 2008, 22, 2308–2341. [Google Scholar] [CrossRef] [PubMed]
- Klaus, A.; Birchmeier, W. Wnt signalling and its impact on development and cancer. Nat. Rev. Cancer 2008, 8, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Lien, W.H.; Fuchs, E. Wnt some lose some: Transcriptional governance of stem cells by WNT/β-catenin signaling. Genes Dev. 2014, 28, 1517–1532. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. Wnt/ β-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Polakis, P. The many ways of WNT in cancer. Curr. Opin. Genet. Dev. 2007, 17, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Taketo, M.M. Shutting down wnt signal-activated cancer. Nat. Genet. 2004, 36, 320–322. [Google Scholar] [CrossRef] [PubMed]
- White, B.D.; Chien, A.J.; Dawson, D.W. Dysregulation of Wnt/β-catenin signaling in gastrointestinal cancers. Gastroenterology 2012, 142, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Krausova, M.; Korinek, V. Wnt signaling in adult intestinal stem cells and cancer. Cell. Signal. 2014, 26, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Satoh, S.; Daigo, Y.; Furukawa, Y.; Kato, T.; Miwa, N.; Nishiwaki, T.; Kawasoe, T.; Ishiguro, H.; Fujita, M.; Tokino, T.; et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat. Genet. 2000, 24, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Guan, C.N.; Chen, X.M.; Lou, H.Q.; Liao, X.H.; Chen, B.Y.; Zhang, P.W. Clinical significance of axin and beta-catenin protein expression in primary hepatocellular carcinomas. Asian Pac. J. Cancer Prev.: APJCP 2012, 13, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Ishizaki, Y.; Ikeda, S.; Fujimori, M.; Shimizu, Y.; Kurihara, T.; Itamoto, T.; Kikuchi, A.; Okajima, M.; Asahara, T. Immunohistochemical analysis and mutational analyses of beta-catenin, axin family and APC genes in hepatocellular carcinomas. Int. J. Oncol. 2004, 24, 1077–1083. [Google Scholar] [PubMed]
- Clevers, H. Axin and hepatocellular carcinomas. Nat. Genet. 2000, 24, 206–208. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, V.; Leigh, I.M. WNT signaling in cutaneous squamous cell carcinoma: A future treatment strategy? J. Investig. Dermatol. 2016, 136, 1760–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, E.F.; Gat, U.; McNiff, J.M.; Fuchs, E. A common human skin tumour is caused by activating mutations in beta-catenin. Nat. Genet. 1999, 21, 410–413. [Google Scholar] [PubMed]
- Shulewitz, M.; Soloviev, I.; Wu, T.; Koeppen, H.; Polakis, P.; Sakanaka, C. Repressor roles for TCF-4 and SFRP1 in WNT signaling in breast cancer. Oncogene 2006, 25, 4361–4369. [Google Scholar] [CrossRef] [PubMed]
- Schlange, T.; Matsuda, Y.; Lienhard, S.; Huber, A.; Hynes, N.E. Autocrine wnt signaling contributes to breast cancer cell proliferation via the canonical wnt pathway and EGFR transactivation. Breast Cancer Res.: BCR 2007, 9, R63. [Google Scholar] [CrossRef] [PubMed]
- Lindvall, C.; Bu, W.; Williams, B.O.; Li, Y. Wnt signaling, stem cells, and the cellular origin of breast cancer. Stem Cell Rev. 2007, 3, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.C.; Verheyen, E.M.; Zeng, Y.A. Mammary development and breast cancer: A WNT perspective. Cancers 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Davies, G.; Jiang, W.G.; Mason, M.D. The interaction between beta-catenin, GSK3β and APC after motogen induced cell-cell dissociation, and their involvement in signal transduction pathways in prostate cancer. Int. J. Oncol. 2001, 18, 843–847. [Google Scholar] [PubMed]
- Hu, B.R.; Fairey, A.S.; Madhav, A.; Yang, D.; Li, M.; Groshen, S.; Stephens, C.; Kim, P.H.; Virk, N.; Wang, L.; et al. AXIN2 expression predicts prostate cancer recurrence and regulates invasion and tumor growth. Prostate 2016, 76, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Brown, A.; Papkoff, J.; Scambler, P.; Shackleford, G.; McMahon, A.; Moon, R.; Varmus, H. A new nomenclature for INT-1 and related genes: The Wnt gene family. Cell 1991. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, C.V.; Alonso, M.C.; Johnston, P.; Phillips, R.G.; Lawrence, P.A. Phenocopies induced with antisense RNA identify the wingless gene. Cell 1987, 50, 659–663. [Google Scholar] [CrossRef]
- Rijsewijk, F.; Schuermann, M.; Wagenaar, E.; Parren, P.; Weigel, D.; Nusse, R. The Drosophila homolog of the mouse mammary oncogene int-1 is identical to the segment polarity gene wingless. Cell 1987, 50, 649–657. [Google Scholar] [CrossRef]
- Nusslein-Volhard, C.; Wieschaus, E. Mutations affecting segment number and polarity in drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Varmus, H.E. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell 1982, 31, 99–109. [Google Scholar] [CrossRef]
- Vinson, C.R.; Conover, S.; Adler, P.N. A Drosophila tissue polarity locus encodes a protein containing seven potential transmembrane domains. Nature 1989, 338, 263–264. [Google Scholar] [CrossRef] [PubMed]
- Tamai, K.; Semenov, M.; Kato, Y.; Spokony, R.; Liu, C.; Katsuyama, Y.; Hess, F.; Saint-Jeannet, J.P.; He, X. LDL-receptor-related proteins in WNT signal transduction. Nature 2000, 407, 530–535. [Google Scholar] [PubMed]
- Wehrli, M.; Dougan, S.T.; Caldwell, K.; O’Keefe, L.; Schwartz, S.; Vaizel-Ohayon, D.; Schejter, E.; Tomlinson, A.; DiNardo, S. Arrow encodes an LDL-receptor-related protein essential for wingless signalling. Nature 2000, 407, 527–530. [Google Scholar] [PubMed]
- Mikels, A.J.; Nusse, R. WNTs as ligands: Processing, secretion and reception. Oncogene 2006, 25, 7461–7468. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; He, X. Wnt/β-catenin signaling: New (and old) players and new insights. Curr. Opin. Cell Biol. 2008, 20, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Rubin, B.; Bodine, P.V.; Billiard, J. WNT5A induces homodimerization and activation of ROR2 receptor tyrosine kinase. Journal of cellular biochemistry 2008, 105, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Carmon, K.S.; Gong, X.; Lin, Q.; Thomas, A.; Liu, Q. R-spondins function as ligands of the orphan receptors LGR4 and LGR5 to regulate wnt/β-catenin signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 11452–11457. [Google Scholar] [CrossRef] [PubMed]
- de Lau, W.; Barker, N.; Low, T.Y.; Koo, B.K.; Li, V.S.; Teunissen, H.; Kujala, P.; Haegebarth, A.; Peters, P.J.; van de Wetering, M.; et al. LGR5 homologues associate with wnt receptors and mediate R-spondin signalling. Nature 2011, 476, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Holland, J.D.; Klaus, A.; Garratt, A.N.; Birchmeier, W. Wnt signaling in stem and cancer stem cells. Curr. Opin. Cell Biol. 2013, 25, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, M.; Geis, K.; Sheldahl, L.C.; Pukrop, T.; Moon, R.T.; Wedlich, D. Antagonistic regulation of convergent extension movements in xenopus by WNT/β-catenin and WNT/Ca2+ signaling. Mech. Dev. 2001, 106, 61–76. [Google Scholar] [CrossRef]
- Oishi, I.; Suzuki, H.; Onishi, N.; Takada, R.; Kani, S.; Ohkawara, B.; Koshida, I.; Suzuki, K.; Yamada, G.; Schwabe, G.C.; et al. The receptor tyrosine kinase ROR2 is involved in non-canonical WNT5A/JNK signalling pathway. Genes Cells: Devot. Mol. Cell. Mech. 2003, 8, 645–654. [Google Scholar] [CrossRef]
- Schambony, A.; Wedlich, D. Wnt-5a/ROR2 regulate expression of xpapc through an alternative noncanonical signaling pathway. Dev. Cell 2007, 12, 779–792. [Google Scholar] [CrossRef] [PubMed]
- Kraus, C.; Liehr, T.; Hulsken, J.; Behrens, J.; Birchmeier, W.; Grzeschik, K.H.; Ballhausen, W.G. Localization of the human β-catenin gene (CTNNB1) to 3p21: A region implicated in tumor development. Genomics 1994, 23, 272–274. [Google Scholar] [CrossRef] [PubMed]
- Huber, A.H.; Weis, W.I. The structure of the beta-catenin/e-cadherin complex and the molecular basis of diverse ligand recognition by β-catenin. Cell 2001, 105, 391–402. [Google Scholar] [CrossRef]
- Willert, K.; Nusse, R. β-catenin: A key mediator of wnt signaling. Curr. Opin. Genet. Dev. 1998, 8, 95–102. [Google Scholar] [CrossRef]
- Behrens, J.; von Kries, J.P.; Kuhl, M.; Bruhn, L.; Wedlich, D.; Grosschedl, R.; Birchmeier, W. Functional interaction of β-catenin with the transcription factor LEF-1. Nature 1996, 382, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, M.; van de Wetering, M.; Oosterwegel, M.; Peterson-Maduro, J.; Godsave, S.; Korinek, V.; Roose, J.; Destree, O.; Clevers, H. XTCF-3 transcription factor mediates beta-catenin-induced axis formation in xenopus embryos. Cell 1996, 86, 391–399. [Google Scholar] [CrossRef]
- Huber, O.; Korn, R.; McLaughlin, J.; Ohsugi, M.; Herrmann, B.G.; Kemler, R. Nuclear localization of beta-catenin by interaction with transcription factor LEF-1. Mech. Dev. 1996, 59, 3–10. [Google Scholar] [CrossRef]
- Riese, J.; Yu, X.; Munnerlyn, A.; Eresh, S.; Hsu, S.C.; Grosschedl, R.; Bienz, M. LEF-1, a nuclear factor coordinating signaling inputs from wingless and decapentaplegic. Cell 1997, 88, 777–787. [Google Scholar] [CrossRef]
- Kemler, R. From cadherins to catenins: Cytoplasmic protein interactions and regulation of cell adhesion. Trends Genet.: TIG 1993, 9, 317–321. [Google Scholar] [CrossRef]
- Rimm, D.L.; Koslov, E.R.; Kebriaei, P.; Cianci, C.D.; Morrow, J.S. Alpha 1(E)-catenin is an actin-binding and -bundling protein mediating the attachment of F-actin to the membrane adhesion complex. Proc. Natl. Acad. Sci. USA 1995, 92, 8813–8817. [Google Scholar] [CrossRef] [PubMed]
- Drees, F.; Pokutta, S.; Yamada, S.; Nelson, W.J.; Weis, W.I. Alpha-Catenin is a molecular switch that binds E-cadherin-β-catenin and regulates actin-filament assembly. Cell 2005, 123, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Pokutta, S.; Drees, F.; Weis, W.I.; Nelson, W.J. Deconstructing the cadherin-catenin-actin complex. Cell 2005, 123, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Meng, W.; Takeichi, M. Adherens junction: Molecular architecture and regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a002899. [Google Scholar] [CrossRef] [PubMed]
- Barth, A.I.; Nathke, I.S.; Nelson, W.J. Cadherins, catenins and apc protein: Interplay between cytoskeletal complexes and signaling pathways. Curr. Opin. Cell Biol. 1997, 9, 683–690. [Google Scholar] [CrossRef]
- Behrens, J.; Jerchow, B.A.; Wurtele, M.; Grimm, J.; Asbrand, C.; Wirtz, R.; Kuhl, M.; Wedlich, D.; Birchmeier, W. Functional interaction of an axin homolog, conductin, with β-catenin, APC, and GSK3beta. Science 1998, 280, 596–599. [Google Scholar] [CrossRef] [PubMed]
- Salomon, D.; Sacco, P.A.; Roy, S.G.; Simcha, I.; Johnson, K.R.; Wheelock, M.J.; Ben-Ze'ev, A. Regulation of β-catenin levels and localization by overexpression of plakoglobin and inhibition of the ubiquitin-proteasome system. J. Cell Biol. 1997, 139, 1325–1335. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, Y.; Semenov, M.; Han, C.; Baeg, G.H.; Tan, Y.; Zhang, Z.; Lin, X.; He, X. Control of β-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 2002, 108, 837–847. [Google Scholar] [CrossRef]
- Zeng, X.; Tamai, K.; Doble, B.; Li, S.; Huang, H.; Habas, R.; Okamura, H.; Woodgett, J.; He, X. A dual-kinase mechanism for WNT co-receptor phosphorylation and activation. Nature 2005, 438, 873–877. [Google Scholar] [CrossRef] [PubMed]
- Aberle, H.; Bauer, A.; Stappert, J.; Kispert, A.; Kemler, R. Beta-Catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997, 16, 3797–3804. [Google Scholar] [CrossRef] [PubMed]
- Roose, J.; Molenaar, M.; Peterson, J.; Hurenkamp, J.; Brantjes, H.; Moerer, P.; van de Wetering, M.; Destree, O.; Clevers, H. The Xenopus Wnt effector XTCF-3 interacts with groucho-related transcriptional repressors. Nature 1998, 395, 608–612. [Google Scholar] [PubMed]
- Davidson, G.; Wu, W.; Shen, J.; Bilic, J.; Fenger, U.; Stannek, P.; Glinka, A.; Niehrs, C. Casein kinase 1 gamma couples wnt receptor activation to cytoplasmic signal transduction. Nature 2005, 438, 867–872. [Google Scholar] [CrossRef] [PubMed]
- Bilic, J.; Huang, Y.L.; Davidson, G.; Zimmermann, T.; Cruciat, C.M.; Bienz, M.; Niehrs, C. Wnt induces LRP6 signalosomes and promotes dishevelled-dependent LRP6 phosphorylation. Science 2007, 316, 1619–1622. [Google Scholar] [CrossRef] [PubMed]
- Daniels, D.L.; Weis, W.I. Beta-catenin directly displaces groucho/TLE repressors from TCF/LEF in wnt-mediated transcription activation. Nat. Struct. Mol. Biol. 2005, 12, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Kramps, T.; Peter, O.; Brunner, E.; Nellen, D.; Froesch, B.; Chatterjee, S.; Murone, M.; Zullig, S.; Basler, K. Wnt/Wingless signaling requires BCL9/legless-mediated recruitment of pygopus to the nuclear beta-catenin-TCF complex. Cell 2002, 109, 47–60. [Google Scholar] [CrossRef]
- Brembeck, F.H.; Schwarz-Romond, T.; Bakkers, J.; Wilhelm, S.; Hammerschmidt, M.; Birchmeier, W. Essential role of BCL9–2 in the switch between beta-catenin’s adhesive and transcriptional functions. Genes Dev. 2004, 18, 2225–2230. [Google Scholar] [CrossRef] [PubMed]
- De la Roche, M.; Bienz, M. Wingless-Independent association of pygopus with DTCF target genes. Curr. Biol.: CB 2007, 17, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Belenkaya, T.Y.; Han, C.; Standley, H.J.; Lin, X.; Houston, D.W.; Heasman, J. Pygopus encodes a nuclear protein essential for wingless/wnt signaling. Development 2002, 129, 4089–4101. [Google Scholar] [PubMed]
- Hecht, A.; Vleminckx, K.; Stemmler, M.P.; van Roy, F.; Kemler, R. The P300/CBP acetyltransferases function as transcriptional coactivators of β-catenin in vertebrates. EMBO J. 2000, 19, 1839–1850. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Rodova, M.; Miska, E.A.; Calvet, J.P.; Kouzarides, T. Acetylation of β-catenin by creb-binding protein (CBP). J. Biol. Chem. 2002, 277, 25562–25567. [Google Scholar] [CrossRef] [PubMed]
- Mosimann, C.; Hausmann, G.; Basler, K. Parafibromin/hyrax activates wnt/wg target gene transcription by direct association with β-catenin/armadillo. Cell 2006, 125, 327–341. [Google Scholar] [CrossRef] [PubMed]
- He, T.C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of C-Myc as a target of the APC pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef]
- D’Amico, M.; Hulit, J.; Amanatullah, D.F.; Zafonte, B.T.; Albanese, C.; Bouzahzah, B.; Fu, M.; Augenlicht, L.H.; Donehower, L.A.; Takemaru, K.; et al. The integrin-linked kinase regulates the cyclin D1 gene through glycogen synthase kinase 3β and camp-responsive element-binding protein-dependent pathways. J. Biol. Chem. 2000, 275, 32649–32657. [Google Scholar] [CrossRef] [PubMed]
- Shtutman, M.; Zhurinsky, J.; Simcha, I.; Albanese, C.; D’Amico, M.; Pestell, R.; Ben-Ze’ev, A. The cyclin d1 gene is a target of the β-catenin/LEF-1 pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 5522–5527. [Google Scholar] [CrossRef] [PubMed]
- Moreau, M.; Mourah, S.; Dosquet, C. Beta-Catenin and NF-KAPPAB cooperate to regulate the UPA/UPAR system in cancer cells. Int. J. Cancer 2011, 128, 1280–1292. [Google Scholar] [CrossRef] [PubMed]
- Bisson, I.; Prowse, D.M. Wnt signaling regulates self-renewal and differentiation of prostate cancer cells with stem cell characteristics. Cell Res. 2009, 19, 683–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, W.; Tinsley, H.N.; Keeton, A.; Qu, Z.; Piazza, G.A.; Li, Y. Suppression of wnt/β-catenin signaling inhibits prostate cancer cell proliferation. Eur. J. Pharmacol. 2009, 602, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Ha, S.; Logan, S.K. Divergent androgen receptor and beta-catenin signaling in prostate cancer cells. PLoS ONE 2015, 10, e0141589. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Liu, J.; Lu, J.F.; Tzelepi, V.; Yang, J.; Starbuck, M.W.; Diao, L.; Wang, J.; Efstathiou, E.; Vazquez, E.S.; et al. Activation of β-catenin signaling in androgen receptor-negative prostate cancer cells. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 2012, 18, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Kazanskaya, O.; Glinka, A.; del Barco Barrantes, I.; Stannek, P.; Niehrs, C.; Wu, W. R-spondin2 is a secreted activator of wntβ-catenin signaling and is required for xenopus myogenesis. Dev. Cell. 2004, 7, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; van Es, J.H.; Kuipers, J.; Kujala, P.; van den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene LGR5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Haegebarth, A.; Clevers, H. Wnt signaling, LGR5, and stem cells in the intestine and skin. Am. J. Pathol. 2009, 174, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Grun, D.; Vaillant, M.; Pieri, V.; Diederich, N.J. Response to letter of the editor by tomoyuki kawada regarding the article “contributory factors to caregiver burden in parkinson disease” by grun et al. J. Am. Med. Dir. Assoc. 2016, 17, 1060–1061. [Google Scholar] [CrossRef] [PubMed]
- De Lau, W.; Peng, W.C.; Gros, P.; Clevers, H. The r-spondin/LGR5/RNF43 module: Regulator of wnt signal strength. Genes Dev. 2014, 28, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Glinka, A.; Dolde, C.; Kirsch, N.; Huang, Y.L.; Kazanskaya, O.; Ingelfinger, D.; Boutros, M.; Cruciat, C.M.; Niehrs, C. LGR4 and LGR5 are R-spondin receptors mediating Wnt/β-catenin and Wnt/PCP signalling. EMBO Rep. 2011, 12, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Mii, Y.; Taira, M. Secreted wnt “inhibitors” are not just inhibitors: Regulation of extracellular Wnt by secreted frizzled-related proteins. Dev. Growth Differ. 2011, 53, 911–923. [Google Scholar] [CrossRef] [PubMed]
- Bovolenta, P.; Esteve, P.; Ruiz, J.M.; Cisneros, E.; Lopez-Rios, J. Beyond Wnt inhibition: New functions of secreted frizzled-related proteins in development and disease. J. Cell Sci. 2008, 121, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Niehrs, C. Function and biological roles of the dickkopf family of wnt modulators. Oncogene 2006, 25, 7469–7481. [Google Scholar] [CrossRef] [PubMed]
- Semenov, M.; Tamai, K.; He, X. Sost is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J. Biol. Chem. 2005, 280, 26770–26775. [Google Scholar] [CrossRef] [PubMed]
- Lintern, K.B.; Guidato, S.; Rowe, A.; Saldanha, J.W.; Itasaki, N. Characterization of wise protein and its molecular mechanism to interact with both Wnt and bmp signals. J. Biol. Chem. 2009, 284, 23159–23168. [Google Scholar] [CrossRef] [PubMed]
- Herr, P.; Hausmann, G.; Basler, K. Wnt secretion and signalling in human disease. Trends Mol. Med. 2012, 18, 483–493. [Google Scholar] [CrossRef] [PubMed]
- McEwen, D.G.; Peifer, M. Wnt signaling: Moving in a new direction. Curr. Biol.: CB 2000, 10, R562–R564. [Google Scholar] [CrossRef]
- Veeman, M.T.; Axelrod, J.D.; Moon, R.T. A second canon. Functions and mechanisms of β-catenin-independent wnt signaling. Dev. Cell 2003, 5, 367–377. [Google Scholar] [CrossRef]
- Sheldahl, L.C.; Slusarski, D.C.; Pandur, P.; Miller, J.R.; Kuhl, M.; Moon, R.T. Dishevelled activates Ca2+ FLUX, PKC, and CAMKII in vertebrate embryos. J. Cell Biol. 2003, 161, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Kohn, A.D.; Moon, R.T. Wnt and calcium signaling: β-catenin-independent pathways. Cell Calcium 2005, 38, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Ishitani, T.; Kishida, S.; Hyodo-Miura, J.; Ueno, N.; Yasuda, J.; Waterman, M.; Shibuya, H.; Moon, R.T.; Ninomiya-Tsuji, J.; Matsumoto, K. The TAK1-NLK mitogen-activated protein kinase cascade functions in the wnt-5a/Ca(2+) pathway to antagonize wnt/β-catenin signaling. Mol. Cell. Biol. 2003, 23, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Rao, T.P.; Kuhl, M. An updated overview on wnt signaling pathways: A prelude for more. Circ. Res. 2010, 106, 1798–1806. [Google Scholar] [CrossRef] [PubMed]
- Hogan, P.G.; Chen, L.; Nardone, J.; Rao, A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003, 17, 2205–2232. [Google Scholar] [CrossRef] [PubMed]
- Feske, S.; Okamura, H.; Hogan, P.G.; Rao, A. Ca2+/calcineurin signalling in cells of the immune system. Biochem. Biophys. Res. Commun. 2003, 311, 1117–1132. [Google Scholar] [CrossRef] [PubMed]
- Manda, K.R.; Tripathi, P.; Hsi, A.C.; Ning, J.; Ruzinova, M.B.; Liapis, H.; Bailey, M.; Zhang, H.; Maher, C.A.; Humphrey, P.A.; et al. NFATC1 promotes prostate tumorigenesis and overcomes pten loss-induced senescence. Oncogene 2016, 35, 3282–3292. [Google Scholar] [CrossRef] [PubMed]
- Bengoa-Vergniory, N.; Kypta, R.M. Canonical and noncanonical wnt signaling in neural stem/progenitor cells. Cell. Mol. Life Sci.: CMLS 2015, 72, 4157–4172. [Google Scholar] [CrossRef] [PubMed]
- Acebron, S.P.; Karaulanov, E.; Berger, B.S.; Huang, Y.L.; Niehrs, C. Mitotic wnt signaling promotes protein stabilization and regulates cell size. Mol. Cell 2014, 54, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Orte, E.; Saenz-Narciso, B.; Moreno, S.; Cabello, J. Multiple functions of the noncanonical wnt pathway. Trends Genet.: TIG 2013, 29, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Weidinger, G.; Moon, R.T. When wnts antagonize wnts. J. Cell Biol. 2003, 162, 753–755. [Google Scholar] [CrossRef] [PubMed]
- Timms, B.G. Prostate development: A historical perspective. Differ. Res. Biol. Divers. 2008, 76, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.M.; Abate-Shen, C. Molecular genetics of prostate cancer: New prospects for old challenges. Genes Dev. 2010, 24, 1967–2000. [Google Scholar] [CrossRef] [PubMed]
- Sugimura, Y.; Cunha, G.R.; Donjacour, A.A. Morphogenesis of ductal networks in the mouse prostate. Biol. Reprod. 1986, 34, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.C.; Joyner, A.L. Hedgehog signaling in prostate epithelial-mesenchymal growth regulation. Dev. Biol. 2015, 400, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Abate-Shen, C.; Shen, M.M. Molecular genetics of prostate cancer. Genes Dev. 2000, 14, 2410–2434. [Google Scholar] [CrossRef] [PubMed]
- Staack, A.; Donjacour, A.A.; Brody, J.; Cunha, G.R.; Carroll, P. Mouse urogenital development: A practical approach. Differ. Res. Biol. Divers. 2003, 71, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Takeda, H.; Nakamoto, T.; Kokontis, J.; Chodak, G.W.; Chang, C. Autoregulation of androgen receptor expression in rodent prostate: Immunohistochemical and in situ hybridization analysis. Biochem. Biophys. Res. Commun. 1991, 177, 488–496. [Google Scholar] [CrossRef]
- Cooke, P.S.; Young, P.; Cunha, G.R. Androgen receptor expression in developing male reproductive organs. Endocrinology 1991, 128, 2867–2873. [Google Scholar] [CrossRef] [PubMed]
- Cunha, G.R.; Ricke, W.; Thomson, A.; Marker, P.C.; Risbridger, G.; Hayward, S.W.; Wang, Y.Z.; Donjacour, A.A.; Kurita, T. Hormonal, cellular, and molecular regulation of normal and neoplastic prostatic development. J. Steroid Biochem. Mol. Biol. 2004, 92, 221–236. [Google Scholar] [CrossRef] [PubMed]
- Cunha, G.R. The role of androgens in the epithelio-mesenchymal interactions involved in prostatic morphogenesis in embryonic mice. Anat. Rec. 1973, 175, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Cunha, G.R.; Donjacour, A.A.; Cooke, P.S.; Mee, S.; Bigsby, R.M.; Higgins, S.J.; Sugimura, Y. The endocrinology and developmental biology of the prostate. Endocr. Rev. 1987, 8, 338–362. [Google Scholar] [CrossRef] [PubMed]
- Cunha, G.R. Mesenchymal-Epithelial interactions: Past, present, and future. Differ. Res. Biol. Divers. 2008, 76, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Marker, P.C.; Donjacour, A.A.; Dahiya, R.; Cunha, G.R. Hormonal, cellular, and molecular control of prostatic development. Dev. Biol. 2003, 253, 165–174. [Google Scholar] [CrossRef]
- Mehta, V.; Abler, L.L.; Keil, K.P.; Schmitz, C.T.; Joshi, P.S.; Vezina, C.M. Atlas of wnt and R-spondin gene expression in the developing male mouse lower urogenital tract. Dev. Dyn.: Off. Publ. Am. Assoc. Anat. 2011, 240, 2548–2560. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hayward, S.; Cao, M.; Thayer, K.; Cunha, G. Cell differentiation lineage in the prostate. Differ. Res. Biol. Divers. 2001, 68, 270–279. [Google Scholar] [CrossRef]
- Hayward, S.W.; Baskin, L.S.; Haughney, P.C.; Cunha, A.R.; Foster, B.A.; Dahiya, R.; Prins, G.S.; Cunha, G.R. Epithelial development in the rat ventral prostate, anterior prostate and seminal vesicle. Acta Anat. 1996, 155, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Rittenhouse, H.G.; Finlay, J.A.; Mikolajczyk, S.D.; Partin, A.W. Human kallikrein 2 (HK2) and prostate-specific antigen (PSA): Two closely related, but distinct, kallikreins in the prostate. Crit. Rev. Clin. Lab. Sci. 1998, 35, 275–368. [Google Scholar] [CrossRef] [PubMed]
- Muniyan, S.; Chaturvedi, N.K.; Dwyer, J.G.; Lagrange, C.A.; Chaney, W.G.; Lin, M.F. Human prostatic acid phosphatase: Structure, function and regulation. Int. J. Mol. Sci. 2013, 14, 10438–10464. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; van der Laak, J.; Smedts, F.; Schoots, C.; Verhofstad, A.; de la Rosette, J.; Schalken, J. Neuroendocrine cells during human prostate development: Does neuroendocrine cell density remain constant during fetal as well as postnatal life? Prostate 2000, 42, 116–123. [Google Scholar] [CrossRef]
- Cohen, R.J.; Glezerson, G.; Taylor, L.F.; Grundle, H.A.; Naude, J.H. The neuroendocrine cell population of the human prostate gland. J. Urol. 1993, 150, 365–368. [Google Scholar] [PubMed]
- Van Leenders, G.; Dijkman, H.; Hulsbergen-van de Kaa, C.; Ruiter, D.; Schalken, J. Demonstration of intermediate cells during human prostate epithelial differentiation in situ and in vitro using triple-staining confocal scanning microscopy. Lab. Investig. J. Tech. Methods Pathol. 2000, 80, 1251–1258. [Google Scholar] [CrossRef]
- Uzgare, A.R.; Isaacs, J.T. Enhanced redundancy in AKT and mitogen-activated protein kinase-induced survival of malignant versus normal prostate epithelial cells. Cancer Res. 2004, 64, 6190–6199. [Google Scholar] [CrossRef] [PubMed]
- Xin, L.; Lukacs, R.U.; Lawson, D.A.; Cheng, D.; Witte, O.N. Self-renewal and multilineage differentiation in vitro from murine prostate stem cells. Stem Cells 2007, 25, 2760–2769. [Google Scholar] [CrossRef] [PubMed]
- Kwon, O.J.; Xin, L. Prostate epithelial stem and progenitor cells. Am. J. Clin. Exp. Urol. 2014, 2, 209–218. [Google Scholar] [PubMed]
- Prins, G.S.; Putz, O. Molecular signaling pathways that regulate prostate gland development. Differ. Res. Biol. Divers. 2008, 76, 641–659. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.J.; Hoffman, B.G.; Ruiz de Algara, T.; Helgason, C.D. Sage reveals expression of wnt signalling pathway members during mouse prostate development. Gene Exp. Patterns: GEP 2006, 6, 310–324. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, C.C.; Nelson, P.S. Gene expression profiling in the developing prostate. Differ. Res. Biol. Divers. 2008, 76, 624–640. [Google Scholar] [CrossRef] [PubMed]
- Joesting, M.S.; Cheever, T.R.; Volzing, K.G.; Yamaguchi, T.P.; Wolf, V.; Naf, D.; Rubin, J.S.; Marker, P.C. Secreted frizzled related protein 1 is a paracrine modulator of epithelial branching morphogenesis, proliferation, and secretory gene expression in the prostate. Dev. Biol. 2008, 317, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.E.; Wang, X.D.; Ernst, J.A.; Polakis, P.; Gao, W.Q. Regulation of epithelial branching morphogenesis and cancer cell growth of the prostate by wnt signaling. PLoS ONE 2008, 3, e2186. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Pu, Y.; Hu, W.Y.; Birch, L.; Luccio-Camelo, D.; Yamaguchi, T.; Prins, G.S. The role of Wnt5a in prostate gland development. Dev. Biol. 2009, 328, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Allgeier, S.H.; Lin, T.M.; Vezina, C.M.; Moore, R.W.; Fritz, W.A.; Chiu, S.Y.; Zhang, C.; Peterson, R.E. Wnt5a selectively inhibits mouse ventral prostate development. Dev. Biol. 2008, 324, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Gat, U.; DasGupta, R.; Degenstein, L.; Fuchs, E. De novo hair follicle morphogenesis and hair tumors in mice expressing a truncated beta-catenin in skin. Cell 1998, 95, 605–614. [Google Scholar] [CrossRef]
- Hatsell, S.; Rowlands, T.; Hiremath, M.; Cowin, P. β-Catenin and TCFS in mammary development and cancer. J. Mammary Gland Biol. Neoplasia 2003, 8, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Chu, E.Y.; Watt, B.; Zhang, Y.; Gallant, N.M.; Andl, T.; Yang, S.H.; Lu, M.M.; Piccolo, S.; Schmidt-Ullrich, R.; et al. Wnt/β-Catenin signaling directs multiple stages of tooth morphogenesis. Dev. Biol. 2008, 313, 210–224. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Wang, Y.; Jiang, M.; Bierie, B.; Roy-Burman, P.; Shen, M.M.; Taketo, M.M.; Wills, M.; Matusik, R.J. Activation of β-catenin in mouse prostate causes hgpin and continuous prostate growth after castration. Prostate 2009, 69, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Simons, B.W.; Hurley, P.J.; Huang, Z.; Ross, A.E.; Miller, R.; Marchionni, L.; Berman, D.M.; Schaeffer, E.M. Wnt signaling though β-catenin is required for prostate lineage specification. Dev. Biol. 2012, 371, 246–255. [Google Scholar] [CrossRef] [PubMed]
- English, H.F.; Santen, R.J.; Isaacs, J.T. Response of glandular versus basal rat ventral prostatic epithelial cells to androgen withdrawal and replacement. Prostate 1987, 11, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Xin, L.; Ide, H.; Kim, Y.; Dubey, P.; Witte, O.N. In vivo regeneration of murine prostate from dissociated cell populations of postnatal epithelia and urogenital sinus mesenchyme. Proc. Natl. Acad. Sci. USA 2003, 100, 11896–11903. [Google Scholar] [CrossRef] [PubMed]
- Garraway, I.P.; Sun, W.; Tran, C.P.; Perner, S.; Zhang, B.; Goldstein, A.S.; Hahm, S.A.; Haider, M.; Head, C.S.; Reiter, R.E.; et al. Human prostate sphere-forming cells represent a subset of basal epithelial cells capable of glandular regeneration in vivo. Prostate 2010, 70, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Lawson, D.A.; Zong, Y.; Memarzadeh, S.; Xin, L.; Huang, J.; Witte, O.N. Basal epithelial stem cells are efficient targets for prostate cancer initiation. Proc. Natl. Acad. Sci. USA 2010, 107, 2610–2615. [Google Scholar] [CrossRef] [PubMed]
- Xin, L.; Lawson, D.A.; Witte, O.N. The Sca-1 cell surface marker enriches for a prostate-regenerating cell subpopulation that can initiate prostate tumorigenesis. Proc. Natl. Acad. Sci. USA 2005, 102, 6942–6947. [Google Scholar] [CrossRef] [PubMed]
- Burger, P.E.; Xiong, X.; Coetzee, S.; Salm, S.N.; Moscatelli, D.; Goto, K.; Wilson, E.L. Sca-1 expression identifies stem cells in the proximal region of prostatic ducts with high capacity to reconstitute prostatic tissue. Proc. Natl. Acad. Sci. USA 2005, 102, 7180–7185. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, A.S.; Lawson, D.A.; Cheng, D.; Sun, W.; Garraway, I.P.; Witte, O.N. Trop2 identifies a subpopulation of murine and human prostate basal cells with stem cell characteristics. Proc. Natl. Acad. Sci. USA 2008, 105, 20882–20887. [Google Scholar] [CrossRef] [PubMed]
- Lawson, D.A.; Xin, L.; Lukacs, R.U.; Cheng, D.; Witte, O.N. Isolation and functional characterization of murine prostate stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Jiao, J.; Hindoyan, A.; Wang, S.; Tran, L.M.; Goldstein, A.S.; Lawson, D.; Chen, D.; Li, Y.; Guo, C.; Zhang, B.; et al. Identification of CD166 as a surface marker for enriching prostate stem/progenitor and cancer initiating cells. PLoS ONE 2012, 7, e42564. [Google Scholar] [CrossRef] [PubMed]
- Richardson, G.D.; Robson, C.N.; Lang, S.H.; Neal, D.E.; Maitland, N.J.; Collins, A.T. Cd133, a novel marker for human prostatic epithelial stem cells. J. Cell Sci. 2004, 117, 3539–3545. [Google Scholar] [CrossRef] [PubMed]
- Leong, K.G.; Wang, B.E.; Johnson, L.; Gao, W.Q. Generation of a prostate from a single adult stem cell. Nature 2008, 456, 804–808. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Kruithof-de Julio, M.; Economides, K.D.; Walker, D.; Yu, H.; Halili, M.V.; Hu, Y.P.; Price, S.M.; Abate-Shen, C.; Shen, M.M. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature 2009, 461, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.A.; Shen, M.M. Revisiting the concept of cancer stem cells in prostate cancer. Oncogene 2011, 30, 1261–1271. [Google Scholar] [CrossRef] [PubMed]
- Drost, J.; Karthaus, W.R.; Gao, D.; Driehuis, E.; Sawyers, C.L.; Chen, Y.; Clevers, H. Organoid culture systems for prostate epithelial and cancer tissue. Nat. Protocols 2016, 11, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Karthaus, W.R.; Iaquinta, P.J.; Drost, J.; Gracanin, A.; van Boxtel, R.; Wongvipat, J.; Dowling, C.M.; Gao, D.; Begthel, H.; Sachs, N.; et al. Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell 2014, 159, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Blum, R.; Gupta, R.; Burger, P.E.; Ontiveros, C.S.; Salm, S.N.; Xiong, X.; Kamb, A.; Wesche, H.; Marshall, L.; Cutler, G.; et al. Molecular signatures of prostate stem cells reveal novel signaling pathways and provide insights into prostate cancer. PLoS ONE 2009, 4, e5722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blum, R.; Gupta, R.; Burger, P.E.; Ontiveros, C.S.; Salm, S.N.; Xiong, X.; Kamb, A.; Wesche, H.; Marshall, L.; Cutler, G.; et al. Molecular signatures of the primitive prostate stem cell niche reveal novel mesenchymal-epithelial signaling pathways. PLoS ONE 2010, 5, e13024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolova, T.; Wu, M.; Brumbarov, K.; Alt, R.; Opitz, H.; Boheler, K.R.; Cross, M.; Wobus, A.M. Wnt-conditioned media differentially affect the proliferation and differentiation of cord blood-derived CD133+ cells in vitro. Differ. Res. Biol. Divers. 2007, 75, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Singla, D.K.; Schneider, D.J.; LeWinter, M.M.; Sobel, B.E. Wnt3a but not wnt11 supports self-renewal of embryonic stem cells. Biochem. Biophys. Res. Commun. 2006, 345, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Lukacs, R.U.; Memarzadeh, S.; Wu, H.; Witte, O.N. BMI-1 is a crucial regulator of prostate stem cell self-renewal and malignant transformation. Cell Stem Cell 2010, 7, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Heinlein, C.A.; Chang, C. Androgen receptor in prostate cancer. Endocr. Rev. 2004, 25, 276–308. [Google Scholar] [CrossRef] [PubMed]
- Heinlein, C.A.; Chang, C. Androgen receptor (AR) coregulators: An overview. Endocr. Rev. 2002, 23, 175–200. [Google Scholar] [CrossRef] [PubMed]
- Jenster, G.; van der Korput, H.A.; van Vroonhoven, C.; van der Kwast, T.H.; Trapman, J.; Brinkmann, A.O. Domains of the human androgen receptor involved in steroid binding, transcriptional activation, and subcellular localization. Mol. Endocrinol. 1991, 5, 1396–1404. [Google Scholar] [CrossRef] [PubMed]
- MacLean, H.E.; Warne, G.L.; Zajac, J.D. Localization of functional domains in the androgen receptor. J. Steroid Biochem. Mol. Biol. 1997, 62, 233–242. [Google Scholar] [CrossRef]
- Ferraldeschi, R.; Welti, J.; Luo, J.; Attard, G.; de Bono, J.S. Targeting the androgen receptor pathway in castration-resistant prostate cancer: Progresses and prospects. Oncogene 2015, 34, 1745–1757. [Google Scholar] [CrossRef] [PubMed]
- Helsen, C.; Kerkhofs, S.; Clinckemalie, L.; Spans, L.; Laurent, M.; Boonen, S.; Vanderschueren, D.; Claessens, F. Structural basis for nuclear hormone receptor DNA binding. Mol. Cell. Endocrinol. 2012, 348, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Clinckemalie, L.; Vanderschueren, D.; Boonen, S.; Claessens, F. The hinge region in androgen receptor control. Mol. Cell. Endocrinol. 2012, 358, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Dittmar, K.D.; Banach, M.; Galigniana, M.D.; Pratt, W.B. The role of DNAJ-like proteins in glucocorticoid receptor.HSP90 heterocomplex assembly by the reconstituted HSP90.P60.Hsp70 foldosome complex. J. Biol. Chem. 1998, 273, 7358–7366. [Google Scholar] [CrossRef] [PubMed]
- Prescott, J.; Coetzee, G.A. Molecular chaperones throughout the life cycle of the androgen receptor. Cancer Lett. 2006, 231, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Chmelar, R.; Buchanan, G.; Need, E.F.; Tilley, W.; Greenberg, N.M. Androgen receptor coregulators and their involvement in the development and progression of prostate cancer. Int. J. Cancer 2007, 120, 719–733. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.K.; Lavrovsky, Y.; Song, C.S.; Chen, S.; Jung, M.H.; Velu, N.K.; Bi, B.Y.; Chatterjee, B. Regulation of androgen action. Vitam. Horm. 1999, 55, 309–352. [Google Scholar] [PubMed]
- Lee, D.K.; Chang, C. Molecular communication between androgen receptor and general transcription machinery. J. Steroid Biochem. Mol. Biol. 2003, 84, 41–49. [Google Scholar] [CrossRef]
- Lee, D.K.; Chang, C. Endocrine mechanisms of disease: Expression and degradation of androgen receptor: Mechanism and clinical implication. J. Clin. Endocrinol. Metab. 2003, 88, 4043–4054. [Google Scholar] [CrossRef] [PubMed]
- Simental, J.A.; Sar, M.; Lane, M.V.; French, F.S.; Wilson, E.M. Transcriptional activation and nuclear targeting signals of the human androgen receptor. J. Biol. Chem. 1991, 266, 510–518. [Google Scholar] [PubMed]
- Zhou, Z.X.; Sar, M.; Simental, J.A.; Lane, M.V.; Wilson, E.M. A ligand-dependent bipartite nuclear targeting signal in the human androgen receptor. Requirement for the DNA-binding domain and modulation by NH2-terminal and carboxyl-terminal sequences. J. Biol. Chem. 1994, 269, 13115–13123. [Google Scholar] [PubMed]
- Gelmann, E.P. Molecular biology of the androgen receptor. J. Clin. Oncol.: Off. J. Am. Soc. Clin. Oncol. 2002, 20, 3001–3015. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Lee, L.W.; Minges, J.T.; Wilson, E.M. Dependence of selective gene activation on the androgen receptor NH2- and COOH-terminal interaction. J. Biol. Chem. 2002, 277, 25631–25639. [Google Scholar] [CrossRef] [PubMed]
- Bennett, N.C.; Gardiner, R.A.; Hooper, J.D.; Johnson, D.W.; Gobe, G.C. Molecular cell biology of androgen receptor signalling. Int. J. Biochem. Cell Biol. 2010, 42, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Gioeli, D.; Ficarro, S.B.; Kwiek, J.J.; Aaronson, D.; Hancock, M.; Catling, A.D.; White, F.M.; Christian, R.E.; Settlage, R.E.; Shabanowitz, J.; et al. Androgen receptor phosphorylation. Regulation and identification of the phosphorylation sites. J. Biol. Chem. 2002, 277, 29304–29314. [Google Scholar] [CrossRef] [PubMed]
- Blok, L.J.; de Ruiter, P.E.; Brinkmann, A.O. Forskolin-Induced dephosphorylation of the androgen receptor impairs ligand binding. Biochemistry 1998, 37, 3850–3857. [Google Scholar] [CrossRef] [PubMed]
- Gaughan, L.; Logan, I.R.; Neal, D.E.; Robson, C.N. Regulation of androgen receptor and histone deacetylase 1 by MDM2-mediated ubiquitylation. Nucleic Acids Res. 2005, 33, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.L.; Shi, Y.; Petrovics, G.; Sun, C.; Makarem, M.; Zhang, W.; Sesterhenn, I.A.; McLeod, D.G.; Sun, L.; Moul, J.W.; et al. PMEPA1, an androgen-regulated NEDD4-binding protein, exhibits cell growth inhibitory function and decreased expression during prostate cancer progression. Cancer Res. 2003, 63, 4299–4304. [Google Scholar] [PubMed]
- Richter, E.; Srivastava, S.; Dobi, A. Androgen receptor and prostate cancer. Prostate Cancer Prostatic Dis. 2007, 10, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Terry, S.; Yang, X.; Chen, M.W.; Vacherot, F.; Buttyan, R. Multifaceted interaction between the androgen and wnt signaling pathways and the implication for prostate cancer. J. Cell. Biochem. 2006, 99, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, A.O.; Blok, L.J.; de Ruiter, P.E.; Doesburg, P.; Steketee, K.; Berrevoets, C.A.; Trapman, J. Mechanisms of androgen receptor activation and function. J. Steroid Biochem. Mol. Biol. 1999, 69, 307–313. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Iljin, K.; Wolf, M.; Edgren, H.; Gupta, S.; Kilpinen, S.; Skotheim, R.I.; Peltola, M.; Smit, F.; Verhaegh, G.; Schalken, J.; et al. TMPRSS2 fusions with oncogenic ETS factors in prostate cancer involve unbalanced genomic rearrangements and are associated with HDAC1 and epigenetic reprogramming. Cancer Res. 2006, 66, 10242–10246. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Mani, R.S.; Cao, Q.; Brenner, C.J.; Cao, X.; Wang, X.; Wu, L.; Li, J.; Hu, M.; Gong, Y.; et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell 2010, 17, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Baena, E.; Shao, Z.; Linn, D.E.; Glass, K.; Hamblen, M.J.; Fujiwara, Y.; Kim, J.; Nguyen, M.; Zhang, X.; Godinho, F.J.; et al. ETV1 directs androgen metabolism and confers aggressive prostate cancer in targeted mice and patients. Genes Dev. 2013, 27, 683–698. [Google Scholar] [CrossRef] [PubMed]
- Kyprianou, N.; Isaacs, J.T. Activation of programmed cell death in the rat ventral prostate after castration. Endocrinology 1988, 122, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Prins, G.S.; Birch, L. Immunocytochemical analysis of androgen receptor along the ducts of the separate rat prostate lobes after androgen withdrawal and replacement. Endocrinology 1993, 132, 169–178. [Google Scholar] [PubMed]
- Kim, D.; Gregory, C.W.; French, F.S.; Smith, G.J.; Mohler, J.L. Androgen receptor expression and cellular proliferation during transition from androgen-dependent to recurrent growth after castration in the cwr22 prostate cancer xenograft. Am. J. Pathol. 2002, 160, 219–226. [Google Scholar] [CrossRef]
- Titus, M.A.; Schell, M.J.; Lih, F.B.; Tomer, K.B.; Mohler, J.L. Testosterone and dihydrotestosterone tissue levels in recurrent prostate cancer. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 2005, 11, 4653–4657. [Google Scholar] [CrossRef]
- Stanbrough, M.; Bubley, G.J.; Ross, K.; Golub, T.R.; Rubin, M.A.; Penning, T.M.; Febbo, P.G.; Balk, S.P. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006, 66, 2815–2825. [Google Scholar] [CrossRef] [PubMed]
- Locke, J.A.; Guns, E.S.; Lubik, A.A.; Adomat, H.H.; Hendy, S.C.; Wood, C.A.; Ettinger, S.L.; Gleave, M.E.; Nelson, C.C. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res. 2008, 68, 6407–6415. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, R.L.; Potthoff, M.J.; Haberland, M.; Qi, X.; Matsuzaki, S.; Humphries, K.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Maintenance of cardiac energy metabolism by histone deacetylase 3 in mice. J. Clin. Investig. 2008, 118, 3588–3597. [Google Scholar] [CrossRef] [PubMed]
- Bubendorf, L.; Kononen, J.; Koivisto, P.; Schraml, P.; Moch, H.; Gasser, T.C.; Willi, N.; Mihatsch, M.J.; Sauter, G.; Kallioniemi, O.P. Survey of gene amplifications during prostate cancer progression by high-throughout fluorescence in situ hybridization on tissue microarrays. Cancer Res. 1999, 59, 803–806. [Google Scholar] [PubMed]
- Haapala, K.; Kuukasjarvi, T.; Hyytinen, E.; Rantala, I.; Helin, H.J.; Koivisto, P.A. Androgen receptor amplification is associated with increased cell proliferation in prostate cancer. Hum. Pathol. 2007, 38, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Linja, M.J.; Savinainen, K.J.; Saramaki, O.R.; Tammela, T.L.; Vessella, R.L.; Visakorpi, T. Amplification and overexpression of androgen receptor gene in hormone-refractory prostate cancer. Cancer Res. 2001, 61, 3550–3555. [Google Scholar] [PubMed]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Visakorpi, T.; Hyytinen, E.; Koivisto, P.; Tanner, M.; Keinanen, R.; Palmberg, C.; Palotie, A.; Tammela, T.; Isola, J.; Kallioniemi, O.P. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat. Genet. 1995, 9, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Koivisto, P.; Kononen, J.; Palmberg, C.; Tammela, T.; Hyytinen, E.; Isola, J.; Trapman, J.; Cleutjens, K.; Noordzij, A.; Visakorpi, T.; et al. Androgen receptor gene amplification: A possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res. 1997, 57, 314–319. [Google Scholar] [PubMed]
- Miyoshi, Y.; Uemura, H.; Fujinami, K.; Mikata, K.; Harada, M.; Kitamura, H.; Koizumi, Y.; Kubota, Y. Fluorescence in situ hybridization evaluation of C-Myc and androgen receptor gene amplification and chromosomal anomalies in prostate cancer in japanese patients. Prostate 2000, 43, 225–232. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.A.; Chen, Y.F.; Balbas, M.D.; Wongvipat, J.; Socci, N.D.; Viale, A.; Kim, K.; Sawyers, C.L. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc. Natl. Acad. Sci. USA 2010, 107, 16759–16765. [Google Scholar] [CrossRef] [PubMed]
- Dehm, S.M.; Tindall, D.J. Alternatively spliced androgen receptor variants. Endocr.-Relat. Cancer 2011, 18, R183–R196. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Lu, C.; Mostaghel, E.A.; Yegnasubramanian, S.; Gurel, M.; Tannahill, C.; Edwards, J.; Isaacs, W.B.; Nelson, P.S.; Bluemn, E.; et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012, 72, 3457–3462. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Fedor, H.L.; Lotan, T.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, E.M.; Marchionni, L.; Huang, Z.; Simons, B.; Blackman, A.; Yu, W.; Parmigiani, G.; Berman, D.M. Androgen-induced programs for prostate epithelial growth and invasion arise in embryogenesis and are reactivated in cancer. Oncogene 2008, 27, 7180–7191. [Google Scholar] [CrossRef] [PubMed]
- Mulholland, D.J.; Dedhar, S.; Coetzee, G.A.; Nelson, C.C. Interaction of nuclear receptors with the wnt/β-catenin/TCF signaling axis: Wnt you like to know? Endocr. Rev. 2005, 26, 898–915. [Google Scholar] [CrossRef] [PubMed]
- Rubinfeld, B.; Souza, B.; Albert, I.; Muller, O.; Chamberlain, S.H.; Masiarz, F.R.; Munemitsu, S.; Polakis, P. Association of the apc gene product with β-catenin. Science 1993, 262, 1731–1734. [Google Scholar] [CrossRef] [PubMed]
- Kharaishvili, G.; Simkova, D.; Makharoblidze, E.; Trtkova, K.; Kolar, Z.; Bouchal, J. Wnt signaling in prostate development and carcinogenesis. Biomed. Papers Med. Fac. Univ. Palacky Olomouc Czechoslov. 2011, 155, 11–18. [Google Scholar] [CrossRef]
- Polakis, P. Wnt signaling in cancer. Cold Spring Harb. Perspect. Biol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Polakis, P. Wnt signaling and cancer. Genes Dev. 2000, 14, 1837–1851. [Google Scholar] [CrossRef] [PubMed]
- Voeller, H.J.; Truica, C.I.; Gelmann, E.P. β-catenin mutations in human prostate cancer. Cancer Res. 1998, 58, 2520–2523. [Google Scholar] [PubMed]
- Chesire, D.R.; Ewing, C.M.; Sauvageot, J.; Bova, G.S.; Isaacs, W.B. Detection and analysis of β-catenin mutations in prostate cancer. Prostate 2000, 45, 323–334. [Google Scholar] [CrossRef]
- de la Taille, A.; Rubin, M.A.; Chen, M.W.; Vacherot, F.; de Medina, S.G.; Burchardt, M.; Buttyan, R.; Chopin, D. β-catenin-related anomalies in apoptosis-resistant and hormone-refractory prostate cancer cells. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 2003, 9, 1801–1807. [Google Scholar]
- Chen, G.; Shukeir, N.; Potti, A.; Sircar, K.; Aprikian, A.; Goltzman, D.; Rabbani, S.A. Up-regulation of wnt-1 and β-catenin production in patients with advanced metastatic prostate carcinoma: Potential pathogenetic and prognostic implications. Cancer 2004, 101, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wang, J.; Sadar, M.D. Crosstalk between the androgen receptor and beta-catenin in castrate-resistant prostate cancer. Cancer Res. 2008, 68, 9918–9927. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, L.; Rizzo, C.A.; Spires, T.E.; Platero, J.S.; Wu, Q.; Lin, T.A.; Gottardis, M.M.; Attar, R.M. The androgen receptor can signal through wnt/β-catenin in prostate cancer cells as an adaptation mechanism to castration levels of androgens. BMC Cell Biol. 2008, 9, 4. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, N.N.; Shao, S.; Hoang, B.H.; Mercola, D.; Zi, X. Wnt signaling in castration-resistant prostate cancer: Implications for therapy. Am. J. Clin. Exp. Urol. 2014, 2, 27–44. [Google Scholar] [PubMed]
- Truica, C.I.; Byers, S.; Gelmann, E.P. β-Catenin affects androgen receptor transcriptional activity and ligand specificity. Cancer Res. 2000, 60, 4709–4713. [Google Scholar] [PubMed]
- Yang, F.; Li, X.; Sharma, M.; Sasaki, C.Y.; Longo, D.L.; Lim, B.; Sun, Z. Linking β-catenin to androgen-signaling pathway. J. Biol. Chem. 2002, 277, 11336–11344. [Google Scholar] [CrossRef] [PubMed]
- Song, L.N.; Herrell, R.; Byers, S.; Shah, S.; Wilson, E.M.; Gelmann, E.P. β-Catenin binds to the activation function 2 region of the androgen receptor and modulates the effects of the n-terminal domain and TIF2 on ligand-dependent transcription. Mol. Cell. Biol. 2003, 23, 1674–1687. [Google Scholar] [CrossRef] [PubMed]
- Masiello, D.; Chen, S.Y.; Xu, Y.; Verhoeven, M.C.; Choi, E.; Hollenberg, A.N.; Balk, S.P. Recruitment of beta-catenin by wild-type or mutant androgen receptors correlates with ligand-stimulated growth of prostate cancer cells. Mol. Endocrinol. 2004, 18, 2388–2401. [Google Scholar] [CrossRef] [PubMed]
- Chesire, D.R.; Ewing, C.M.; Gage, W.R.; Isaacs, W.B. In vitro evidence for complex modes of nuclear β-catenin signaling during prostate growth and tumorigenesis. Oncogene 2002, 21, 2679–2694. [Google Scholar] [CrossRef] [PubMed]
- Mulholland, D.J.; Cheng, H.; Reid, K.; Rennie, P.S.; Nelson, C.C. The androgen receptor can promote β-catenin nuclear translocation independently of adenomatous polyposis coli. J. Biol. Chem. 2002, 277, 17933–17943. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, L.; Zhang, M.; Melamed, J.; Liu, X.; Reiter, R.; Wei, J.; Peng, Y.; Zou, X.; Pellicer, A.; et al. LEF1 in androgen-independent prostate cancer: Regulation of androgen receptor expression, prostate cancer growth, and invasion. Cancer Res. 2009, 69, 3332–3338. [Google Scholar] [CrossRef] [PubMed]
- Pawlowski, J.E.; Ertel, J.R.; Allen, M.P.; Xu, M.; Butler, C.; Wilson, E.M.; Wierman, M.E. Liganded androgen receptor interaction with β-catenin: Nuclear co-localization and modulation of transcriptional activity in neuronal cells. J. Biol. Chem. 2002, 277, 20702–20710. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, W.; Zhang, Y.; Yuan, X.; Xu, K.; Yu, J.; Chen, Z.; Beroukhim, R.; Wang, H.; Lupien, M.; et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell 2009, 138, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Chesire, D.R.; Dunn, T.A.; Ewing, C.M.; Luo, J.; Isaacs, W.B. Identification of aryl hydrocarbon receptor as a putative wnt/β-catenin pathway target gene in prostate cancer cells. Cancer Res. 2004, 64, 2523–2533. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Symes, A.J.; Kane, C.A.; Freeman, A.; Nariculam, J.; Munson, P.; Thrasivoulou, C.; Masters, J.R.; Ahmed, A. A novel role for wnt/Ca2+ signaling in actin cytoskeleton remodeling and cell motility in prostate cancer. PLoS ONE 2010, 5, e10456. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Oue, N.; Sato, A.; Hasegawa, Y.; Matsubara, A.; Yasui, W.; Kikuchi, A. Wnt5a signaling is involved in the aggressiveness of prostate cancer and expression of metalloproteinase. Oncogene 2010, 29, 2036–2046. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, D.T.; Zheng, Y.; Wittner, B.S.; Lee, R.J.; Zhu, H.; Broderick, K.T.; Desai, R.; Fox, D.B.; Brannigan, B.W.; Trautwein, J.; et al. RNA-seq of single prostate CTCS implicates noncanonical wnt signaling in antiandrogen resistance. Science 2015, 349, 1351–1356. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.C.; Kodjabachian, L.; Rebbert, M.L.; Rattner, A.; Smallwood, P.M.; Samos, C.H.; Nusse, R.; Dawid, I.B.; Nathans, J. A new secreted protein that binds to wnt proteins and inhibits their activities. Nature 1999, 398, 431–436. [Google Scholar] [PubMed]
- Jones, S.E.; Jomary, C. Secreted frizzled-related proteins: Searching for relationships and patterns. BioEssays 2002, 24, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Kypta, R. Secreted antagonists of the wnt signalling pathway. J. Cell Sci. 2003, 116, 2627–2634. [Google Scholar] [CrossRef] [PubMed]
- O'Hurley, G.; Perry, A.S.; O’Grady, A.; Loftus, B.; Smyth, P.; O’Leary, J.J.; Sheils, O.; Fitzpatrick, J.M.; Hewitt, S.M.; Lawler, M.; et al. The role of secreted frizzled-related protein 2 expression in prostate cancer. Histopathology 2011, 59, 1240–1248. [Google Scholar] [CrossRef] [PubMed]
- Wissmann, C.; Wild, P.J.; Kaiser, S.; Roepcke, S.; Stoehr, R.; Woenckhaus, M.; Kristiansen, G.; Hsieh, J.C.; Hofstaedter, F.; Hartmann, A.; et al. WIF1, a component of the wnt pathway, is down-regulated in prostate, breast, lung, and bladder cancer. J. Pathol. 2003, 201, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Zi, X.; Guo, Y.; Simoneau, A.R.; Hope, C.; Xie, J.; Holcombe, R.F.; Hoang, B.H. Expression of FRZB/secreted frizzled-related protein 3, a secreted wnt antagonist, in human androgen-independent prostate cancer PC-3 cells suppresses tumor growth and cellular invasiveness. Cancer Res. 2005, 65, 9762–9770. [Google Scholar] [CrossRef] [PubMed]
- Yee, D.S.; Tang, Y.; Li, X.; Liu, Z.; Guo, Y.; Ghaffar, S.; McQueen, P.; Atreya, D.; Xie, J.; Simoneau, A.R.; et al. The wnt inhibitory factor 1 restoration in prostate cancer cells was associated with reduced tumor growth, decreased capacity of cell migration and invasion and a reversal of epithelial to mesenchymal transition. Mol. Cancer 2010. [Google Scholar] [CrossRef] [PubMed]
- Thiele, S.; Rauner, M.; Goettsch, C.; Rachner, T.D.; Benad, P.; Fuessel, S.; Erdmann, K.; Hamann, C.; Baretton, G.B.; Wirth, M.P.; et al. Expression profile of wnt molecules in prostate cancer and its regulation by aminobisphosphonates. J. Cell. Biochem. 2011, 112, 1593–1600. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.L.; Daignault, S.D.; Shah, R.B.; Pienta, K.J.; Keller, E.T. Dickkopf-1 expression increases early in prostate cancer development and decreases during progression from primary tumor to metastasis. Prostate 2008, 68, 1396–1404. [Google Scholar] [CrossRef] [PubMed]
- Lawson, D.A.; Witte, O.N. Stem cells in prostate cancer initiation and progression. J. Clin. Investig. 2007, 117, 2044–2050. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Dai, J.; Zhang, H.; Sottnik, J.L.; Keller, J.M.; Escott, K.J.; Sanganee, H.J.; Yao, Z.; McCauley, L.K.; Keller, E.T. Activation of the wnt pathway through AR79, a GSK3BETA inhibitor, promotes prostate cancer growth in soft tissue and bone. Mol. Cancer Res.: MCR 2013, 11, 1597–1610. [Google Scholar] [CrossRef] [PubMed]
- Van den Hoogen, C.; van der Horst, G.; Cheung, H.; Buijs, J.T.; Lippitt, J.M.; Guzman-Ramirez, N.; Hamdy, F.C.; Eaton, C.L.; Thalmann, G.N.; Cecchini, M.G.; et al. High aldehyde dehydrogenase activity identifies tumor-initiating and metastasis-initiating cells in human prostate cancer. Cancer Res. 2010, 70, 5163–5173. [Google Scholar] [CrossRef] [PubMed]
- Trerotola, M.; Rathore, S.; Goel, H.L.; Li, J.; Alberti, S.; Piantelli, M.; Adams, D.; Jiang, Z.; Languino, L.R. CD133, trop-2 and alpha2beta1 integrin surface receptors as markers of putative human prostate cancer stem cells. Am. J. Transl. Res. 2010, 2, 135–144. [Google Scholar] [PubMed]
- Yun, E.J.; Zhou, J.; Lin, C.J.; Hernandez, E.; Fazli, L.; Gleave, M.; Hsieh, J.T. Targeting cancer stem cells in castration-resistant prostate cancer. Clin. Cancer Res. 2016, 22, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Rajan, P.; Sudbery, I.M.; Villasevil, M.E.; Mui, E.; Fleming, J.; Davis, M.; Ahmad, I.; Edwards, J.; Sansom, O.J.; Sims, D.; et al. Next-generation sequencing of advanced prostate cancer treated with androgen-deprivation therapy. Eur. Urol. 2014, 66, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Madar, A.; David, G.; Garabedian, M.J.; Dasgupta, R.; Logan, S.K. Inhibition of androgen receptor and beta-catenin activity in prostate cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 15710–15715. [Google Scholar] [CrossRef] [PubMed]
- Panet-Raymond, V.; Gottlieb, B.; Beitel, L.K.; Pinsky, L.; Trifiro, M.A. Interactions between androgen and estrogen receptors and the effects on their transactivational properties. Mol. Cell. Endocrinol. 2000, 167, 139–150. [Google Scholar] [CrossRef]
- Hickey, T.E.; Robinson, J.L.; Carroll, J.S.; Tilley, W.D. Minireview: The androgen receptor in breast tissues: Growth inhibitor, tumor suppressor, oncogene? Mol. Endocrinol. 2012, 26, 1252–1267. [Google Scholar] [CrossRef] [PubMed]
- Peters, A.A.; Ingman, W.V.; Tilley, W.D.; Butler, L.M. Differential effects of exogenous androgen and an androgen receptor antagonist in the peri- and postpubertal murine mammary gland. Endocrinology 2011, 152, 3728–3737. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Ng, S.; Adesanya-Famuiya, O.; Anderson, K.; Bondy, C.A. Testosterone inhibits estrogen-induced mammary epithelial proliferation and suppresses estrogen receptor expression. FASEB J. 2000, 14, 1725–1730. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.R.; Walters, K.A.; Desai, R.; Zhou, H.; Handelsman, D.J.; Simanainen, U. Androgen receptor inactivation resulted in acceleration in pubertal mammary gland growth, upregulation of eralpha expression, and wnt/β-catenin signaling in female mice. Endocrinology 2014, 155, 4951–4963. [Google Scholar] [CrossRef] [PubMed]
- Peters, A.A.; Buchanan, G.; Ricciardelli, C.; Bianco-Miotto, T.; Centenera, M.M.; Harris, J.M.; Jindal, S.; Segara, D.; Jia, L.; Moore, N.L.; et al. Androgen receptor inhibits estrogen receptor-alpha activity and is prognostic in breast cancer. Cancer Res. 2009, 69, 6131–6140. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Koo, J.; Park, H.S.; Kim, J.H.; Choi, S.Y.; Lee, J.H.; Park, B.W.; Lee, K.S. Expression of androgen receptors in primary breast cancer. Ann. Oncol. 2010, 21, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Dorgan, J.F.; Stanczyk, F.Z.; Kahle, L.L.; Brinton, L.A. Prospective case-control study of premenopausal serum estradiol and testosterone levels and breast cancer risk. Breast Cancer Res. 2010. [Google Scholar] [CrossRef] [PubMed]
- Zeleniuch-Jacquotte, A.; Shore, R.E.; Koenig, K.L.; Akhmedkhanov, A.; Afanasyeva, Y.; Kato, I.; Kim, M.Y.; Rinaldi, S.; Kaaks, R.; Toniolo, P. Postmenopausal levels of oestrogen, androgen, and SHBG and breast cancer: Long-term results of a prospective study. Br. J. Cancer 2004, 90, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, L.O.; Corte, M.D.; Vazquez, J.; Junquera, S.; Sanchez, R.; Alvarez, A.C.; Rodriguez, J.C.; Lamelas, M.L.; Vizoso, F.J. Androgen receptor expresion in breast cancer: Relationship with clinicopathological characteristics of the tumors, prognosis, and expression of metalloproteases and their inhibitors. BMC Cancer 2008. [Google Scholar] [CrossRef] [PubMed]
- Vera-Badillo, F.E.; Templeton, A.J.; de Gouveia, P.; Diaz-Padilla, I.; Bedard, P.L.; Al-Mubarak, M.; Seruga, B.; Tannock, I.F.; Ocana, A.; Amir, E. Androgen receptor expression and outcomes in early breast cancer: A systematic review and meta-analysis. J. Natl. Cancer Inst. 2014. [Google Scholar] [CrossRef] [PubMed]
- Proverbs-Singh, T.; Feldman, J.L.; Morris, M.J.; Autio, K.A.; Traina, T.A. Targeting the androgen receptor in prostate and breast cancer: Several new agents in development. Endocr. Relat. Cancer 2015, 22, R87–R106. [Google Scholar] [CrossRef] [PubMed]
- Safarpour, D.; Pakneshan, S.; Tavassoli, F.A. Androgen receptor (ar) expression in 400 breast carcinomas: Is routine ar assessment justified? Am. J. Cancer Res. 2014, 4, 353–368. [Google Scholar] [PubMed]
- Micello, D.; Marando, A.; Sahnane, N.; Riva, C.; Capella, C.; Sessa, F. Androgen receptor is frequently expressed in HER2-positive, ER/PR-negative breast cancers. Virchows Arch. 2010, 457, 467–476. [Google Scholar] [CrossRef] [PubMed]
- McNamara, K.M.; Moore, N.L.; Hickey, T.E.; Sasano, H.; Tilley, W.D. Complexities of androgen receptor signalling in breast cancer. Endocr. Relat. Cancer 2014, 21, T161–T181. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, B.J. Fluoxymesterone therapy in advanced breast cancer. N. Engl. J. Med. 1958, 259, 673–675. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, R.; Ahn, S.; Cheney, M.D.; Yepuru, M.; Miller, D.D.; Steiner, M.S.; Dalton, J.T. Selective androgen receptor modulators (SARMS) negatively regulate triple-negative breast cancer growth and epithelial:Mesenchymal stem cell signaling. PLoS ONE 2014, 9, e103202. [Google Scholar] [CrossRef] [PubMed]
- Santagata, S.; Thakkar, A.; Ergonul, A.; Wang, B.; Woo, T.; Hu, R.; Harrell, J.C.; McNamara, G.; Schwede, M.; Culhane, A.C.; et al. Taxonomy of breast cancer based on normal cell phenotype predicts outcome. J. Clin. Invest. 2014, 124, 859–870. [Google Scholar] [CrossRef] [PubMed]
- Peters, K.M.; Edwards, S.L.; Nair, S.S.; French, J.D.; Bailey, P.J.; Salkield, K.; Stein, S.; Wagner, S.; Francis, G.D.; Clark, S.J.; et al. Androgen receptor expression predicts breast cancer survival: The role of genetic and epigenetic events. BMC Cancer 2012. [Google Scholar] [CrossRef] [PubMed]
- Chia, K.; O'Brien, M.; Brown, M.; Lim, E. Targeting the androgen receptor in breast cancer. Curr. Oncol. Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Li, L.; Zhang, N.; Liu, J.; Zhang, L.; Gao, H.; Wang, G.; Li, Y.; Zhang, Y.; Li, X.; et al. Androgen and AR contribute to breast cancer development and metastasis: An insight of mechanisms. Oncogene 2016. [Google Scholar] [CrossRef]
- Gucalp, A.; Tolaney, S.; Isakoff, S.J.; Ingle, J.N.; Liu, M.C.; Carey, L.A.; Blackwell, K.; Rugo, H.; Nabell, L.; Forero, A.; et al. Phase II trial of bicalutamide in patients with androgen receptor-positive, estrogen receptor-negative metastatic breast cancer. Clin. Cancer Res. 2013, 19, 5505–5512. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Xu, S.; Zhang, Q.; Zhao, W. The expression and clinical significance of the androgen receptor and E-cadherin in triple-negative breast cancer. Med. Oncol. 2012, 29, 526–533. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Hai, E.; Matsumoto, K.; Ikeda, K.; Tokunaga, S.; Nagahara, H.; Sakurai, K.; Inoue, T.; Nishiguchi, Y. Androgen receptor expression in breast cancer: Relationship with clinicopathological factors and biomarkers. Int. J. Clin. Oncol. 2008, 13, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.E.; Kang, S.H.; Lee, S.J.; Bae, Y.K. Androgen receptor expression predicts decreased survival in early stage triple-negative breast cancer. Ann. Surg. Oncol. 2015, 22, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Cochrane, D.R.; Bernales, S.; Jacobsen, B.M.; Cittelly, D.M.; Howe, E.N.; D'Amato, N.C.; Spoelstra, N.S.; Edgerton, S.M.; Jean, A.; Guerrero, J.; et al. Role of the androgen receptor in breast cancer and preclinical analysis of enzalutamide. Breast Cancer Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Niemeier, L.A.; Dabbs, D.J.; Beriwal, S.; Striebel, J.M.; Bhargava, R. Androgen receptor in breast cancer: Expression in estrogen receptor-positive tumors and in estrogen receptor-negative tumors with apocrine differentiation. Mod. Pathol. 2010, 23, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.P.; Yang, Y.L.; Zhu, H.; Wang, J.; Jia, Y.; Liu, N.; Song, Y.J.; Zan, L.K.; Zhang, X.; Zhou, M.; et al. Expression of the androgen receptor and its correlation with molecular subtypes in 980 chinese breast cancer patients. Breast Cancer 2012, 6, 1–8. [Google Scholar] [PubMed]
- Ni, M.; Chen, Y.; Lim, E.; Wimberly, H.; Bailey, S.T.; Imai, Y.; Rimm, D.L.; Liu, X.S.; Brown, M. Targeting androgen receptor in estrogen receptor-negative breast cancer. Cancer Cell 2011, 20, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed]
- Higa, G.M.; Fell, R.G. Sex hormone receptor repertoire in breast cancer. Int. J. Breast Cancer 2013. [Google Scholar] [CrossRef] [PubMed]
- Leung, B.S.; Fletcher, W.S.; Lindell, T.D.; Wood, D.C.; Krippaechne, W.W. Predictability of response to endocrine ablation in advanced breast carcinoma. A correlation to estrogen receptor and steroid sulfurylation. Arch. Surg. 1973, 106, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, B.J. Systemic effects of androgenic and estrogenic hormones in advanced breast cancer. J. Am. Geriatr. Soc. 1965, 13, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Pietri, E.; Conteduca, V.; Andreis, D.; Massa, I.; Melegari, E.; Sarti, S.; Cecconetto, L.; Schirone, A.; Bravaccini, S.; Serra, P.; et al. Androgen receptor signaling pathways as a target for breast cancer treatment. Endocr. Relat. Cancer 2016, 23, R485–R498. [Google Scholar] [CrossRef] [PubMed]
- Jordan, V.C.; Robinson, S.P. Species-specific pharmacology of antiestrogens: Role of metabolism. Fed. Proc. 1987, 46, 1870–1874. [Google Scholar] [PubMed]
- Boni, C.; Pagano, M.; Panebianco, M.; Bologna, A.; Sierra, N.M.; Gnoni, R.; Formisano, D.; Bisagni, G. Therapeutic activity of testoterone in metastatic breast cancer. Anticancer Res. 2014, 34, 1287–1290. [Google Scholar] [PubMed]
- Ingle, J.N.; Twito, D.I.; Schaid, D.J.; Cullinan, S.A.; Krook, J.E.; Mailliard, J.A.; Tschetter, L.K.; Long, H.J.; Gerstner, J.G.; Windschitl, H.E.; et al. Combination hormonal therapy with tamoxifen plus fluoxymesterone versus tamoxifen alone in postmenopausal women with metastatic breast cancer. An updated analysis. Cancer 1991, 67, 886–891. [Google Scholar] [CrossRef]
- Perrault, D.J.; Logan, D.M.; Stewart, D.J.; Bramwell, V.H.; Paterson, A.H.; Eisenhauer, E.A. Phase ii study of flutamide in patients with metastatic breast cancer. A National Cancer Institute of Canada Clinical Trials Group Study. Invest. New Drugs 1988, 6, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Anestis, A.; Karamouzis, M.V.; Dalagiorgou, G.; Papavassiliou, A.G. Is androgen receptor targeting an emerging treatment strategy for triple negative breast cancer? Cancer Treat. Rev. 2015, 41, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Collins, L.C.; Cole, K.S.; Marotti, J.D.; Hu, R.; Schnitt, S.J.; Tamimi, R.M. Androgen receptor expression in breast cancer in relation to molecular phenotype: Results from the nurses’ health study. Mod. Pathol. 2011, 24, 924–931. [Google Scholar] [CrossRef] [PubMed]
- Fioretti, F.M.; Sita-Lumsden, A.; Bevan, C.L.; Brooke, G.N. Revising the role of the androgen receptor in breast cancer. J. Mol. Endocrinol. 2014, 52, R257–R265. [Google Scholar] [CrossRef] [PubMed]
- Lanzino, M.; De Amicis, F.; McPhaul, M.J.; Marsico, S.; Panno, M.L.; Ando, S. Endogenous coactivator ARA70 interacts with estrogen receptor alpha (eralpha) and modulates the functional eralpha/androgen receptor interplay in mcf-7 cells. J. Biol. Chem. 2005, 280, 20421–20430. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Dawood, S.; Holmes, M.D.; Collins, L.C.; Schnitt, S.J.; Cole, K.; Marotti, J.D.; Hankinson, S.E.; Colditz, G.A.; Tamimi, R.M. Androgen receptor expression and breast cancer survival in postmenopausal women. Clin. Cancer Res. 2011, 17, 1867–1874. [Google Scholar] [CrossRef] [PubMed]
- Castellano, I.; Allia, E.; Accortanzo, V.; Vandone, A.M.; Chiusa, L.; Arisio, R.; Durando, A.; Donadio, M.; Bussolati, G.; Coates, A.S.; et al. Androgen receptor expression is a significant prognostic factor in estrogen receptor positive breast cancers. Breast Cancer Res. Treat. 2010, 124, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Angulo, A.M.; Stemke-Hale, K.; Palla, S.L.; Carey, M.; Agarwal, R.; Meric-Berstam, F.; Traina, T.A.; Hudis, C.; Hortobagyi, G.N.; Gerald, W.L.; et al. Androgen receptor levels and association with pik3ca mutations and prognosis in breast cancer. Clin. Cancer Res. 2009, 15, 2472–2478. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, E.; Hisamatsu, Y.; Taketani, K.; Yamashita, N.; Akiyoshi, S.; Okada, S.; Tanaka, K.; Saeki, H.; Oki, E.; Aishima, S.; et al. Differential impact of the expression of the androgen receptor by age in estrogen receptor-positive breast cancer. Cancer Med. 2013, 2, 763–773. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Koo, J.S.; Kim, M.S.; Park, H.S.; Lee, J.S.; Lee, J.S.; Kim, S.I.; Park, B.W.; Lee, K.S. Androgen receptor expression is significantly associated with better outcomes in estrogen receptor-positive breast cancers. Ann. Oncol. 2011, 22, 1755–1762. [Google Scholar] [CrossRef] [PubMed]
- Chia, K.M.; Liu, J.; Francis, G.D.; Naderi, A. A feedback loop between androgen receptor and ERK signaling in estrogen receptor-negative breast cancer. Neoplasia 2011, 13, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Ni, M.; Chen, Y.; Fei, T.; Li, D.; Lim, E.; Liu, X.S.; Brown, M. Amplitude modulation of androgen signaling by C-Myc. Genes Dev. 2013, 27, 734–748. [Google Scholar] [CrossRef] [PubMed]
- Naderi, A.; Chia, K.M.; Liu, J. Synergy between inhibitors of androgen receptor and mek has therapeutic implications in estrogen receptor-negative breast cancer. Breast Cancer Res. 2011, 13. [Google Scholar] [CrossRef] [PubMed]
- Farmer, P.; Bonnefoi, H.; Becette, V.; Tubiana-Hulin, M.; Fumoleau, P.; Larsimont, D.; Macgrogan, G.; Bergh, J.; Cameron, D.; Goldstein, D.; et al. Identification of molecular apocrine breast tumours by microarray analysis. Oncogene 2005, 24, 4660–4671. [Google Scholar] [CrossRef] [PubMed]
- Doane, A.S.; Danso, M.; Lal, P.; Donaton, M.; Zhang, L.; Hudis, C.; Gerald, W.L. An estrogen receptor-negative breast cancer subset characterized by a hormonally regulated transcriptional program and response to androgen. Oncogene 2006, 25, 3994–4008. [Google Scholar] [CrossRef] [PubMed]
- Cuenca-Lopez, M.D.; Montero, J.C.; Morales, J.C.; Prat, A.; Pandiella, A.; Ocana, A. Phospho-kinase profile of triple negative breast cancer and androgen receptor signaling. BMC Cancer 2014. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Schafer, J.M.; Pendleton, C.S.; Tang, L.; Johnson, K.C.; Chen, X.; Balko, J.M.; Gomez, H.; Arteaga, C.L.; et al. PIK3CA mutations in androgen receptor-positive triple negative breast cancer confer sensitivity to the combination of PI3K and androgen receptor inhibitors. Breast Cancer Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Arce-Salinas, C.; Riesco-Martinez, M.C.; Hanna, W.; Bedard, P.; Warner, E. Complete response of metastatic androgen receptor-positive breast cancer to bicalutamide: Case report and review of the literature. J. Clin. Oncol. 2016, 34, e21–e24. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.K.; Lu, S.Y.; Kang, S.A.; Tan, H.J.; Li, Z.; Adrian Wee, Z.N.; Guan, J.S.; Reddy Chichili, V.P.; Sivaraman, J.; Putti, T.; et al. Wnt signaling promotes breast cancer by blocking itch-mediated degradation of yap/taz transcriptional coactivator wbp2. Cancer Res. 2016, 76, 6278–6289. [Google Scholar] [CrossRef] [PubMed]
- Turashvili, G.; Bouchal, J.; Burkadze, G.; Kolar, Z. Wnt signaling pathway in mammary gland development and carcinogenesis. Pathobiology 2006, 73, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Gavin, B.J.; McMahon, A.P. Differential regulation of the wnt gene family during pregnancy and lactation suggests a role in postnatal development of the mammary gland. Mol. Cell Biol. 1992, 12, 2418–2423. [Google Scholar] [CrossRef] [PubMed]
- Weber-Hall, S.J.; Phippard, D.J.; Niemeyer, C.C.; Dale, T.C. Developmental and hormonal regulation of wnt gene expression in the mouse mammary gland. Differentiation 1994, 57, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Huguet, E.L.; McMahon, J.A.; McMahon, A.P.; Bicknell, R.; Harris, A.L. Differential expression of human wnt genes 2, 3, 4, and 7b in human breast cell lines and normal and disease states of human breast tissue. Cancer Res. 1994, 54, 2615–2621. [Google Scholar] [PubMed]

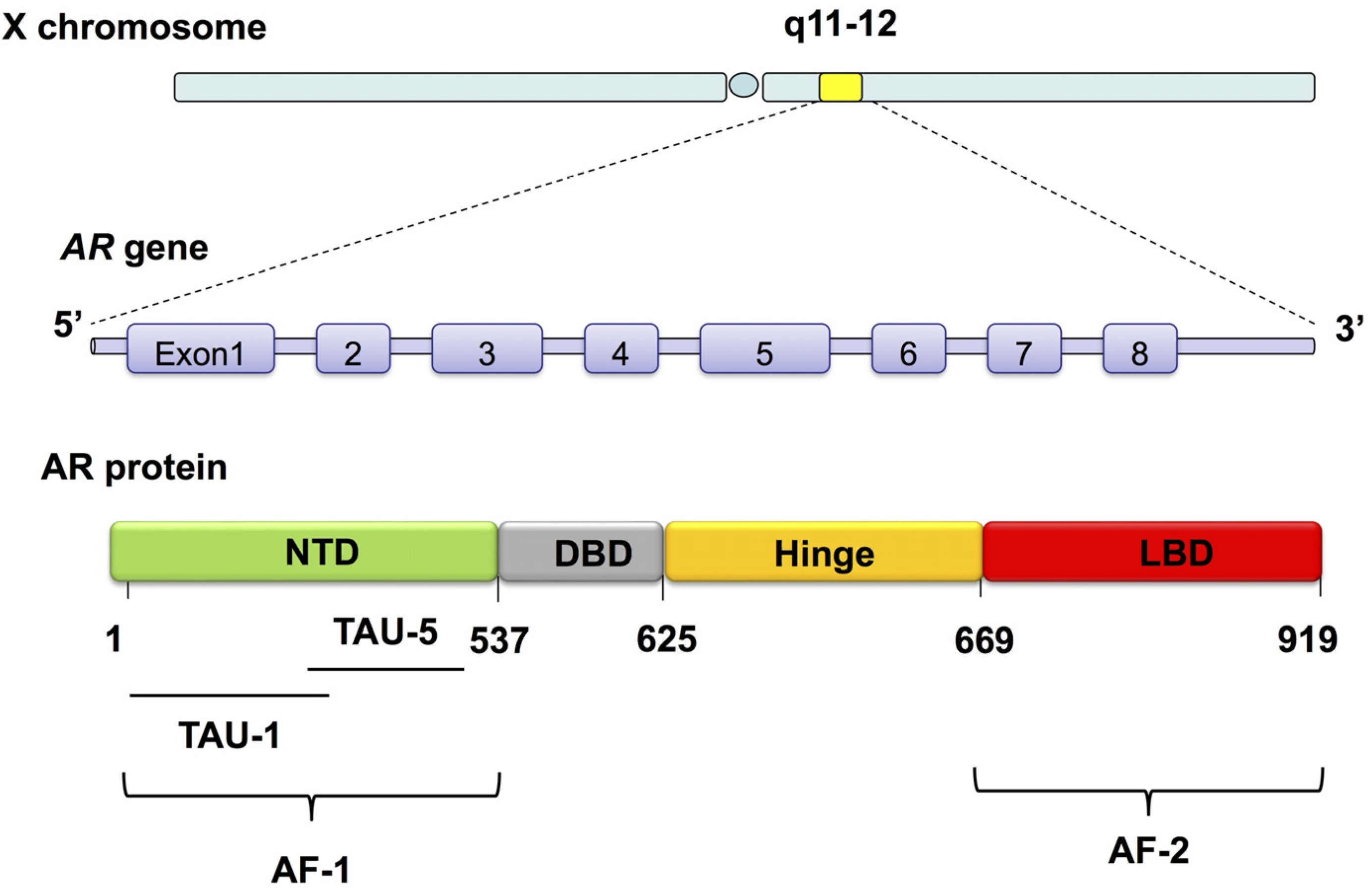

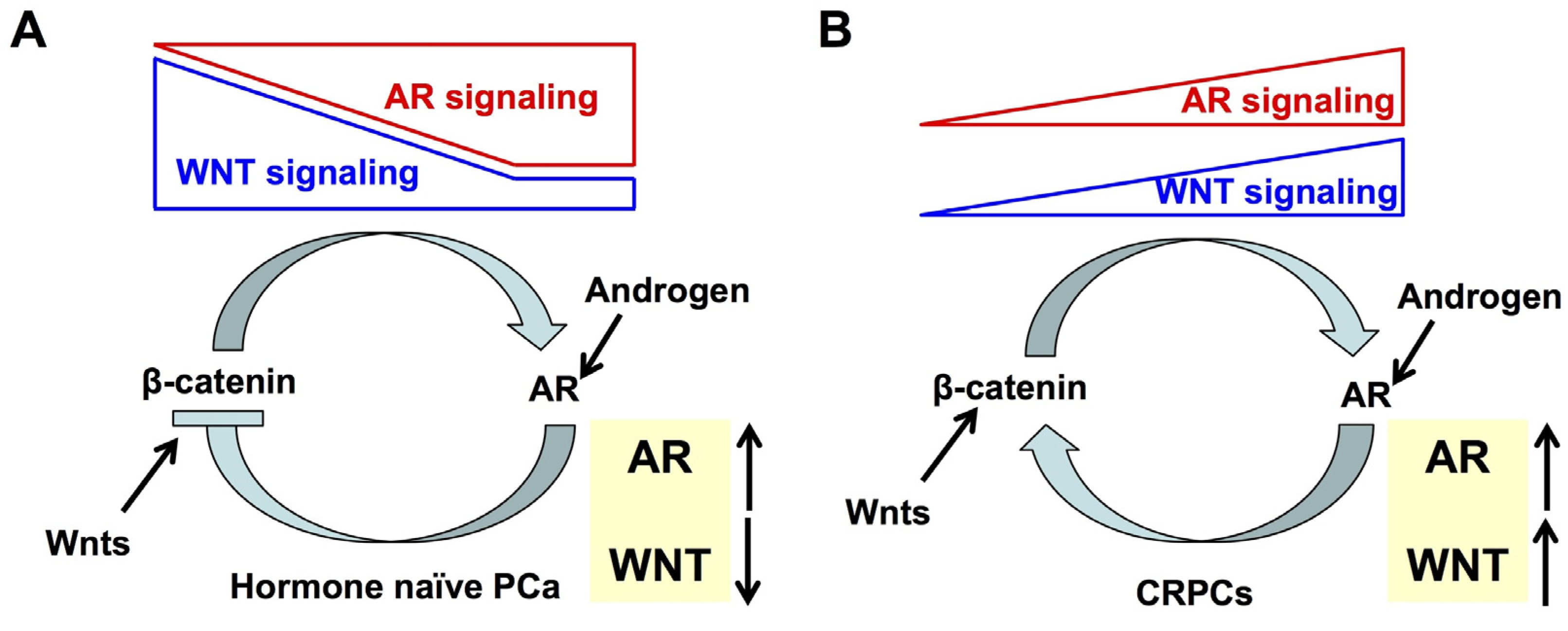
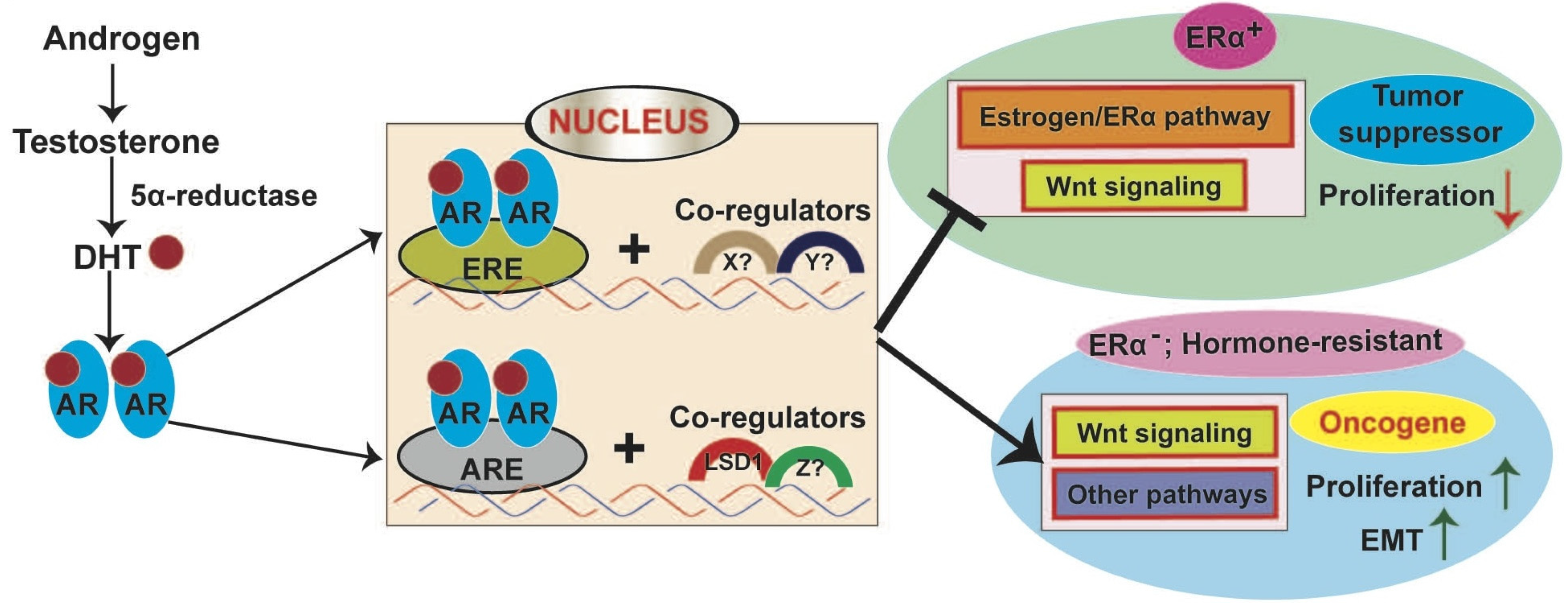
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pakula, H.; Xiang, D.; Li, Z. A Tale of Two Signals: AR and WNT in Development and Tumorigenesis of Prostate and Mammary Gland. Cancers 2017, 9, 14. https://doi.org/10.3390/cancers9020014
Pakula H, Xiang D, Li Z. A Tale of Two Signals: AR and WNT in Development and Tumorigenesis of Prostate and Mammary Gland. Cancers. 2017; 9(2):14. https://doi.org/10.3390/cancers9020014
Chicago/Turabian StylePakula, Hubert, Dongxi Xiang, and Zhe Li. 2017. "A Tale of Two Signals: AR and WNT in Development and Tumorigenesis of Prostate and Mammary Gland" Cancers 9, no. 2: 14. https://doi.org/10.3390/cancers9020014