Characterization of Catalytically Active Octahedral Metal Halide Cluster Complexes

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
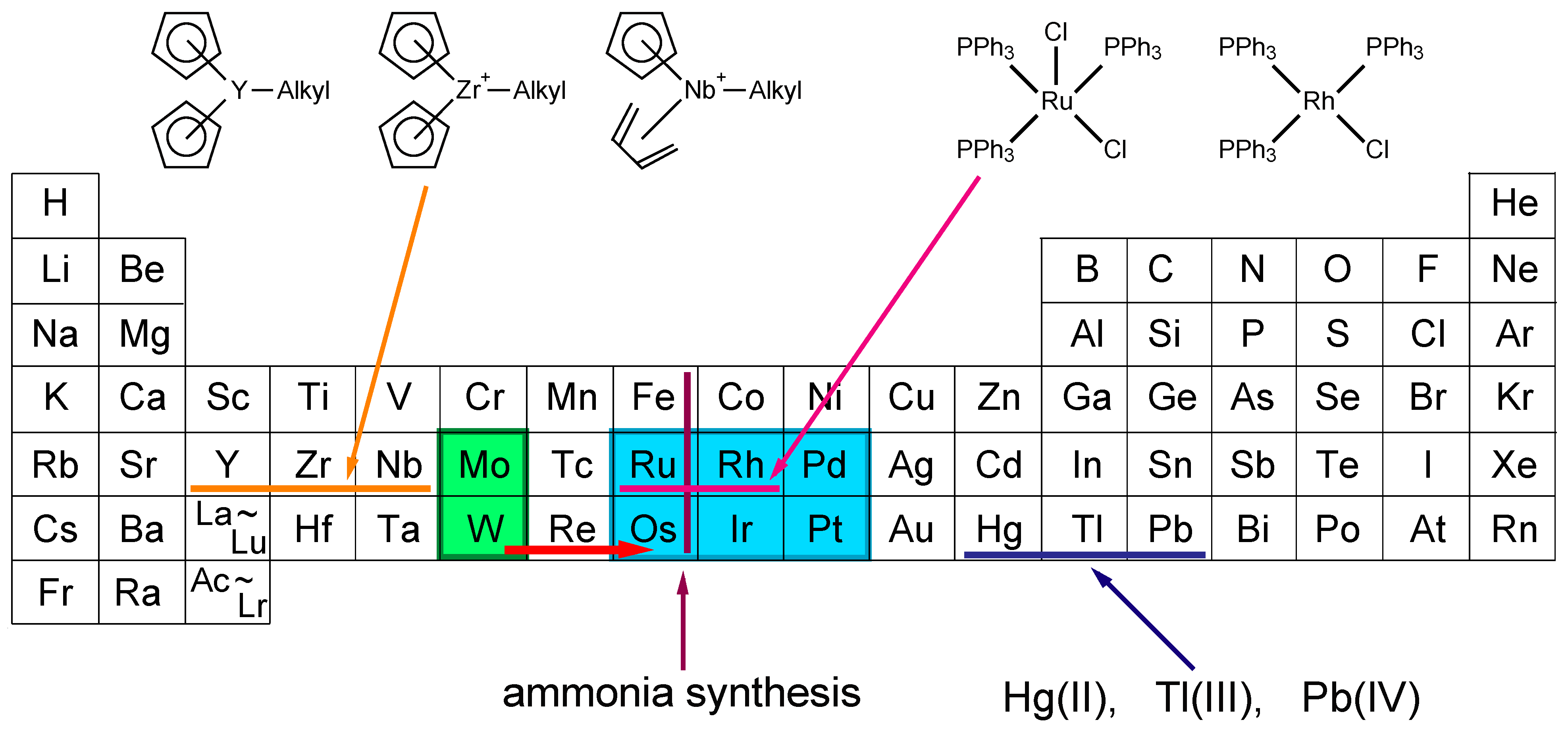
1. Introduction



2. Activation and Characterization of Cluster Complexes Containing Group 6 Metals
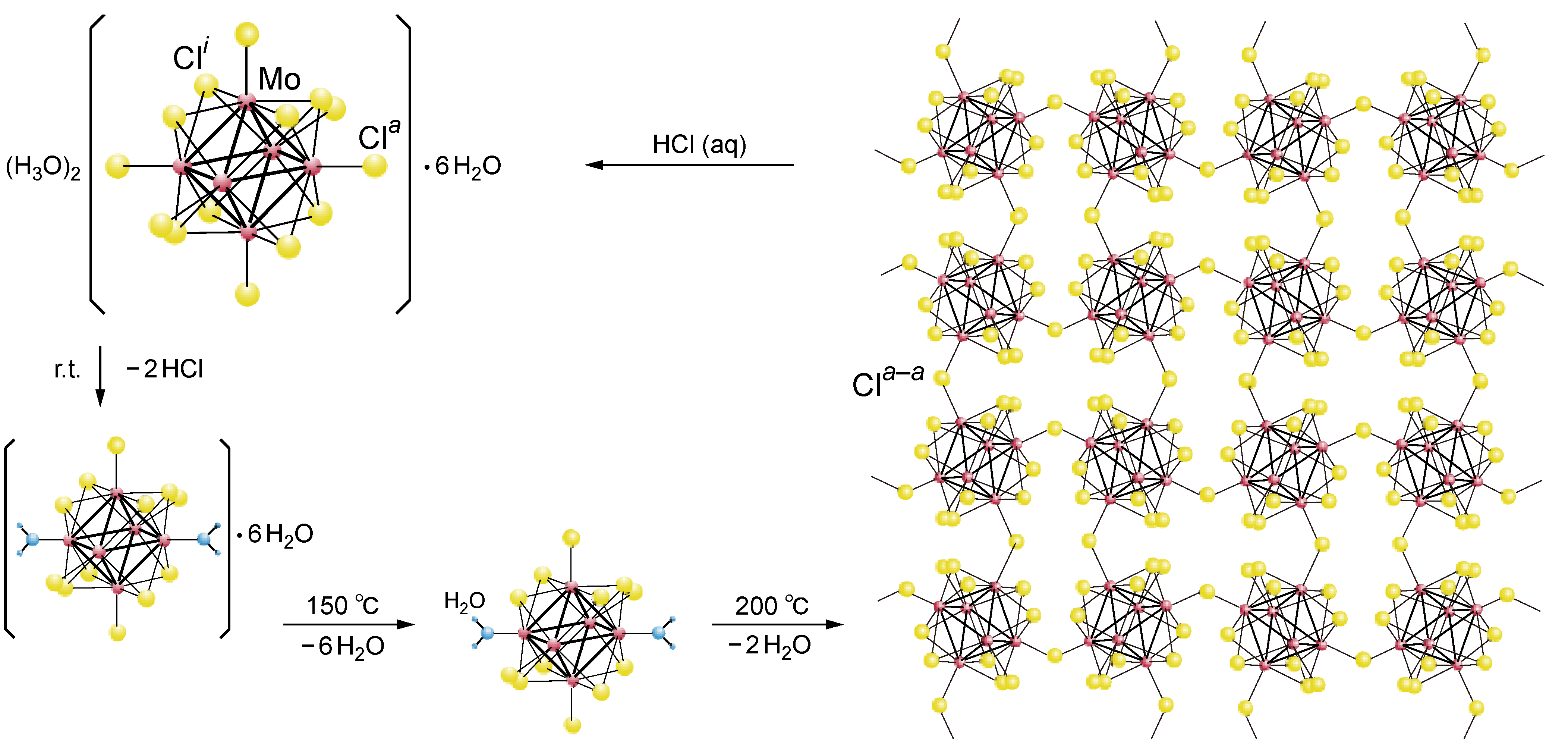
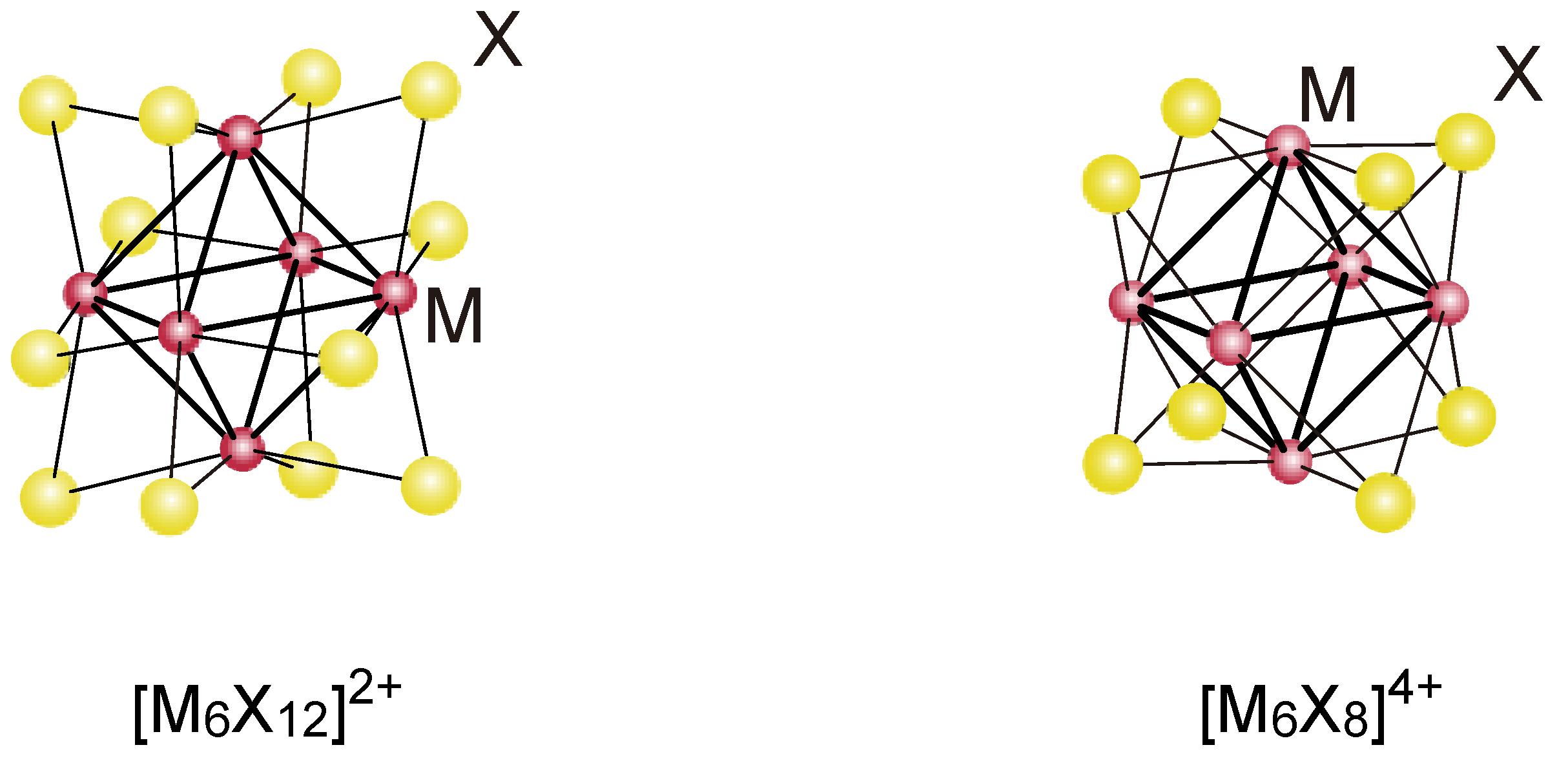
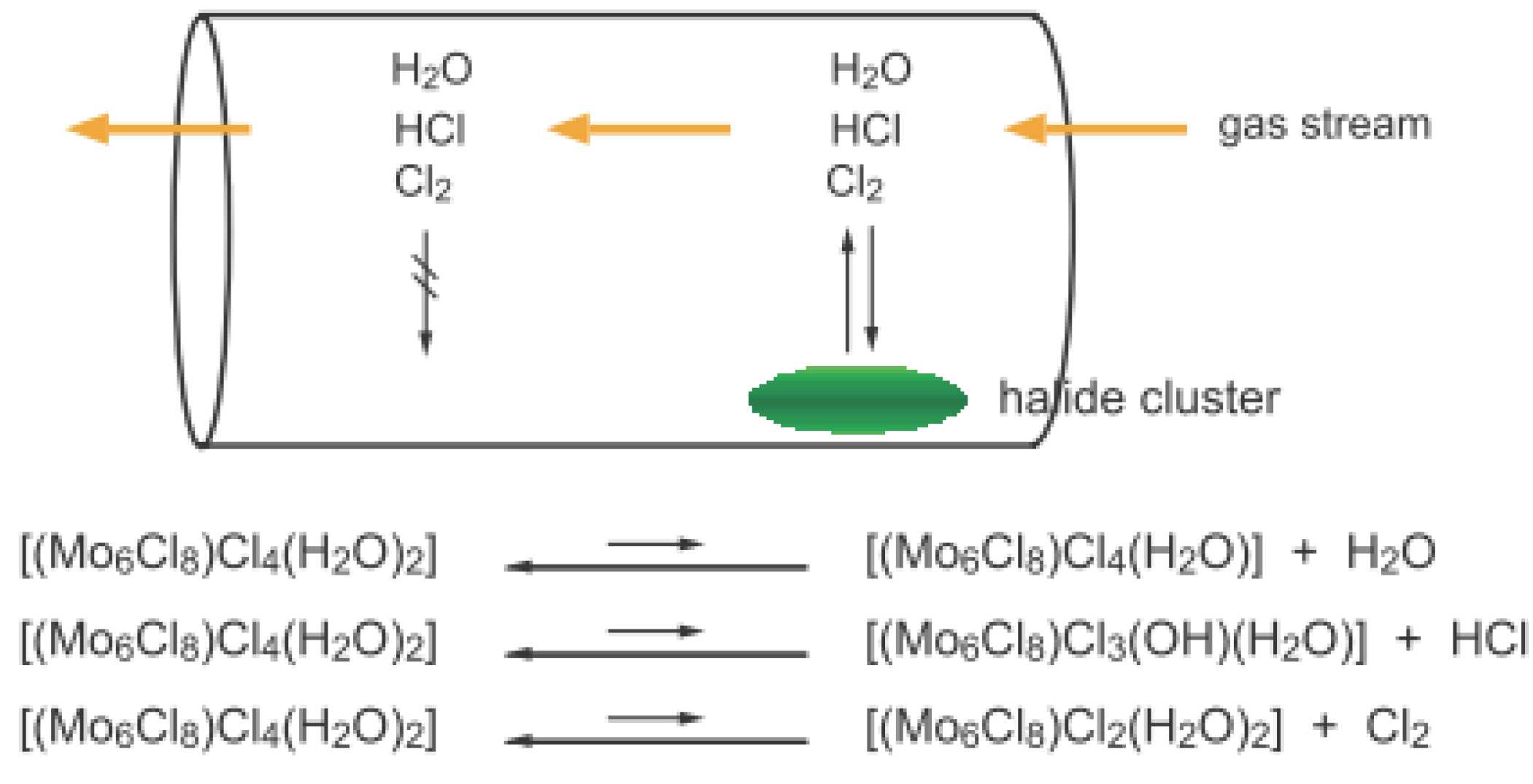
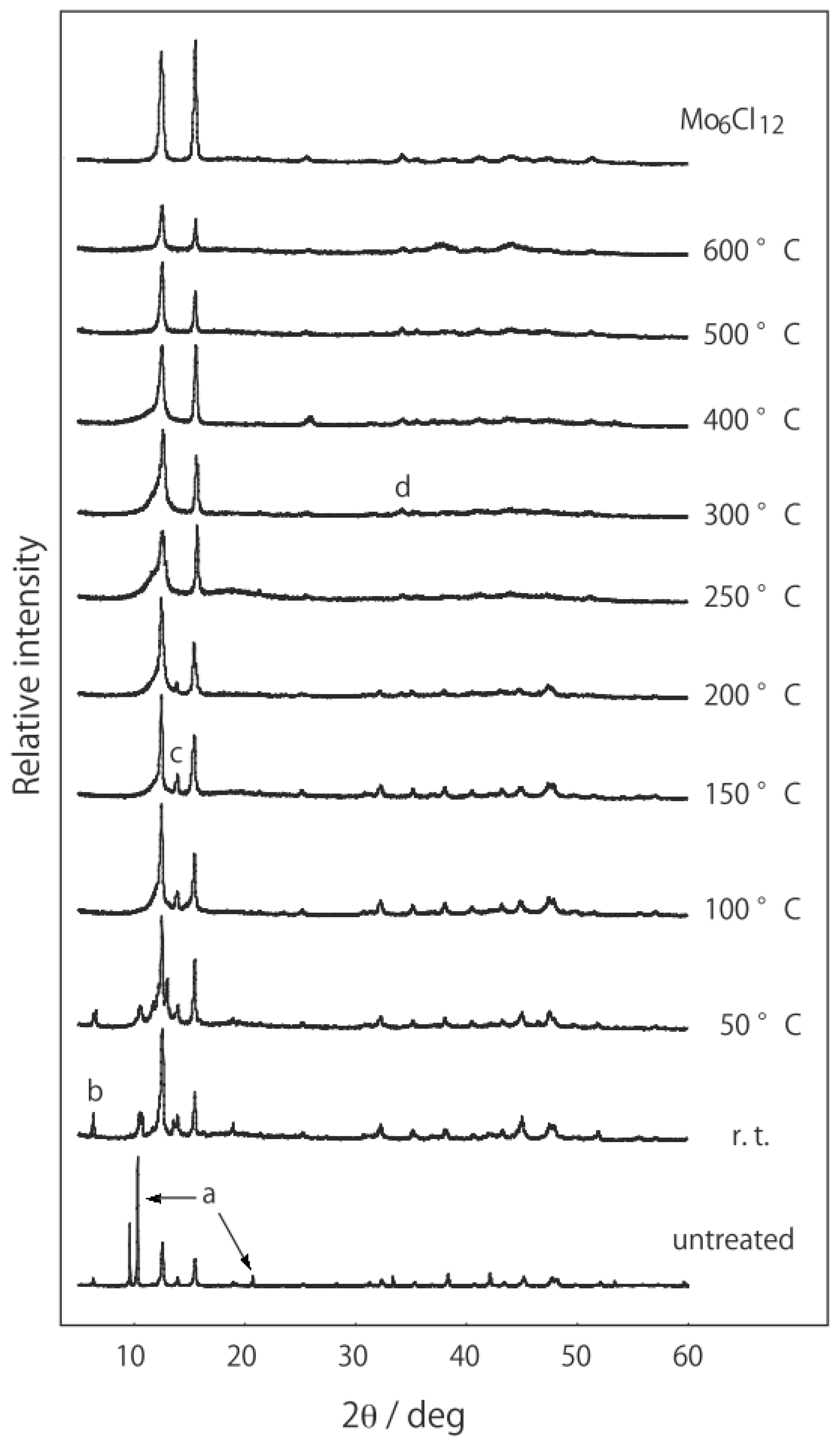
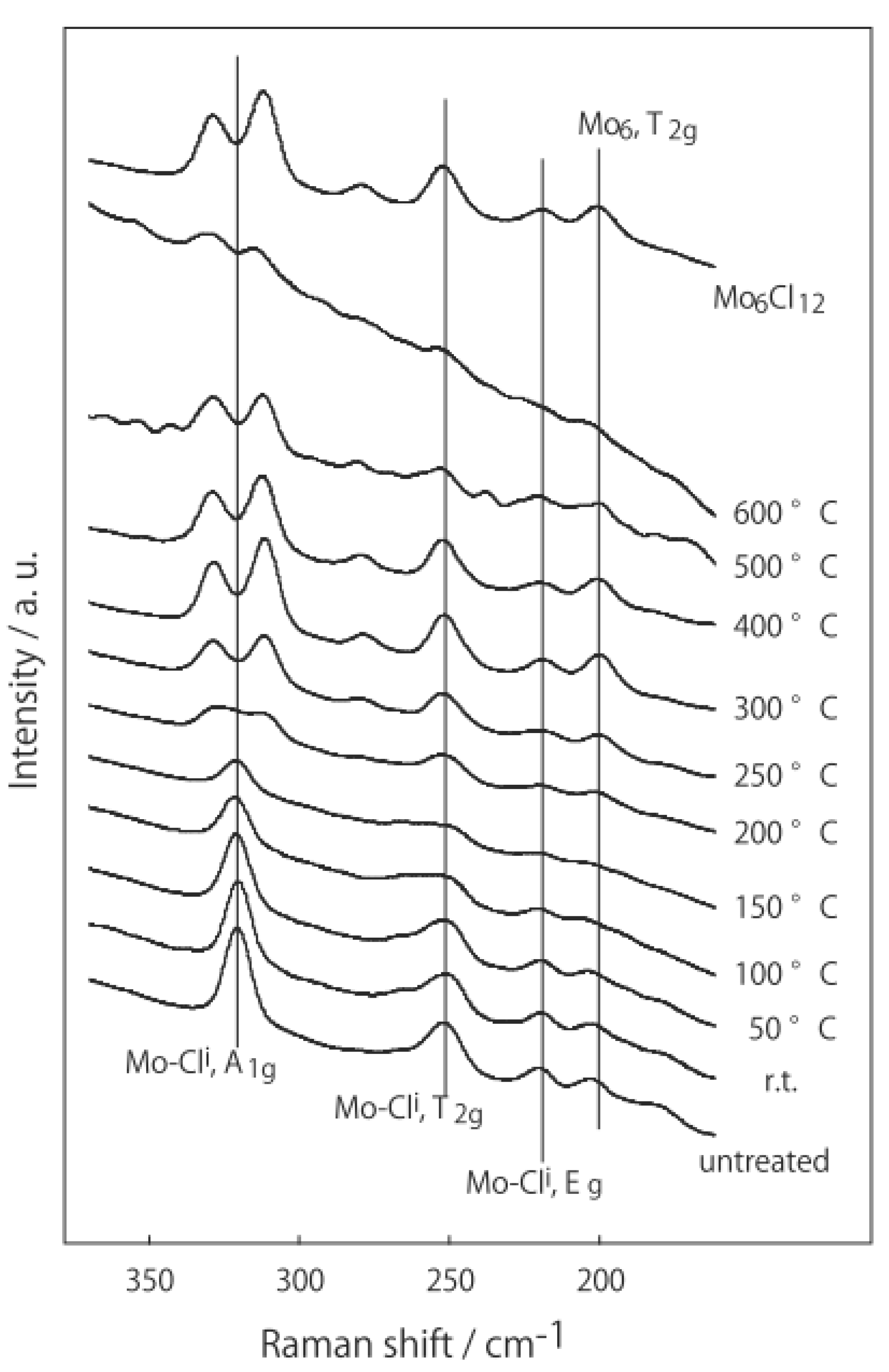
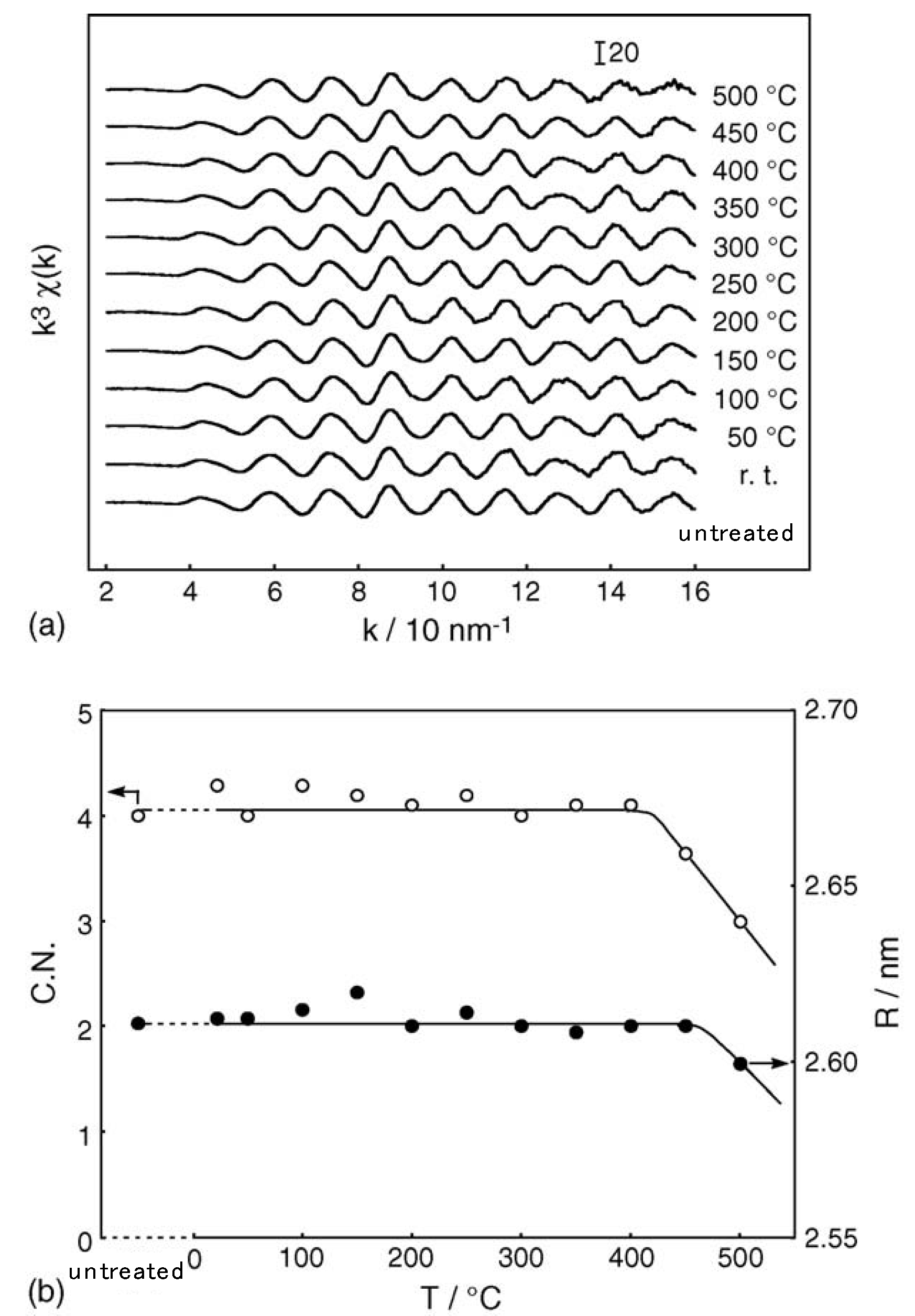
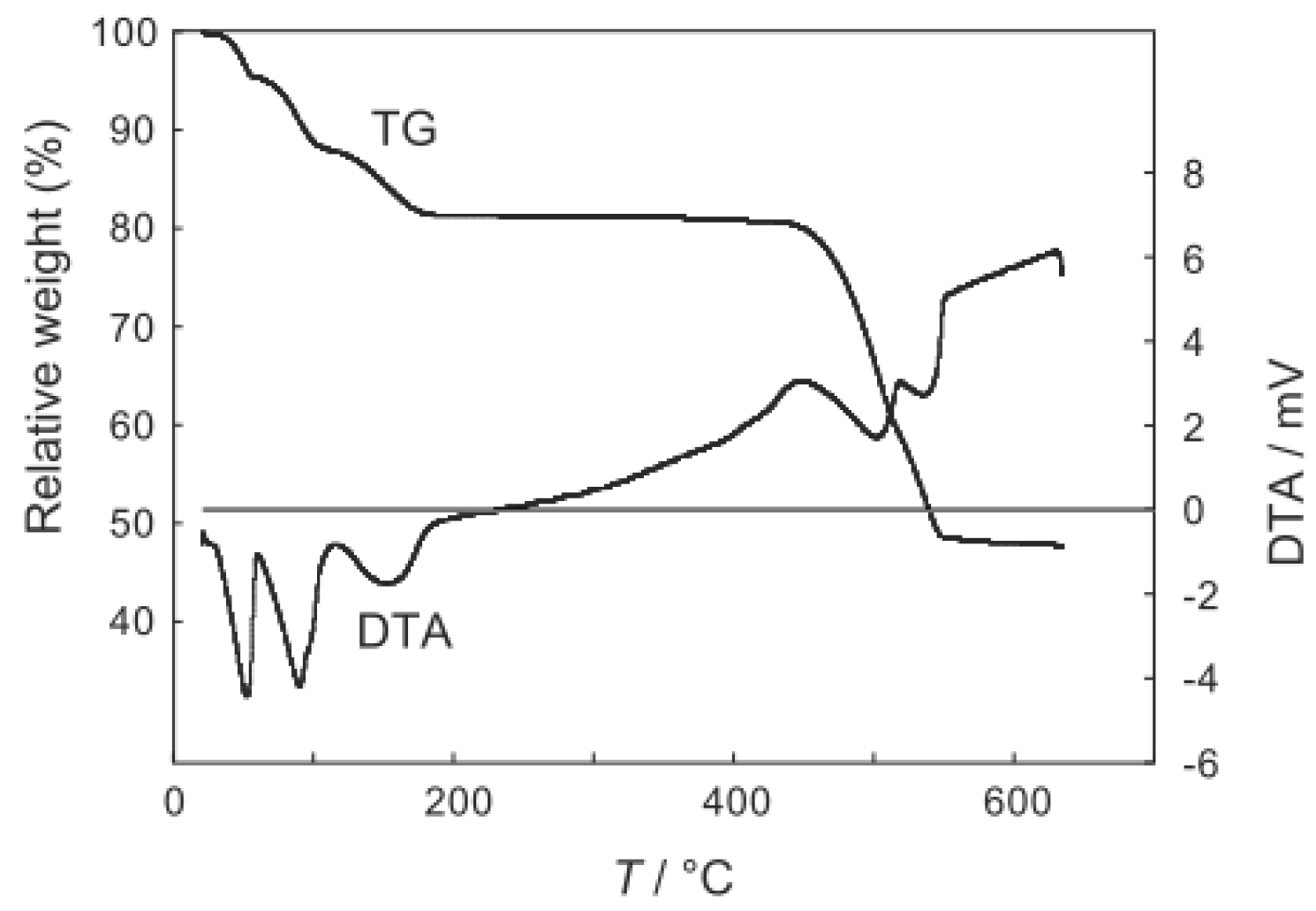
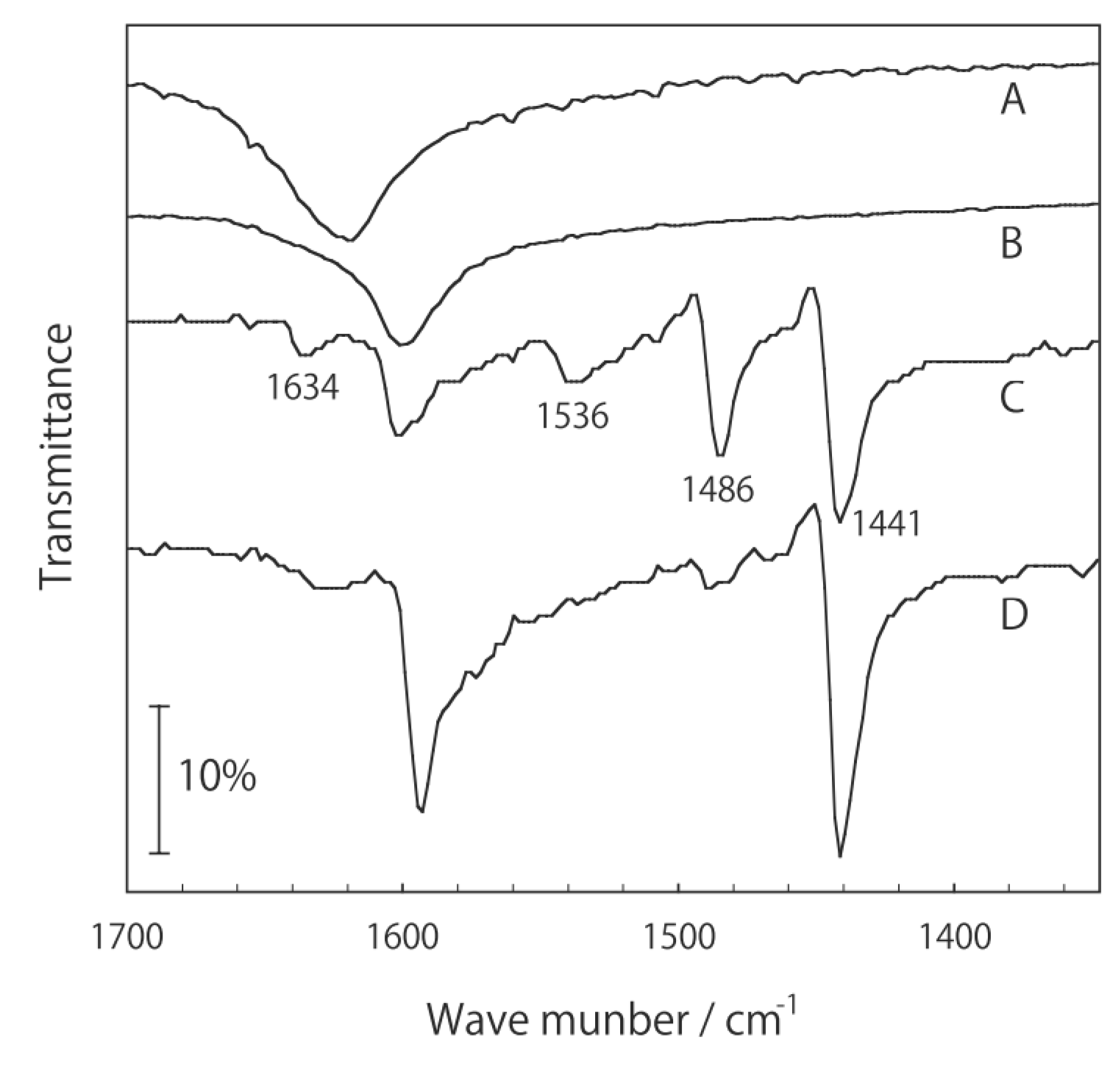
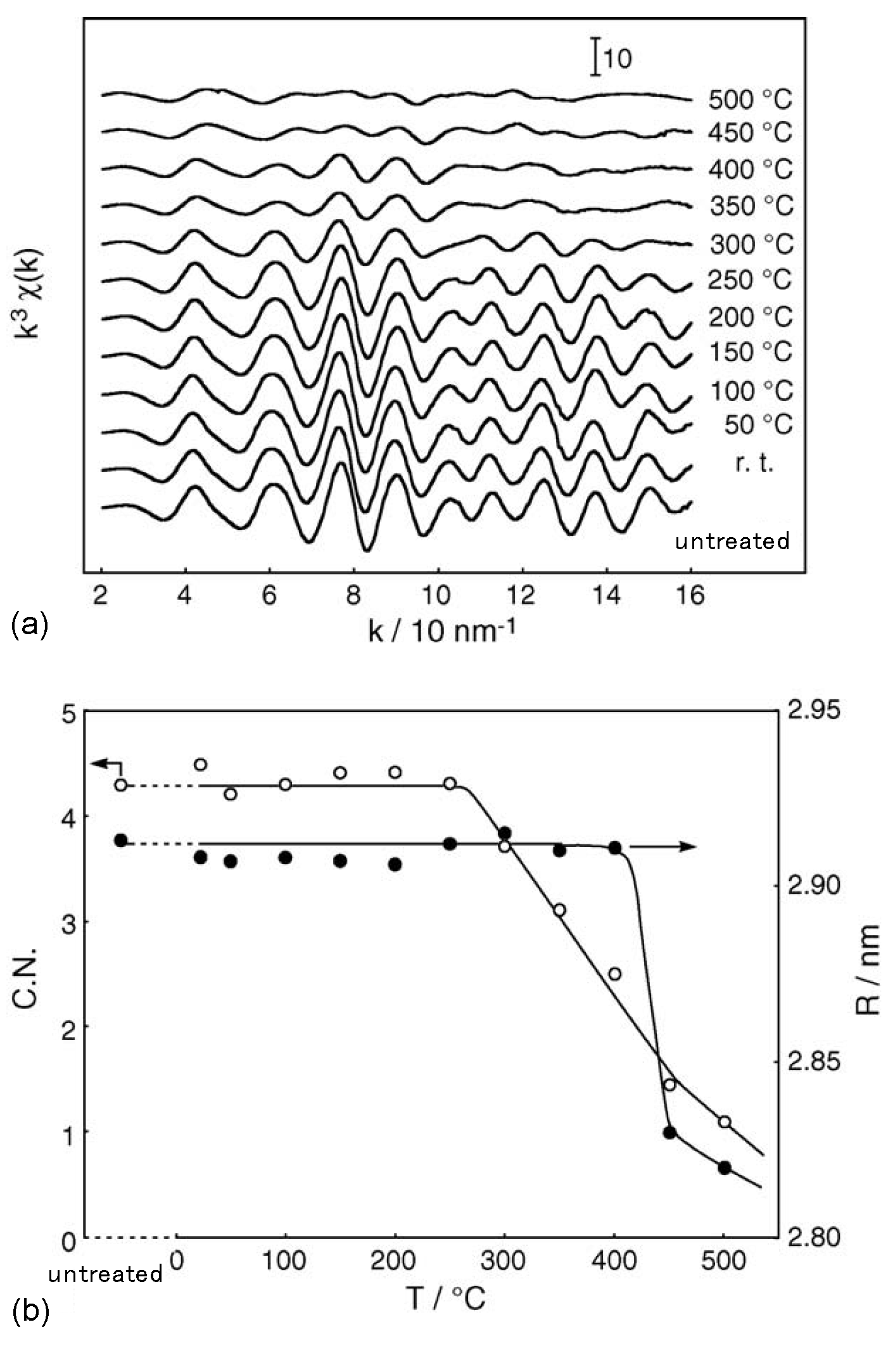
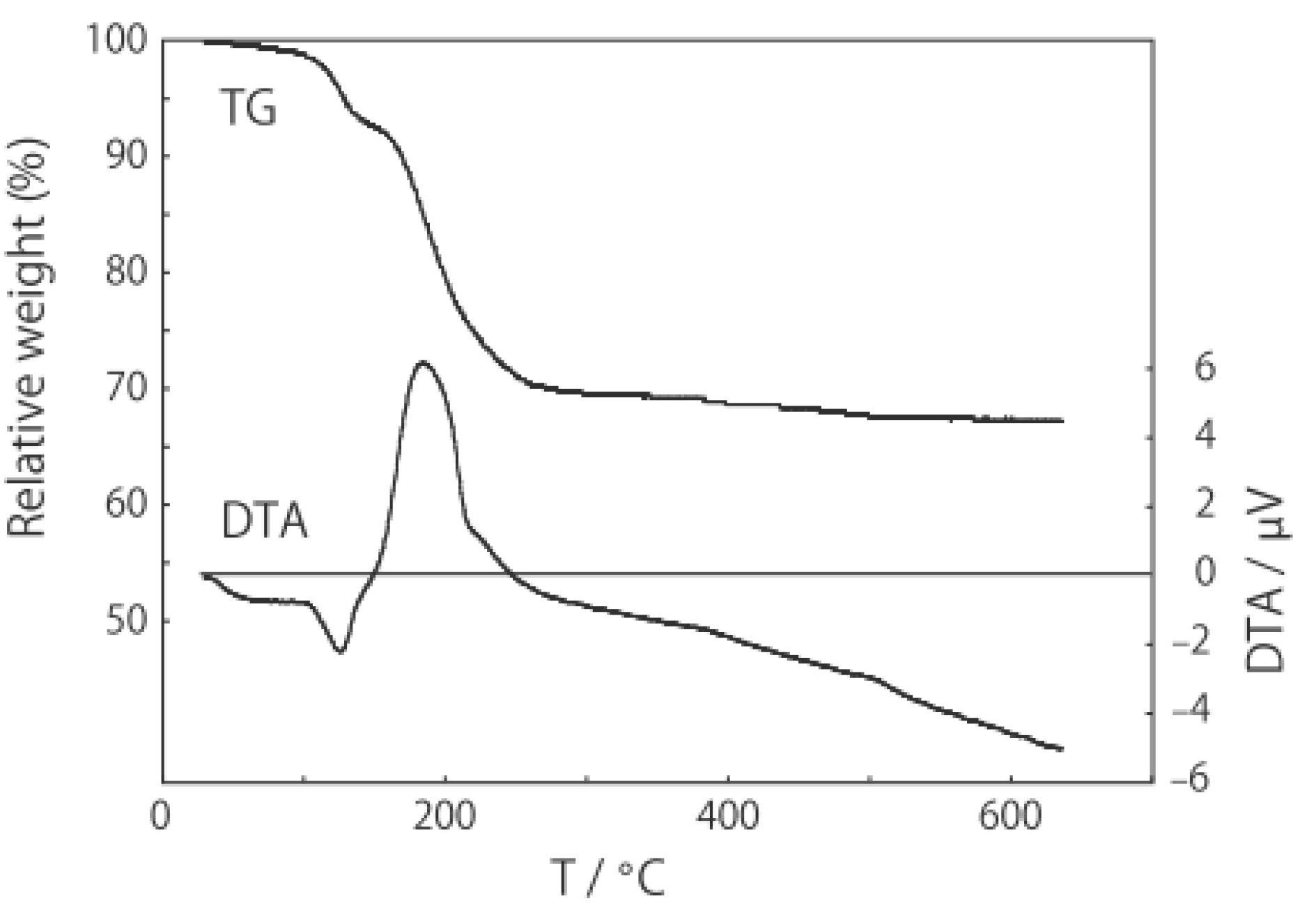
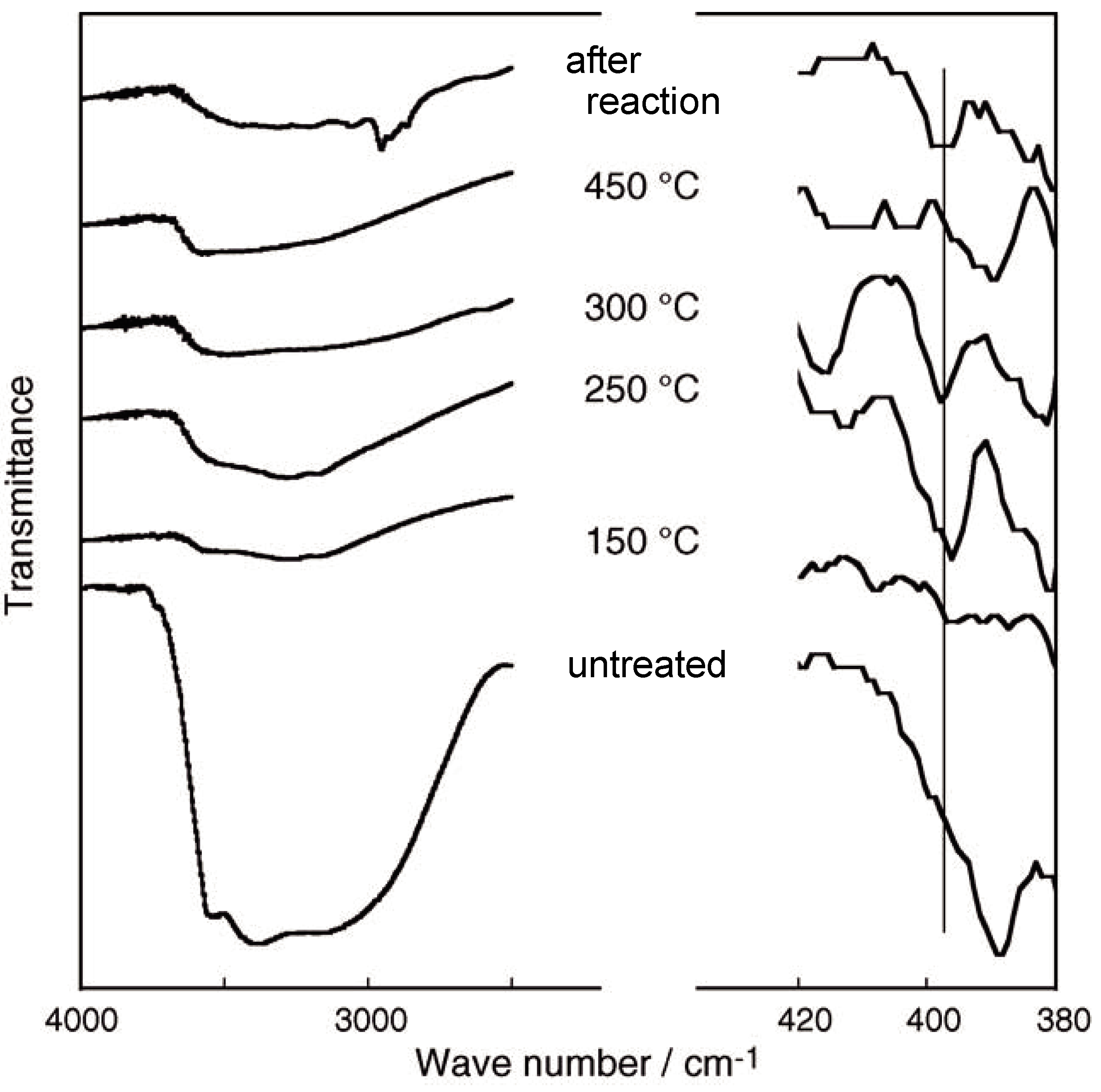
2.1. Retention of the Cluster Framework of the (H3O)2[(Mo6Cl8)Cl6]·6H2O Molybdenum Cluster [20]



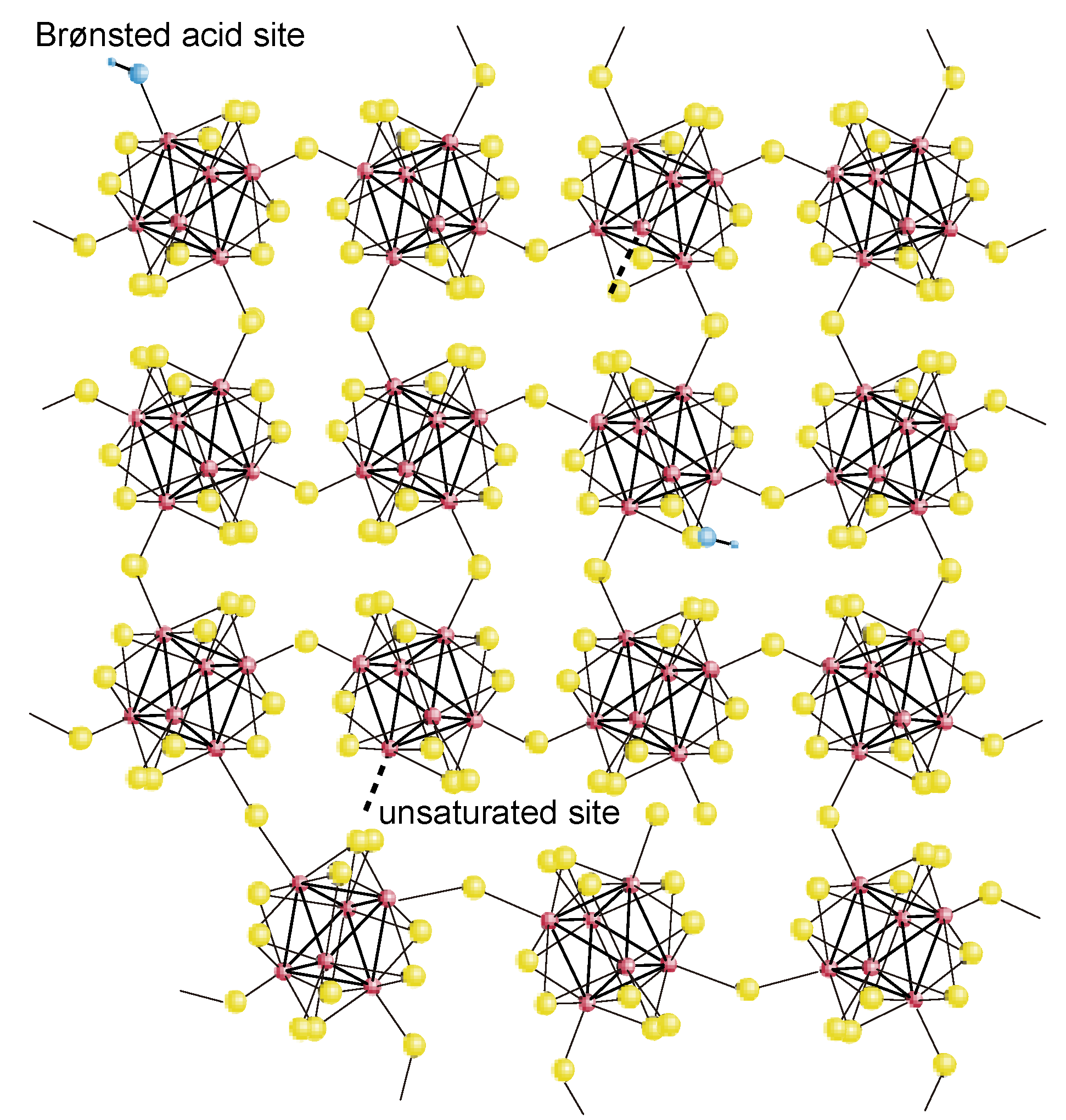
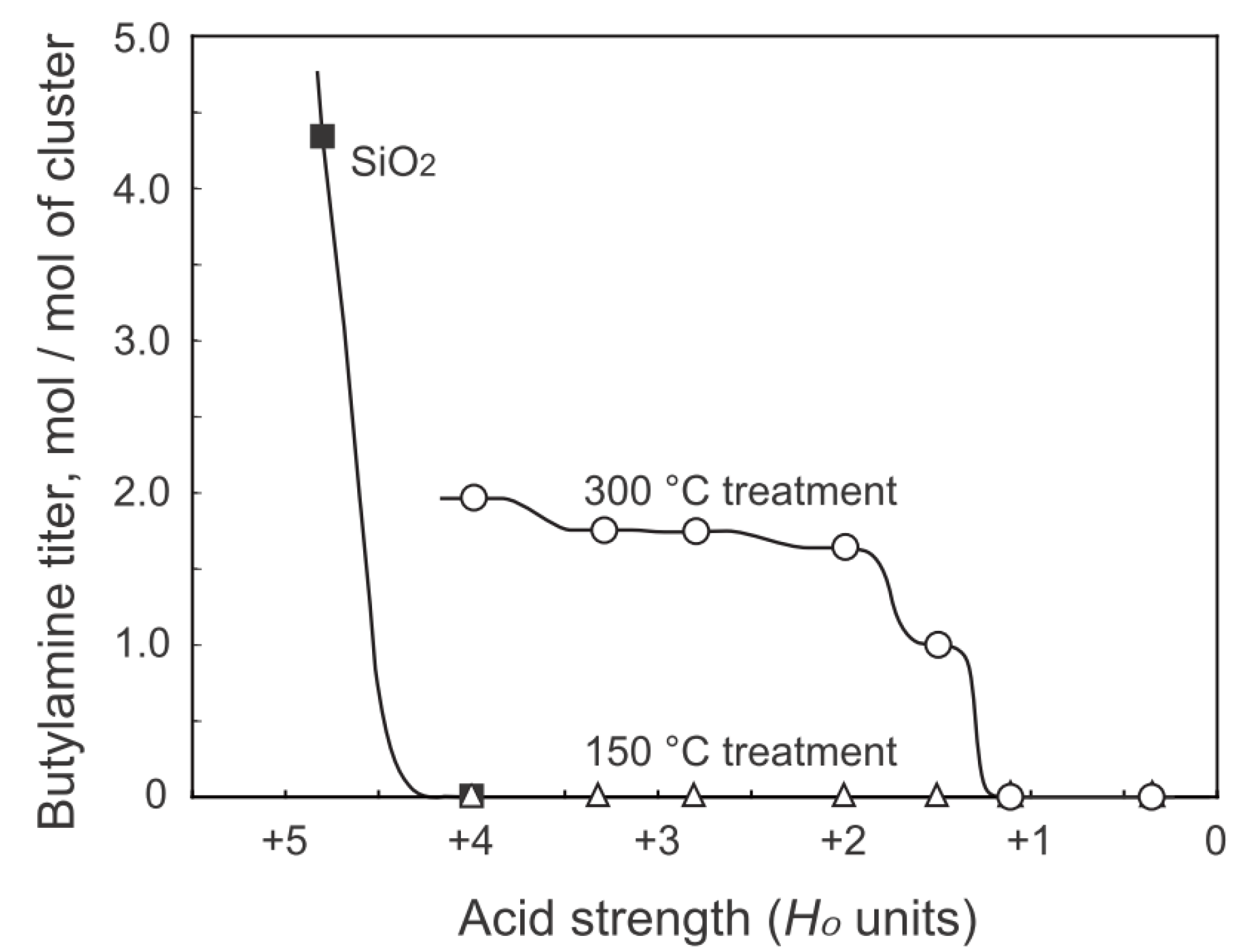
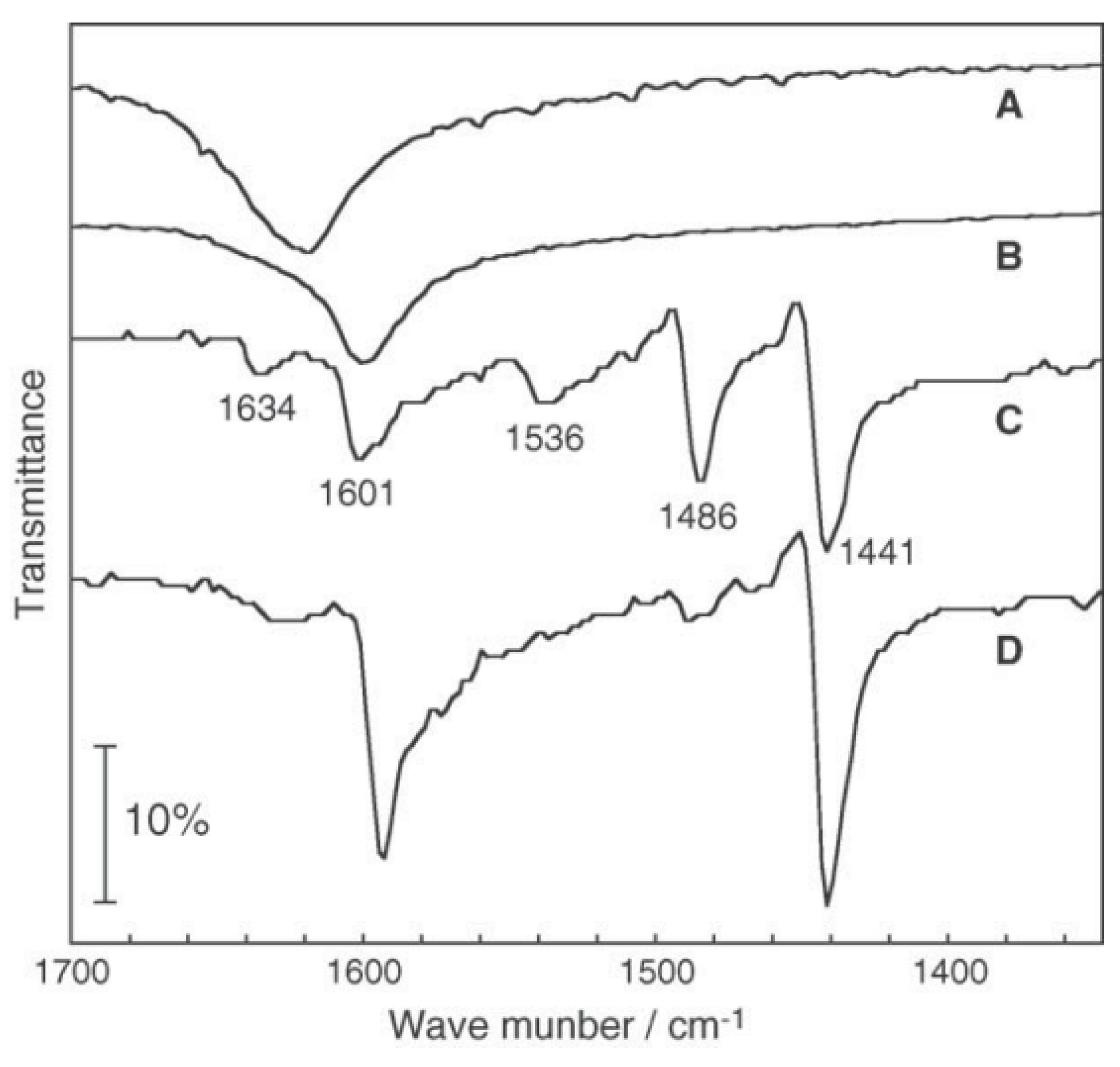
2.2. Active Site of Molybdenum Cluster




2.3. Tungsten Cluster (H3O)2[(W6Cl8)Cl6]·6H2O

3. Activation and Characterization of Cluster Complexes Containing Group 5 Metals
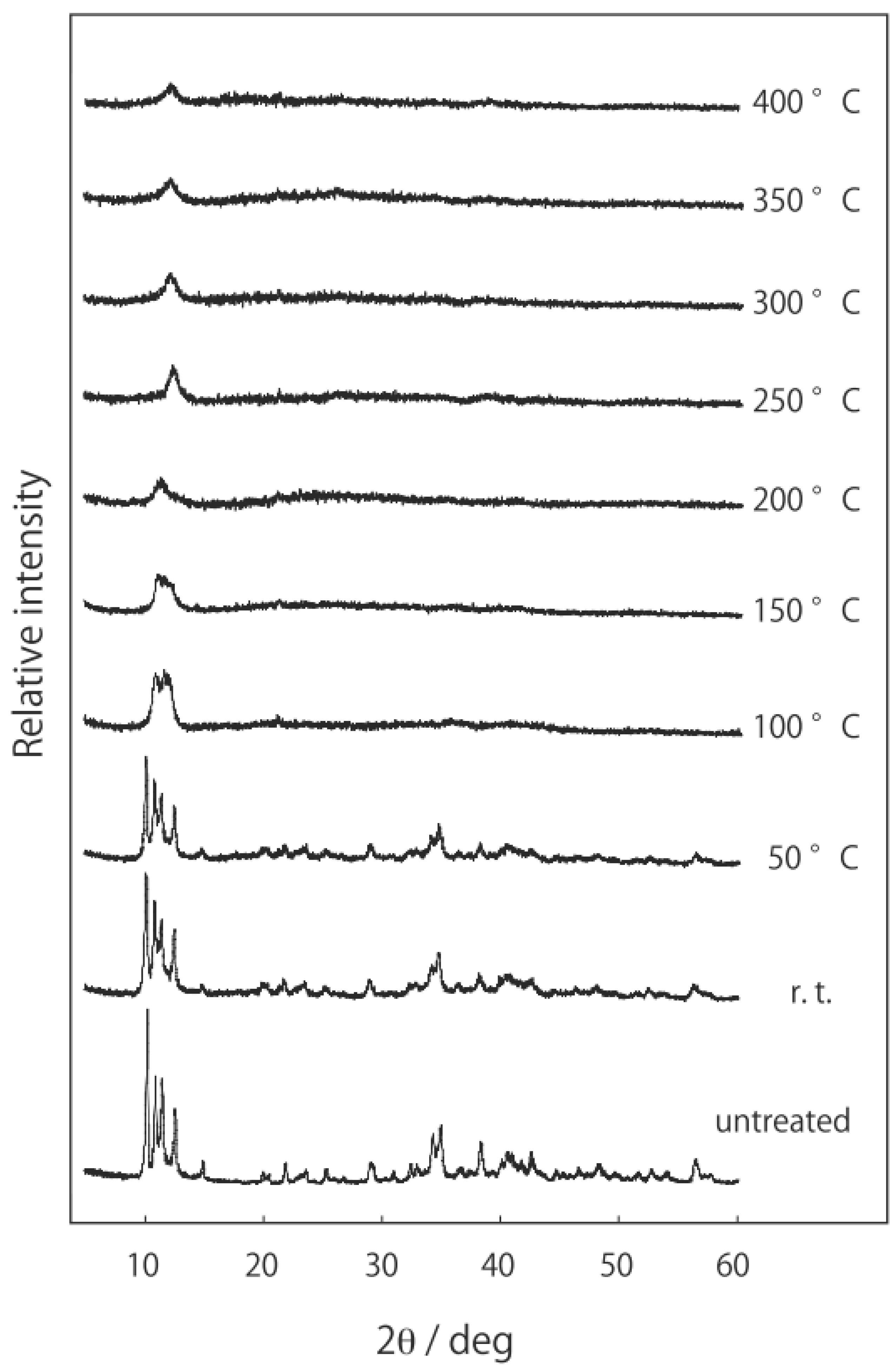
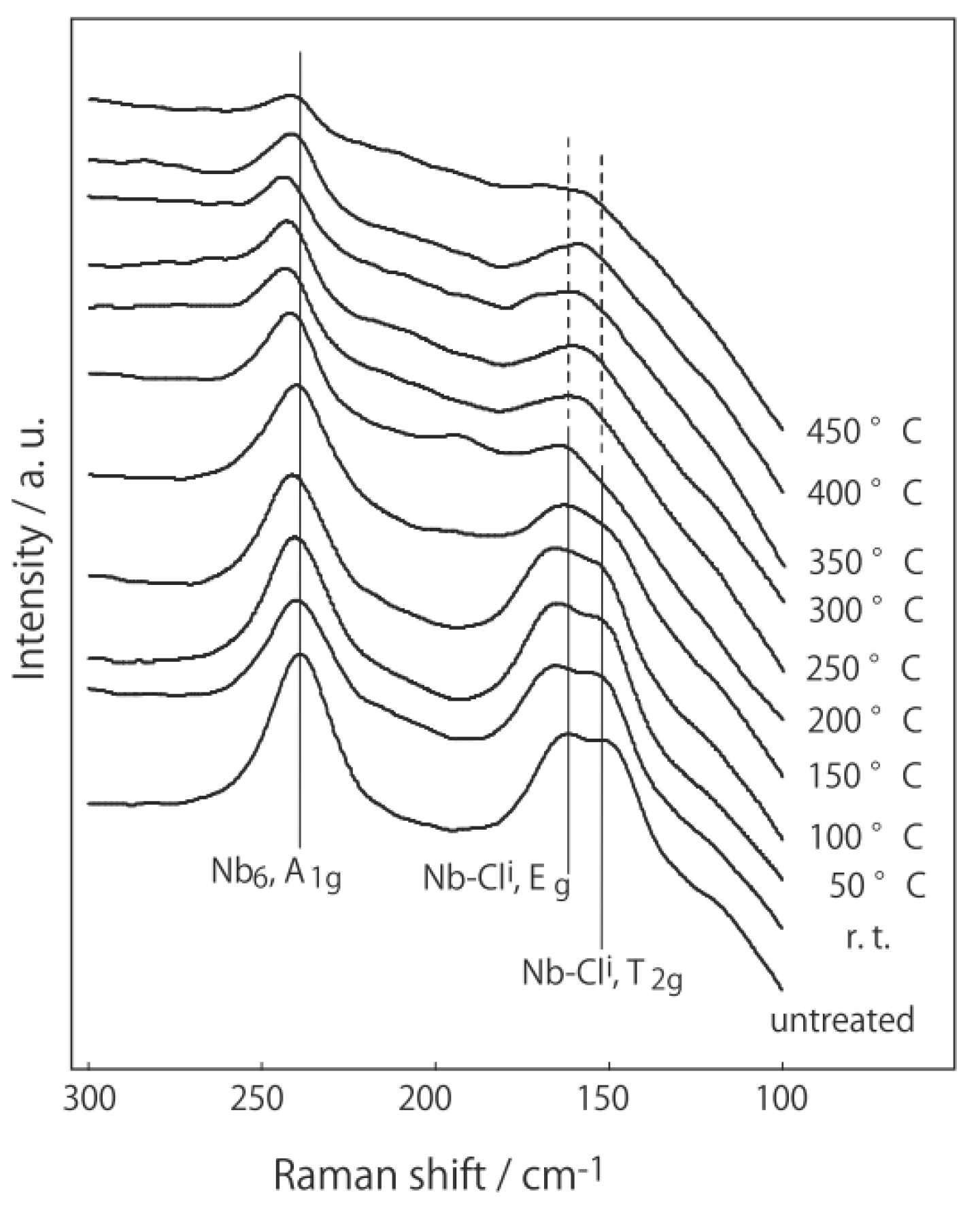
3.1. Retention of the Cluster Framework of the Niobium Cluster [(Nb6Cl12)Cl2(H2O)4]·4H2O [20]



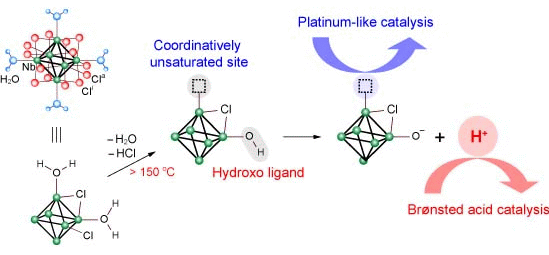
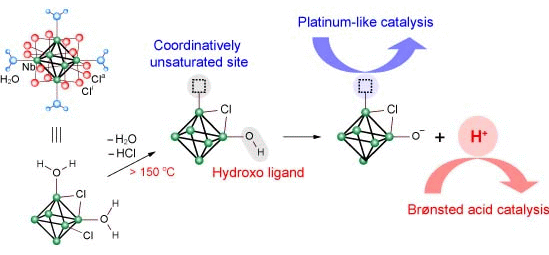
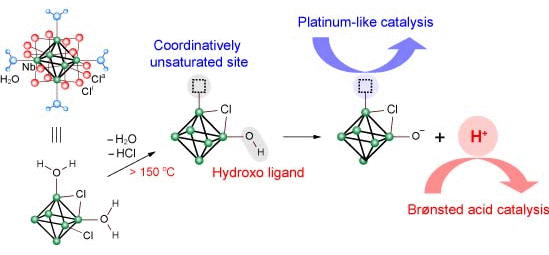
3.2. Active Site of Niobium Cluster



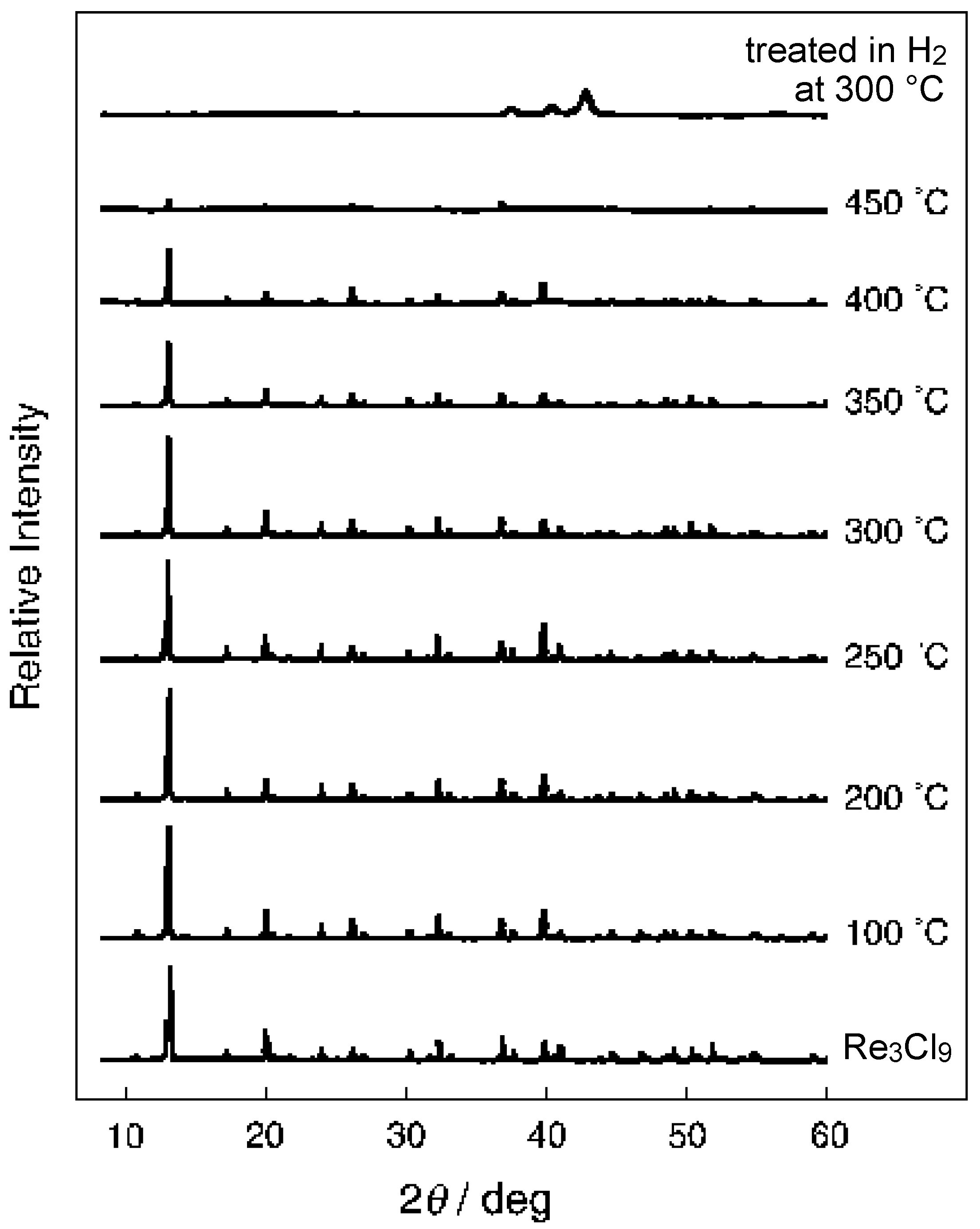
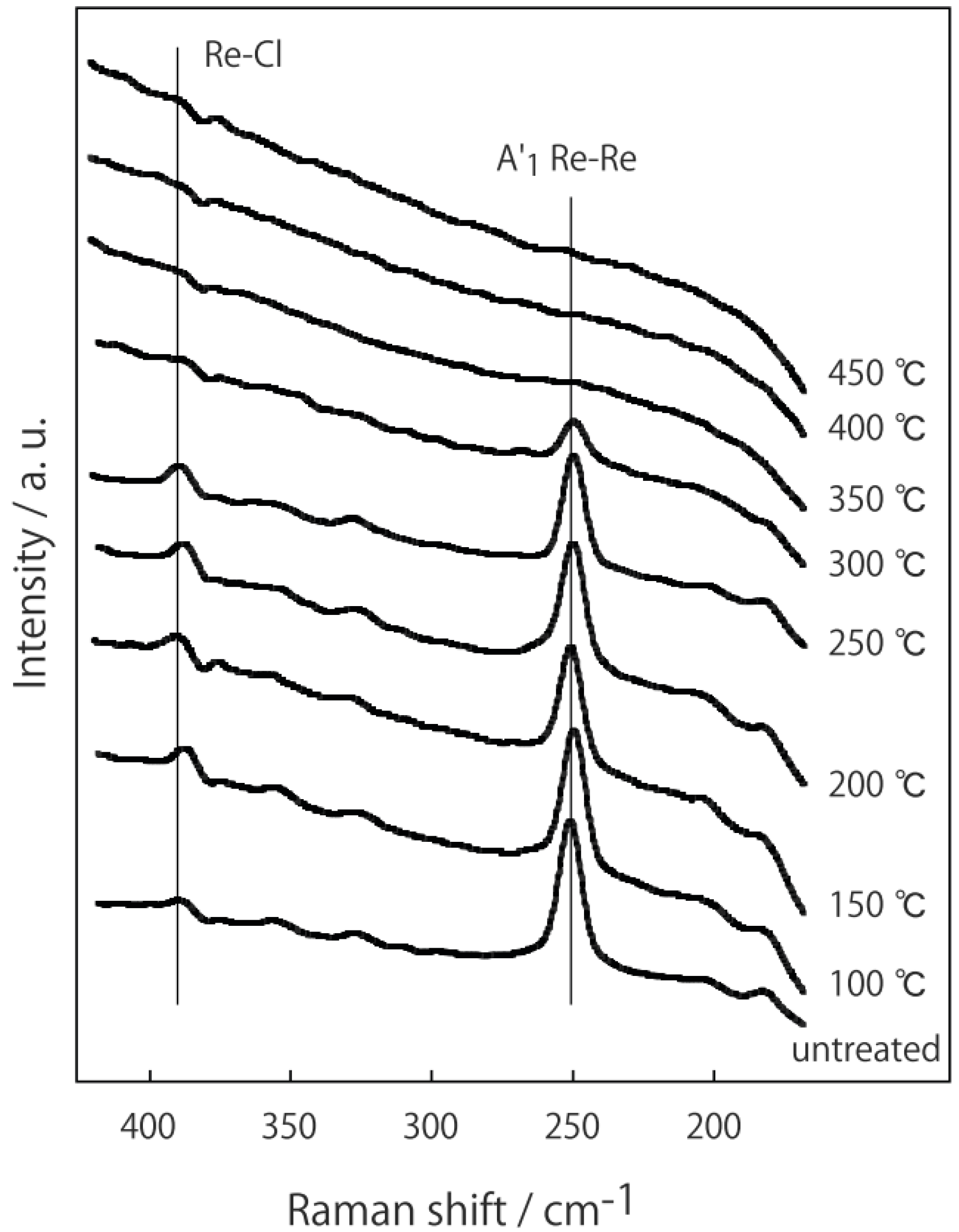
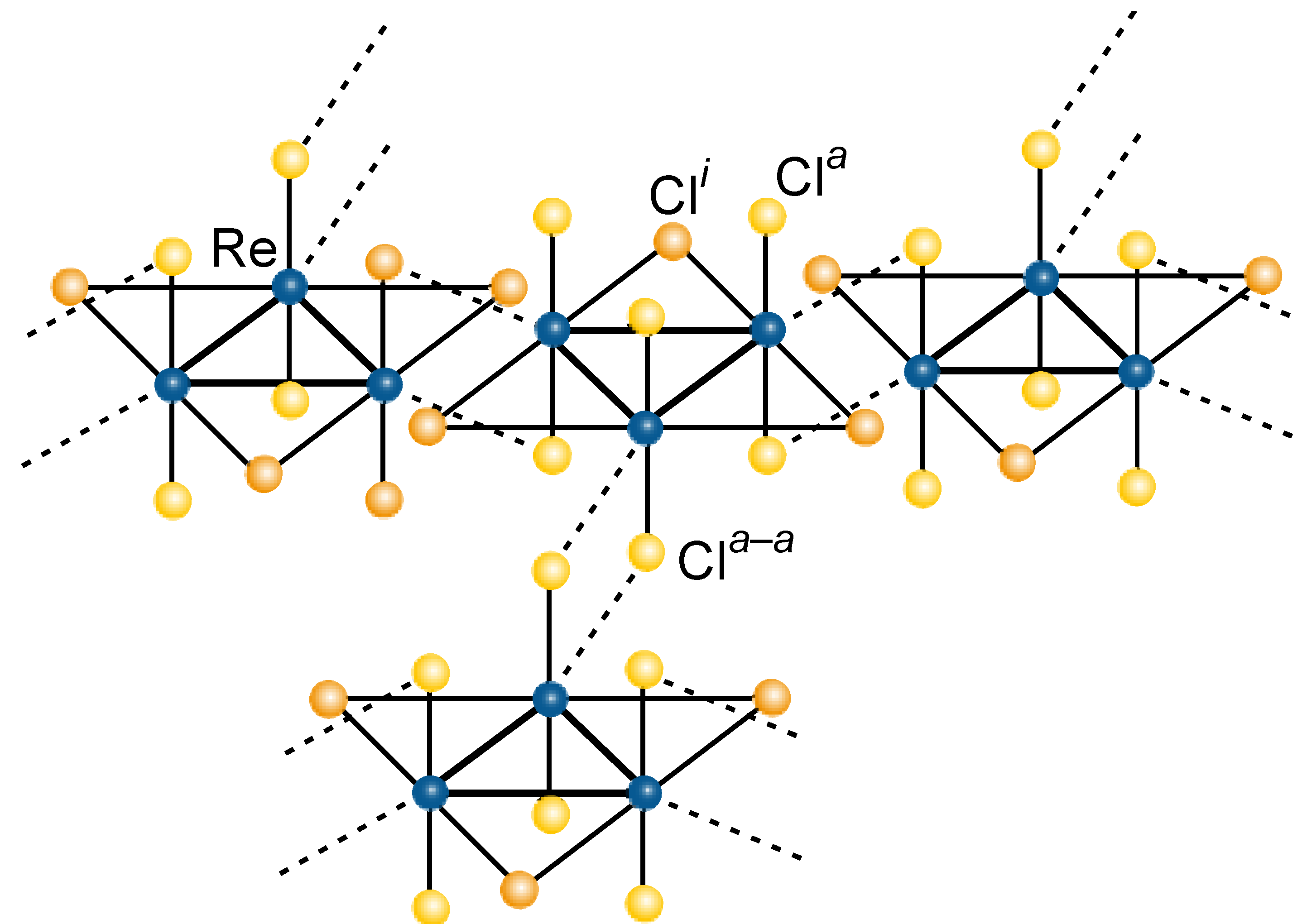
3.3 Rhenium Cluster Re3Cl9 ([Re3Cli3]Cla3Cla-a6/2)



4. Conclusions
Conflicts of Interest
References
- Mingos, D.M.P.; Wales, D.J. Introduction to Cluster Chemistry; Prentice-Hall: Upper Saddle River, NJ, USA, 1990; p. 6. [Google Scholar]
- Gade, L.H.; Johnson, B.F.G.; Lewis, J.; McPartlin, M.; Powell, H.R.; Raithby, P.R.; Wong, W.T. Synthesis and structural characterisation of the osmium cluster dianions [Os17(CO)36]2− and [Os20(CO)40]2−. J. Chem. Soc. Dalton Trans. 1994, 1994, 521–532. [Google Scholar]
- Chihara, T.; Yamazaki, H. Hexaruthenium carbido carbonyl methyl cluster [PPN][Ru6C(CO)16(CH3)] as catalyst precursor for hydrogenation of olefins. Syntheses and structures of unsaturated and saturated hexaruthenium hydrido clusters [PPN][Ru6C(CO)15H] and [PPN][Ru6C(CO)16H]. J. Organomet. Chem. 1994, 473, 273–284. [Google Scholar] [CrossRef]
- Lamb, H.H. Engineering catalyst surfaces with metal carbonyl clusters. Catal. Today 1993, 18, 3–19. [Google Scholar] [CrossRef]
- Braunstein, P.; Rosé, J. Heterometallic clusters for heterogeneous catalysis. In Catalysis by Di- and Polynuclear Metal Cluster Complexes; Adams, R.D., Cotton, F.A., Eds.; Wiley-VCH: New York, NY, USA, 1998; Chapter 13; p. 443. [Google Scholar]
- Gates, B.C. Metal cluster catalysts dispersed on solid supports. In Catalysis by Di- and Polynuclear Metal Cluster Complexes; Adams, R.D., Cotton, F.A., Eds.; Wiley-VCH: New York, NY, USA, 1998; Chapter 14; p. 509. [Google Scholar]
- Zuffa, J.L.; Gladfelter, W.L. On the mechanism of homogeneous catalytic hydrogenation using anion-promoted metal clusters. J. Am. Chem. Soc. 1986, 108, 4669–4671. [Google Scholar] [CrossRef]
- Adams, R.D.; Babin, J.E.; Tasi, M.; Wang, J.G. Catalyst design. The activation of a trinuclear metal cluster complex by metal atom substitution. Organometallics 1988, 7, 755–764. [Google Scholar] [CrossRef]
- Blomstrand, W. Ueber unorganische Haloidverbindungen, die sich wie Radicale verhalten. J. Prakt. Chem. 1859, 77, 88–119. [Google Scholar] [CrossRef]
- Schafer, H.; Schnering, H.G.; Tillack, J.; Kuhnen, F.; Wohrle, H.; Baumann, H. Neue Untersuchungen über die Chloride des Molybdäns. Z. Anorg. Allg. Chem. 1967, 353, 281–310. [Google Scholar] [CrossRef]
- Lee, S.C.; Holm, R.H. Nonmolecular metal chalcogenide/halide solids and their molecular cluster analogues. Angew. Chem. Int. Ed. Engl. 1990, 29, 840–856. [Google Scholar] [CrossRef]
- Cotton, F.A.; Hughbanks, T.; Runyan, C.E., Jr.; Wojtczak, W.A. Halide-suppored octahedral clusters of zirconium: Structural and bonding questions. In Early Transition Metal Clusters with π-Donor Ligands; Chisholm, M.H., Ed.; VCH Publishers: New York, NY, USA, 1995; Chapter 1; p. 1. [Google Scholar]
- Saito, T. Chalcogenide cluster complexes of the early transition metals. In Early Transition Metal Clusters with π-Donor Ligands; Chisholm, M.H., Ed.; VCH Publishers: New York, NY, USA, 1995; Chapter 3; p. 63. [Google Scholar]
- Muetterties, E.L. Metal clusters in catalysis VIII. Reduction of triple bonds. Bull. Soc. Chim. Belg. 1976, 85, 451–470. [Google Scholar] [CrossRef]
- Struss, A.W.; Corbett, J.D. Reaction of hydrogen with metallic and reduced halides. The requirement of delocalized electrons for reaction. Inorg. Chem. 1978, 17, 965–969. [Google Scholar] [CrossRef]
- Imoto, H.; Corbett, J.D.; Cisar, A. Synthesis by hydrogen-driven disproportionation reactions. Synthesis and structure of the hexazirconium dodecahalide clusters Zr6Cl12 and Zr6Br12 and the double salt Zr6Cl12.M2ZrCl6 (M = sodium, potassium, cesium). Inorg. Chem. 1981, 20, 145–151. [Google Scholar] [CrossRef]
- Miller, G.J. Chemistry and properties of novel niobium cluster compounds. J. Alloys Compd. 1995, 229, 93–106. [Google Scholar] [CrossRef]
- Boorman, P.M.; Chong, K.; Jasim, K.S.; Kydd, R.A.; Lewis, J.M. A study of supported molybdenum cluster complexes as catalyst precursors: Part II. Evaluation of the catalytic activity of clusters supported on γ-alumina and fluorided γ-alumina. J. Mol. Catal. 1989, 53, 371–380. [Google Scholar] [CrossRef]
- Prokopuk, N.; Shriver, D.F. The octahedral M6Y8 and M6Y12 clusters of group 4 and 5 transition metals. Adv. Inorg. Chem. 1999, 46, 1–49. [Google Scholar]
- Kamiguchi, S.; Mori, T.; Watanabe, M.; Suzuki, A.; Kodomari, M.; Nomura, M.; Iwasawa, Y.; Chihara, T. Retention of the octahedral metal framework of Nb and Mo halide clusters in catalytic decomposition of phenyl acetate to phenol and ketene. J. Mol. Catal. A 2006, 253, 176–186. [Google Scholar] [CrossRef]
- Guggenberger, L.J.; Sleight, A.W. Structural and bonding characterizations of molybdenum dibromide, Mo6Br12·2H2O. Inorg. Chem. 1969, 8, 2041–2049. [Google Scholar] [CrossRef]
- Nannelli, P.; Block, B.P. Molybdenum(II) halides. Inorg. Synth. 1970, 12, 170. [Google Scholar]
- Kamiguchi, S.; Kondo, K.; Kodomari, M.; Chihara, T. Catalytic ring-attachment isomerization and dealkylation of diethylbenzenes over halide clusters of group 5 and group 6 transition metals. J. Catal. 2004, 223, 54–63. [Google Scholar] [CrossRef]
- Nagashima, S.; Kamiguchi, S.; Ohguchi, S.; Chihara, T. Gas-phase alkylation of pyridine and phenol with alcohols over halide clusters of group 5–7 transition metals as solid acid catalysts. J. Clust. Sci. 2011, 22, 647–660. [Google Scholar] [CrossRef]
- Brosset, C. The structure of complex compounds of bivalent molybdenum. III. X-ray analysis of the structure of the chloro acid in alcohol solution. Arkiv. Kemi. 1949, 1, 353. [Google Scholar]
- Johnson, O. Acidity and polymerization activity of solid acid catalysts. J. Phys. Chem. 1955, 59, 827–831. [Google Scholar]
- Benesi, H.A. Acidity of catalyst surfaces. I. Acid strength from colors of asdorbed indicatios. J. Am. Chem. Soc. 1956, 78, 5490–5494. [Google Scholar] [CrossRef]
- Greenop, M.W.; Thomas, C.B. Nitration of alkylbenzenes catalyzed by mercury(II), thallium(III) and lead(IV). J. Chem. Soc. Perkin Trans. 1995, 2, 1595–1599. [Google Scholar] [CrossRef]
- Mashima, K. Catalytic diversity of diene complexes of niobium and tantalum on polymerizations of ethylene, norbornene, and methyl methacrylate. Macromol. Symp. 2000, 159, 69–76. [Google Scholar] [CrossRef]
- Bennett, M.A.; Matheson, T.W. Catalysis by ruthenium compounds. In Comprehensive Organometallic Chemistry; Wilkinson, G., Stone, F.G.A., Abel, E.W., Eds.; Plenum Press: Oxford, UK, 1982; Volume 4, p. 931. [Google Scholar]
- Chen, X.; Zhang, T.; Zheng, M.; Wu, Z.; Wu, W.; Li, C. The reaction route and active site of catalytic decomposition of hydrazine over molybdenum nitride catalyst. J. Catal. 2004, 224, 473–478. [Google Scholar] [CrossRef]
- Kamiguchi, S.; Noda, M.; Miyagishi, Y.; Nishida, S.; Kodomari, M.; Chihara, T. Catalytic isomerization of 1-hexene to 2-hexene by halide clusters of Nb, Mo, Ta and W possessing an octahedral metal core. J. Mol. Catal. A 2003, 195, 159–171. [Google Scholar] [CrossRef]
- Kamiguchi, S.; Nagashima, S.; Komori, K.; Kodomari, M.; Chihara, T. Thermal activation of molecular tungsten halide clusters with the retention of an octahedral metal framework and the catalytic dehydration of alcohols to olefins as a solid acid catalyst. J. Clust. Sci. 2007, 18, 414–430. [Google Scholar] [CrossRef]
- Kamiguchi, S.; Nishida, S.; Kurokawa, H.; Miura, H.; Chihara, T.T. Formation of Brønsted acid site on halide clusters of group 5 and 6 transition metals. Catalytic methylation and demethylation of methylbenzenes with methanol. J. Mol. Catal. A 2005, 226, 1–9. [Google Scholar]
- Simon, A.; Schnering, H.G.; Wöhrle, H.; Schäfer, H. Nb6Cl14–Synthese, Eigenschaften, Struktur. Z. Anorg. Allg. Chem. 1965, 339, 155. [Google Scholar] [CrossRef]
- Harder, K.; Preetz, W. Schwingungsspektren der Clusterverbindungen (M6Xi12)Xa2·8H2O, M = Nb, Ta; Xi = Cl, Br; Xa = Cl, Br, I. Z. Anorg. Allg. Chem. 1990, 591, 32–40. [Google Scholar] [CrossRef]
- Schoonover, J.R.; Zietlow, T.C.; Clark, D.L.; Heppert, J.A.; Chisholm, M.H.; Gray, H.B.; Sattelberger, A.P.; Woodruff, W.H. Resonance Raman spectra of [M6X8Y6]2− cluster complexes (M = Mo, W; X, Y = Cl, Br, I). Inorg. Chem. 1996, 35, 6606–6613. [Google Scholar] [CrossRef]
- Kamiguchi, S.; Takahashi, I.; Kurokawa, H.; Miura, H.; Chihara, T. Vapor-phase synthesis of 1,2-dihydro-2,2,4-trimethylquinolines from anilines and acetone over group 5–7 metal halide clusters as catalysts. Appl. Catal. A 2006, 309, 70–75. [Google Scholar]
- Kamiguchi, S.; Takaku, S.; Kodomari, M.; Chihara, T. Variable catalytic behavior of Nb, Mo, Ta, W, and Re halide clusters. Isomerization of alkynes to conjugated dienes under nitrogen and hydrogenation to alkenes under hydrogen. J. Mol. Catal. A 2006, 260, 43–48. [Google Scholar] [CrossRef]
- Kamiguchi, S.; Watanabe, M.; Kondo, K.; Kodomari, M.; Chihara, T. Catalytic dehydrohalogenation of alkyl halides by Nb, Mo, Ta, and W halide clusters with an octahedral metal framework and by a Re chloride cluster with a triangular metal framework. J. Mol. Catal. A 2003, 203, 153–163. [Google Scholar] [CrossRef]
- Cotton, F.A.; Wilkinson, G.; Murillo, C.A.; Bochemann, M. Advanced Inorganic Chemistry, 6th ed.; Wiley: New York, NY, USA, 1999; p. 981. [Google Scholar]
- Cotton, F.A.; Lippard, S.J.; Mague, J.T. Preparation and spectra of some adducts of the nonachlorotrirhenium and nonabromotrirhenium groups; evidence for the Re3Br9 group. Inorg. Chem. 1965, 4, 508–514. [Google Scholar] [CrossRef]
- Nagashima, S.; Kamiguchi, S.; Chihara, T. Halide cluster catalysis. Catalytic reactions. Metals submitted for publication. 2014. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kamiguchi, S.; Nagashima, S.; Chihara, T. Characterization of Catalytically Active Octahedral Metal Halide Cluster Complexes. Metals 2014, 4, 84-107. https://doi.org/10.3390/met4020084
Kamiguchi S, Nagashima S, Chihara T. Characterization of Catalytically Active Octahedral Metal Halide Cluster Complexes. Metals. 2014; 4(2):84-107. https://doi.org/10.3390/met4020084
Chicago/Turabian StyleKamiguchi, Satoshi, Sayoko Nagashima, and Teiji Chihara. 2014. "Characterization of Catalytically Active Octahedral Metal Halide Cluster Complexes" Metals 4, no. 2: 84-107. https://doi.org/10.3390/met4020084
APA StyleKamiguchi, S., Nagashima, S., & Chihara, T. (2014). Characterization of Catalytically Active Octahedral Metal Halide Cluster Complexes. Metals, 4(2), 84-107. https://doi.org/10.3390/met4020084