
Parallel and Divergent Evolutionary Solutions for the Optimization of an Engineered Central Metabolism in Methylobacterium extorquens AM1

Abstract
:1. Introduction
2. Materials and Methods
2.1. Strain Construction and Evolution Regime
2.2. Growth Conditions and Measurement of Specific Growth Rate
2.3. Stress Test with Hydrogen Peroxide
2.4. Whole-Genome Re-Sequencing
2.5. Analysis of Microarray Data
3. Results and Discussion
3.1. Overview of Genomic Changes at 600 Generations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | Isolate | Growth Relative to EM 1 | # Novel Expression 1 | Total # Mutations | # Intergenic | # Coding | # IS Elements |
|---|---|---|---|---|---|---|---|
| F1 | CM1727 | 1.96 | 149 | 11 | 2 | 8 | 2 |
| F2 | CM1730 | 2.15 | 12 | 4 | 2 | 1 | 1 |
| F3 | CM1139 | 2.44 | 17 | 11 | 5 | 5 | 2 |
| F4 | CM1145 | 2.50 | 217 | 9 | 4 | 4 | 2 |
| F5 | CM1739 | 2.09 | 42 | 11 | 5 | 5 | 6 |
| F6 | CM1742 | 2.09 | 47 | 11 | 2 | 8 | 3 |
| F7 | CM1745 | 2.16 | 21 | 9 | 2 | 6 | 2 |
| F8 | CM1748 | 2.11 | 197 | 18 | 8 | 8 | 6 |
| Isolate | pCM410 | icuAB | gshA | META1_4902 1 | kefB | rpoA | ATP Synthase 2 | pntAB | META2_0008 3 | META1_3102 4 |
|---|---|---|---|---|---|---|---|---|---|---|
| F1 | + | + | − | + | − | + | + | − | − | − |
| F2 | + | + | + | − | − | − | − | − | − | − |
| F3 | + | + | + | + | − | − | + | + | + | − |
| F4 | + | + | + | + | − | − | − | + | − | − |
| F5 | + | − | + | − | + | − | − | − | + | − |
| F6 | + | − | + | − | + | − | − | − | − | − |
| F7 | + | + | − | − | − | − | − | − | − | + |
| F8 | + | + | − | − | + | + | − | − | − | + |

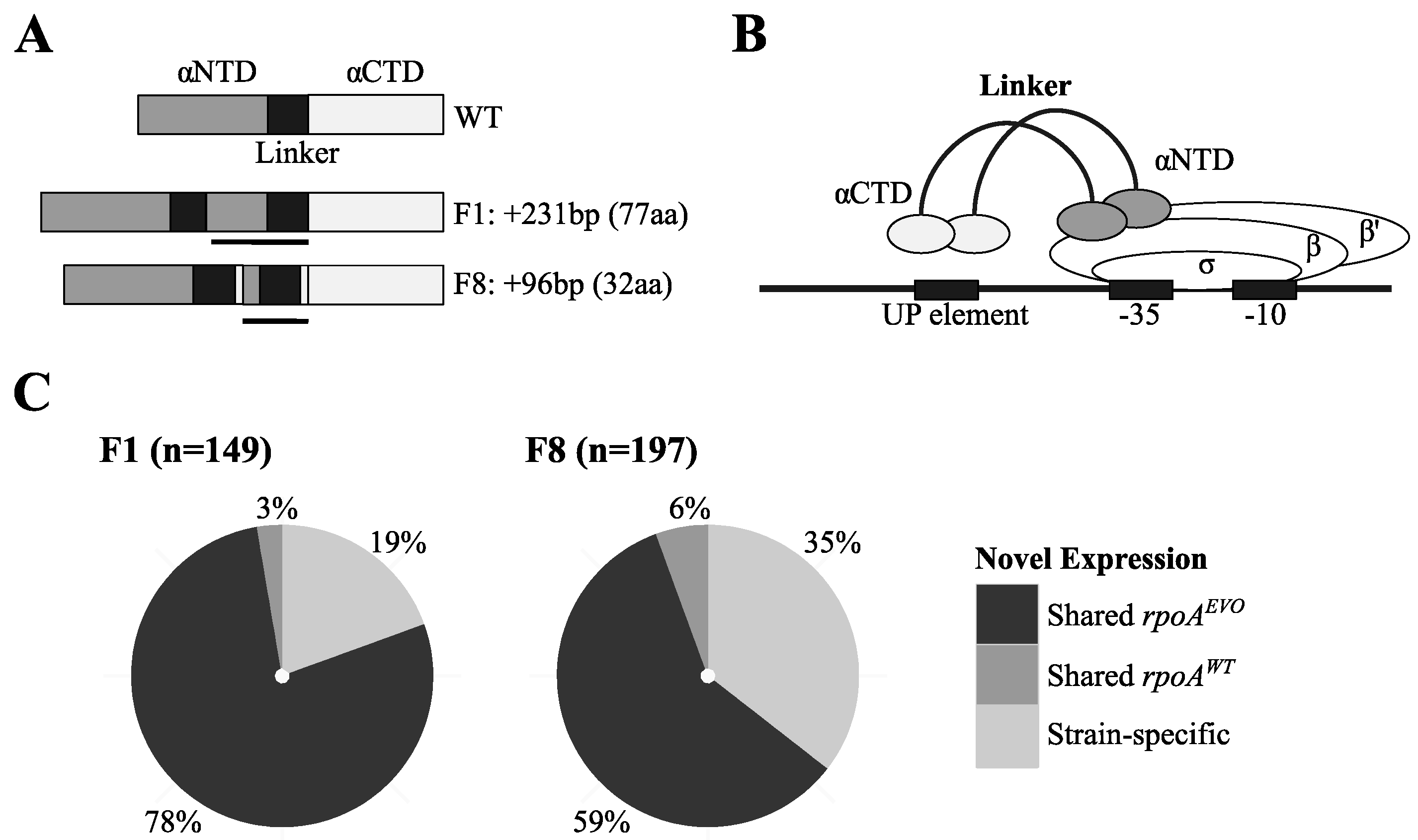
3.2. Unique Expression Profiles Linked to Mutations in RNA Polymerase

| Gene | Locus | Function | Expression Change 1 | Exp F1 2 | Exp F8 2 | Exp Others 2,3 |
|---|---|---|---|---|---|---|
| recO | META1_0821 | DNA repair | Novel | −2.23 | −2.14 | −1.21 |
| dnaK | META2_0894 | Hsp70 chaperone protein | Novel | 2.41 | 2.06 | −1.02 |
| polA | META2_0971 | DNA polymerase | Novel | 2.68 | 2.46 | 1.22 |
| META1_2781 | META1_2781 | putative antioxidant enzyme; Tpx-related thiol peroxidase | Novel | 5.17 | 4.06 | 1.33 |
| xoxF | META1_1740 | C1 metabolism/regulation | Novel | 6.32 | 5.70 | 1.38 |
| mxbD | META1_1753 | C1 metabolism/regulation | Novel | −1.78 | −1.96 | 1.00 |
| fdh4A | META1_2094 | C1 metabolism | Novel | −8.28 | −8.28 | −1.85 |
| mauF | META1_2769 | C1 metabolism | Novel | 7.57 | 6.41 | 1.55 |
| gap | META1_2218 | PHB biosynthesis | Novel | −3.71 | −3.94 | −2.27 |
| META1_0222 | META1_0222 | putative sensor histidine kinase | Unrestored | 1.31 | 1.09 | 1.79 |
| def | META1_1530 | peptide deformylase | Restored | 2.17 | 2.83 | 1.47 |
| ibpA | META1_1514 | heat shock protein | Restored | −2.53 | −3.14 | −1.61 |
| META1_4113 | META1_4113 | hypothetical protein; putative sarcosine oxidase-related | Restored | −18.13 | −10.34 | −3.34 |
| META1_5150 | META1_5150 | hypothetical protein | Restored | −35.75 | −31.56 | −2.69 |
3.3. Investigation of Conditions under which rpoAEVO Was Advantageous
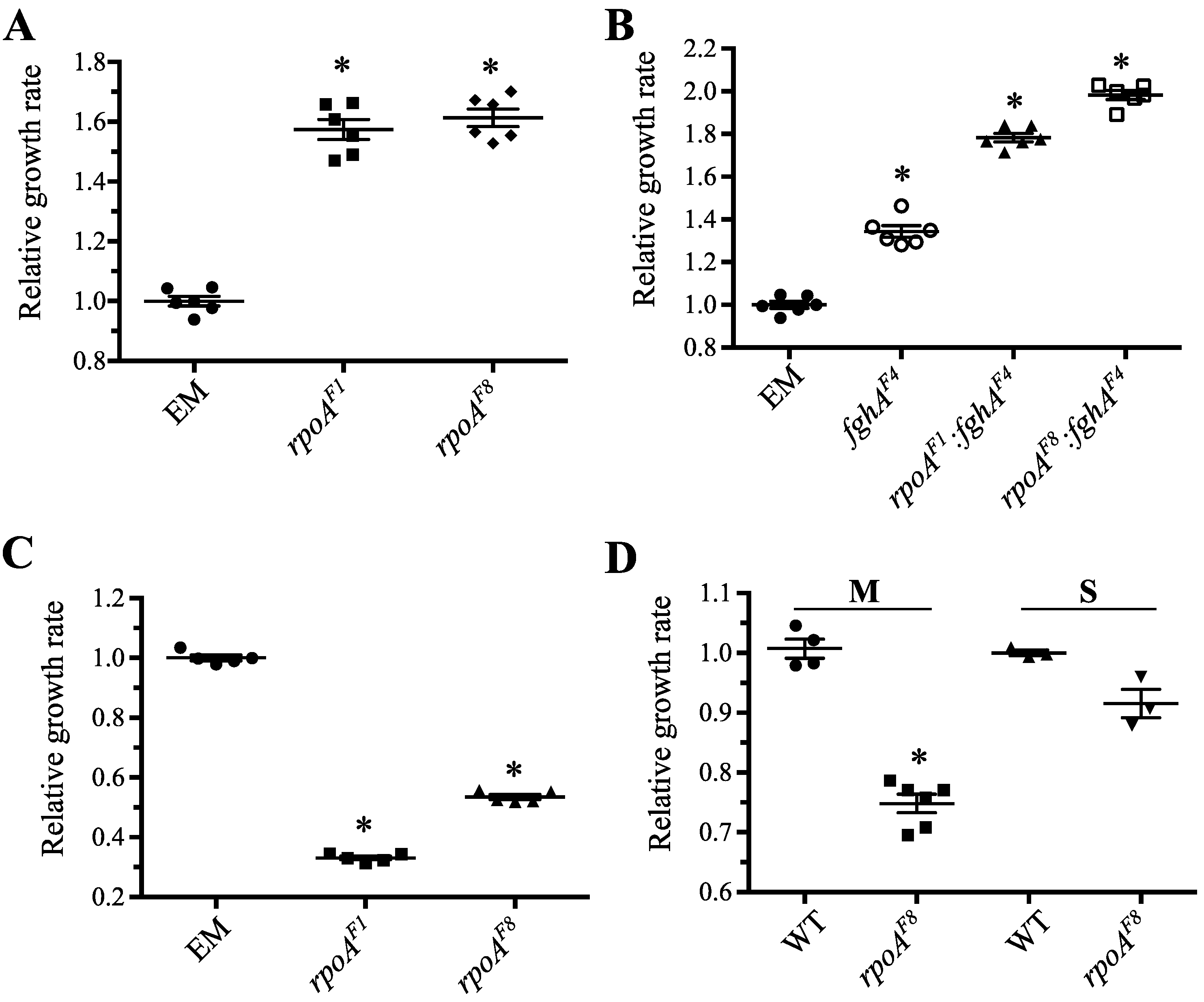
3.3.1. Mutations in rpoA Were Highly Beneficial in EM Grown on Methanol

3.3.2. Mutations in rpoA Remained Advantageous in the Context of Other Beneficial Mutations to the Engineered Pathway
3.3.3. Mutations in rpoA Were Deleterious in EM Grown on Succinate
3.3.4. Mutations in rpoA Are Deleterious in WT Grown on Methanol and Succinate
3.3.5. Mutations in rpoA Offer Protection against Hydrogen Peroxide Stress

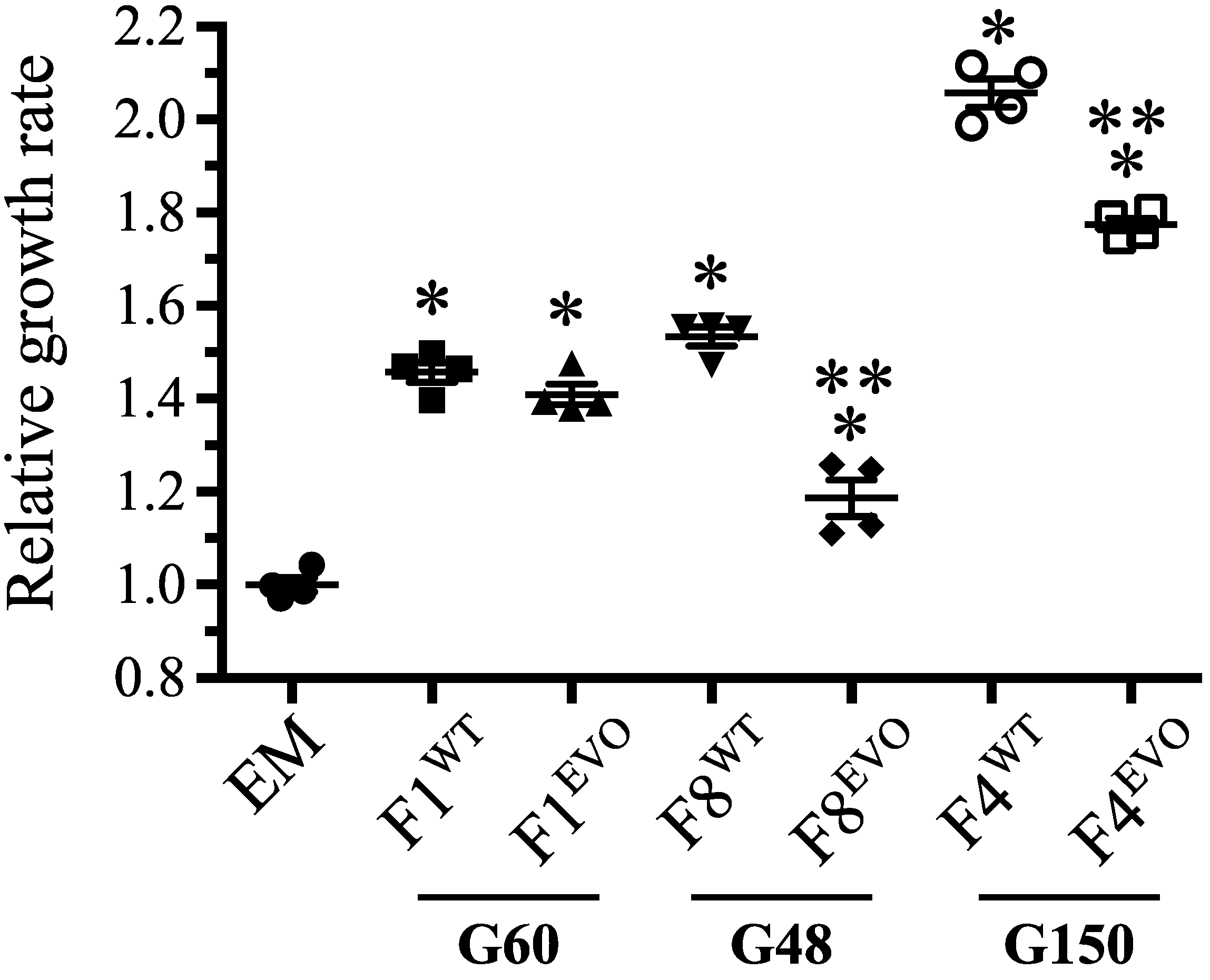
3.4. Complex Evolutionary Dynamics of rpoAEV° Mutations

4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Choi, Y.J.; Bourque, D.; Morel, L.; Groleau, D.; Míguez, C.B. Multicopy integration and expression of heterologous genes in Methylobacterium extorquens ATCC 55366. Appl. Environ. Microbiol. 2006, 72, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Chubiz, L.M.; Purswani, J.; Carroll, S.M.; Marx, C.J. A novel pair of inducible expression vectors for use in Methylobacterium extorquens. BMC Res. Notes 2013, 6, 183. [Google Scholar] [CrossRef] [PubMed]
- Marx, C.J. Development of a broad-host-range sacB-based vector for unmarked allelic exchange. BMC Res. Notes 2008, 1, 1. [Google Scholar] [CrossRef] [PubMed]
- Marx, C.J.; Lidstrom, M.E. Broad-host-range cre-lox system for antibiotic marker recycling in Gram-negative bacteria. BioTechniques 2002, 33, 1062–1067. [Google Scholar] [PubMed]
- Okubo, Y.; Skovran, E.; Guo, X.; Sivam, D.; Lidstrom, M.E. Implementation of microarrays for Methylobacterium extorquens AM1. OMICS 2007, 11, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Schada von Borzyskowski, L.; Remus-Emsermann, M.; Weishaupt, R.; Vorholt, J.A.; Erb, T.J. A set of versatile brick vectors and promoters for the assembly, expression, and integration of synthetic operons in Methylobacterium extorquens AM1 and other Alphaproteobacteria. ACS Synth. Biol. 2014. [Google Scholar] [CrossRef]
- Marx, C.J.; van Dien, S.J.; Lidstrom, M.E. Flux analysis uncovers key role of functional redundancy in formaldehyde metabolism. PLoS Biol. 2005, 3, e16. [Google Scholar] [CrossRef] [PubMed]
- Van Dien, S.J.; Lidstrom, M.E. Stoichiometric model for evaluating the metabolic capabilities of the facultative methylotroph Methylobacterium extorquens AM1, with application to reconstruction of C(3) and C(4) metabolism. Biotechnol. Bioeng. 2002, 78, 296–312. [Google Scholar]
- Peyraud, R.; Schneider, K.; Kiefer, P.; Massou, S.; Vorholt, J.A.; Portais, J.-C. Genome-scale reconstruction and system level investigation of the metabolic network of Methylobacterium extorquens AM1. BMC Syst. Biol. 2011, 5, 189. [Google Scholar] [CrossRef] [PubMed]
- Carroll, S.M.; Marx, C.J. Evolution after introduction of a novel metabolic pathway consistently leads to restoration of wild-type physiology. PLoS Genet. 2013, 9, e1003427. [Google Scholar] [CrossRef] [PubMed]
- Vuilleumier, S.; Chistoserdova, L.; Lee, M.-C.; Bringel, F.; Lajus, A.; Zhou, Y.; Gourion, B.; Barbe, V.; Chang, J.; Cruveiller, S.; et al. Methylobacterium genome sequences: A reference blueprint to investigate microbial metabolism of C1 compounds from natural and industrial sources. PLoS ONE 2009, 4, e5584. [Google Scholar] [CrossRef] [PubMed]
- Marx, C.J.; Bringel, F.; Chistoserdova, L.; Moulin, L.; Farhan Ul Haque, M.; Fleischman, D.E.; Gruffaz, C.; Jourand, P.; Knief, C.; Lee, M.-C.; et al. Complete genome sequences of six strains of the genus Methylobacterium. J. Bacteriol. 2012, 194, 4746–4748. [Google Scholar] [CrossRef] [PubMed]
- Quayle, J.; Peel, D. Methanol and carbon dioxide incorporation by Pseudomonas sp. AM1. Biochem. J. 1960, 76, 3P. [Google Scholar]
- Ochsner, A.M.; Sonntag, F.; Buchhaupt, M.; Schrader, J.; Vorholt, J.A. Methylobacterium extorquens: Methylotrophy and biotechnological applications. Appl. Microbiol. Biotechnol. 2015, 99, 517–534. [Google Scholar] [CrossRef] [PubMed]
- Orita, I.; Nishikawa, K.; Nakamura, S.; Fukui, T. Biosynthesis of polyhydroxyalkanoate copolymers from methanol by Methylobacterium extorquens AM1 and the engineered strains under cobalt-deficient conditions. Appl. Microbiol. Biotechnol. 2014, 98, 3715–3725. [Google Scholar] [CrossRef] [PubMed]
- Sirirote, P.; Yamane, T.; Shimizu, S. Production of L-serine from methanol and glycine by resting cells of a methylotroph under automatically controlled conditions. J. Ferment. Technol. 1986, 64, 389–396. [Google Scholar] [CrossRef]
- Hu, B.; Lidstrom, M.E. Metabolic engineering of Methylobacterium extorquens AM1 for 1-butanol production. Biotechnol. Biofuels 2014, 7, 156. [Google Scholar] [CrossRef] [PubMed]
- Bélanger, L.; Figueira, M.M.; Bourque, D.; Morel, L.; Béland, M.; Laramée, L.; Groleau, D.; Míguez, C.B. Production of heterologous protein by Methylobacterium extorquens in high cell density fermentation. FEMS Microbiol. Lett. 2004, 231, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Choi, Y.J.; Gringorten, J.L.; Gringorten, J.L.; Bélanger, L.; Bélanger, L.; Morel, L.; Morel, L.; Bourque, D.; Bourque, D.; et al. Production of an insecticidal crystal protein from Bacillus thuringiensis by the methylotroph Methylobacterium extorquens. Appl. Environ. Microbiol. 2008, 74, 5178–5182. [Google Scholar] [CrossRef] [PubMed]
- Michener, J.M.; Marx, C.J. After horizontal gene transfer, metabolic pathways may need further optimization. Microbe 2015, 10, 61–67. [Google Scholar]
- Elena, S.F.; Lenski, R.E. Evolution experiments with microorganisms: The dynamics and genetic bases of adaptation. Nat. Rev. Genet. 2003, 4, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Conrad, T.M.; Lewis, N.E.; Palsson, B.Ø. Microbial laboratory evolution in the era of genome-scale science. Mol. Syst. Biol. 2011, 7, 509. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-C.; Marx, C.J. Synchronous waves of failed soft sweeps in the laboratory: Remarkably rampant clonal interference of alleles at a single locus. Genetics 2013, 193, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-C.; Chou, H.-H.; Marx, C.J. Asymmetric, bimodal trade-offs during adaptation of Methylobacterium to distinct growth substrates. Evolution 2009, 63, 2816–2830. [Google Scholar] [CrossRef] [PubMed]
- Chou, H.-H.; Chiu, H.-C.; Delaney, N.F.; Segrè, D.; Marx, C.J. Diminishing returns epistasis among beneficial mutations decelerates adaptation. Science 2011, 332, 1190–1192. [Google Scholar] [CrossRef] [PubMed]
- Marx, C.J. Recovering from a bad start: Rapid adaptation and tradeoffs to growth below a threshold density. BMC Evol. Biol. 2012, 12, 109. [Google Scholar] [CrossRef] [PubMed]
- Agashe, D.; Sane, M.; Phalnikar, K.; Diwan, G.D.; Habibullah, A.; Martinez-Gomez, N.C.; Sahasrabuddhe, V.; Polachek, W.; Wang, J.; Chubiz, L.M.; et al. Large-effect beneficial synonymous mutations mediate rapid and parallel adaptation in a bacterium. Unpublished work. 2015. [Google Scholar]
- Michener, J.K.; Camargo Neves, A.A.; Vuileumier, S.; Bringel, F.; Marx, C.J. Effective use of a horizontally-transferred pathway for dichloromethane catabolism requires post-transfer refinement. eLife 2014, 3. [Google Scholar] [CrossRef]
- Chistoserdova, L.; Vorholt, J.A.; Thauer, R.K.; Lidstrom, M.E. C1 transfer enzymes and coenzymes linking methylotrophic bacteria and methanogenic Archaea. Science 1998, 281, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Marx, C.J.; Chistoserdova, L.; Lidstrom, M.E. Formaldehyde-detoxifying role of the tetrahydromethanopterin-linked pathway in Methylobacterium extorquens AM1. J. Bacteriol. 2003, 185, 7160–7168. [Google Scholar] [CrossRef] [PubMed]
- Chistoserdova, L.; Crowther, G.J.; Vorholt, J.A.; Skovran, E.; Portais, J.C.; Lidstrom, M.E. Identification of a fourth formate dehydrogenase in Methylobacterium extorquens AM1 and confirmation of the essential role of formate oxidation in methylotrophy. J. Bacteriol. 2007, 189, 9076–9081. [Google Scholar] [CrossRef] [PubMed]
- Chistoserdova, L.; Laukel, M.; Portais, J.-C.; Vorholt, J.A.; Lidstrom, M.E. Multiple formate dehydrogenase enzymes in the facultative methylotroph Methylobacterium extorquens AM1 are dispensable for growth on methanol. J. Bacteriol. 2004, 186, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Marx, C.J.; Laukel, M.; Vorholt, J.A.; Lidstrom, M.E. Purification of the formate-tetrahydrofolate ligase from Methylobacterium extorquens AM1 and demonstration of its requirement for methylotrophic growth. J. Bacteriol. 2003, 185, 7169–7175. [Google Scholar] [CrossRef] [PubMed]
- Crowther, G.J.; Kosály, G.; Lidstrom, M.E. Formate as the main branch point for methylotrophic metabolism in Methylobacterium extorquens AM1. J. Bacteriol. 2008, 190, 5057–5062. [Google Scholar] [CrossRef] [PubMed]
- Ras, J.; van Ophem, P.W.; Reijnders, W.N.; van Spanning, R.J.; Duine, J.A.; Stouthamer, A.H.; Harms, N. Isolation, sequencing, and mutagenesis of the gene encoding NAD- and glutathione-dependent formaldehyde dehydrogenase (GD-FALDH) from Paracoccus denitrificans, in which GD-FALDH is essential for methylotrophic growth. J. Bacteriol. 1995, 177, 247–251. [Google Scholar] [PubMed]
- Harms, N.; Ras, J.; Reijnders, W.N.; van Spanning, R.J.; Stouthamer, A.H. S-formylglutathione hydrolase of Paracoccus denitrificans is homologous to human esterase D: A universal pathway for formaldehyde detoxification? J. Bacteriol. 1996, 178, 6296–6299. [Google Scholar] [PubMed]
- Chou, H.-H.; Marx, C.J. Optimization of gene expression through divergent mutational paths. Cell Rep. 2012, 1, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, P.; Buchhaupt, M.; Christen, P.; Kaup, B.; Schrader, J.; Vorholt, J.A. Metabolite profiling uncovers plasmid-induced cobalt limitation under methylotrophic growth conditions. PLoS ONE 2009, 4, e7831. [Google Scholar] [CrossRef] [PubMed]
- Chou, H.-H.; Berthet, J.; Marx, C.J. Fast growth increases the selective advantage of a mutation arising recurrently during evolution under metal limitation. PLoS Genet. 2009, 5, e1000652. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-C.; Marx, C.J. Repeated, selection-driven genome reduction of accessory genes in experimental populations. PLoS Genet. 2012, 8, e1002651. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.W.; Rasche, M.E. Purification, overproduction, and partial characterization of beta-RFAP synthase, a key enzyme in the methanopterin biosynthesis pathway. J. Bacteriol. 2002, 184, 4442–4448. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.G.; Young, L.; Chuang, R.-Y.; Venter, J.C.; Hutchison, C.A.; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Meth. 2009, 6, 343–345. [Google Scholar] [CrossRef]
- Figurski, D.H.; Helinski, D.R. Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc. Natl. Acad. Sci. USA 1979, 76, 1648–1652. [Google Scholar] [CrossRef] [PubMed]
- Delaney, N.F.; Rojas Echenique, J.I.; Marx, C.J. Clarity: An open-source manager for laboratory automation. J. Lab. Autom. 2013, 18, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Delaney, N.F.; Kaczmarek, M.E.; Ward, L.M.; Swanson, P.K.; Lee, M.-C.; Marx, C.J. Development of an optimized medium, strain and high-throughput culturing methods for Methylobacterium extorquens. PLoS ONE 2013, 8, e62957. [Google Scholar] [CrossRef] [PubMed]
- Carroll, S.M.; Harvard University, USA. Unpublished work. 2015.
- Gourion, B.; Francez-Charlot, A.; Vorholt, J.A. PhyR is involved in the general stress response of Methylobacterium extorquens AM1. J. Bacteriol. 2008, 190, 1027–1035. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K. Preparation of genomic DNA from bacteria. Curr. Protoc. Mol. Biol. 2001. [Google Scholar] [CrossRef]
- Barrick, J.E.; Yu, D.S.; Yoon, S.H.; Jeong, H.; Oh, T.K.; Schneider, D.; Lenski, R.E.; Kim, J.F. Genome evolution and adaptation in a long-term experiment with Escherichia coli. Nature 2009, 461, 1243–1247. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Meth. 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Makui, H.; Roig, E.; Cole, S.T.; Helmann, J.D.; Gros, P.; Cellier, M.F. M. Identification of the Escherichia coli K-12 Nramp orthologue (MntH) as a selective divalent metal ion transporter. Mol. Microbiol. 2000, 35, 1065–1078. [Google Scholar] [CrossRef] [PubMed]
- Elmore, M.J.; Lamb, A.J.; Ritchie, G.Y.; Douglas, R.M.; Munro, A.; Gajewska, A.; Booth, I.R. Activation of potassium efflux from Escherichia coli by glutathione metabolites. Mol. Microbiol. 1990, 4, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, G.P.; McLaggan, D.; Booth, I.R. Potassium channel activation by glutathione-S-conjugates in Escherichia coli: Protection against methylglyoxal is mediated by cytoplasmic acidification. Mol. Microbiol. 1995, 17, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Nayak, D.D.; Agashe, D.; Lee, M.-C.; Marx, C.J. Tradeoffs between using methylamine as a carbon versus a nitrogen source generates selection to maintain degenerate metabolic pathways. Unpublished work. 2015. [Google Scholar]
- Zhang, M.; FitzGerald, K.A.; Lidstrom, M.E. Identification of an upstream regulatory sequence that mediates the transcription of mox genes in Methylobacterium extorquens AM1. Microbiology 2005, 151, 3723–3728. [Google Scholar] [CrossRef] [PubMed]
- Meng, W.; Savery, N.J.; Busby, S.J.; Thomas, M.S. The Escherichia coli RNA polymerase alpha subunit linker: Length requirements for transcription activation at CRP-dependent promoters. EMBO J. 2000, 19, 1555–1566. [Google Scholar] [CrossRef] [PubMed]
- Husnain, S.I.; Meng, W.; Busby, S.J.W.; Thomas, M.S. Escherichia coli can tolerate insertions of up to 16 amino acids in the RNA polymerase α subunit inter-domain linker. Biochim. Biophys. Acta 2004, 1678, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Browning, D.F.; Busby, S.J. The regulation of bacterial transcription initiation. Nat. Rev. Microbiol 2004, 2, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Ferenci, T. Maintaining a healthy SPANC balance through regulatory and mutational adaptation. Mol. Microbiol. 2005, 57, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Chou, H.-H.; Delaney, N.F.; Draghi, J.A.; Marx, C.J. Mapping the fitness landscape of gene expression uncovers the cause of antagonism and sign epistasis between adaptive mutations. PLoS Genet. 2014, 10, e1004149. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.I.; Dinh, D.M.; Schneider, D.; Lenski, R.E.; Cooper, T.F. Negative epistasis between beneficial mutations in an evolving bacterial population. Science 2011, 332, 1193–1196. [Google Scholar] [CrossRef] [PubMed]
- Taylor, I.J.; Anthony, C. A biochemical basis for obligate methylotrophy: Properties of a mutant of Pseudomonas AM1 lacking 2-oxoglutarate dehydrogenase. J. Gen. Microbiol. 1976, 93, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Van Dien, S.J.; Okubo, Y.; Hough, M.T.; Korotkova, N.; Taitano, T.; Lidstrom, M.E. Reconstruction of C3 and C4 metabolism in Methylobacterium extorquens AM1 using transposon mutagenesis. Microbiology 2003, 149, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Skovran, E.; Crowther, G.J.; Guo, X.; Yang, S.; Lidstrom, M.E. A systems biology approach uncovers cellular strategies used by Methylobacterium extorquens AM1 during the switch from multi- to single-carbon growth. PLoS ONE 2010, 5, e14091. [Google Scholar] [CrossRef] [PubMed]
- Vorholt, J.A.; Marx, C.J.; Lidstrom, M.E.; Thauer, R.K. Novel formaldehyde-activating enzyme in Methylobacterium extorquens AM1 required for growth on methanol. J. Bacteriol. 2000, 182, 6645–6650. [Google Scholar] [CrossRef] [PubMed]
- Chubiz, L.M.; Lee, M.-C.; Delaney, N.F.; Marx, C.J. FREQ-Seq: A rapid, cost-effective, sequencing-based method to determine allele frequencies directly from mixed populations. 2012; 7, e47959. [Google Scholar]
- Woods, R.J.; Barrick, J.E.; Cooper, T.F.; Shrestha, U.; Kauth, M.R.; Lenski, R.E. Second-order selection for evolvability in a large Escherichia coli population. Science 2011, 331, 1433–1436. [Google Scholar] [CrossRef] [PubMed]
- Tenaillon, O.; Rodriguez-Verdugo, A.; Gaut, R.L.; McDonald, P.; Bennett, A.F.; Long, A.D.; Gaut, B.S. The molecular diversity of adaptive convergence. Science 2012, 335, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Cooper, T.F.; Rozen, D.E.; Lenski, R.E. Parallel changes in gene expression after 20,000 generations of evolution in Escherichia coli. Proc Natl. Acad. Sci. USA 2003, 100, 1072–1077. [Google Scholar] [CrossRef] [PubMed]
- Charusanti, P.; Conrad, T.M.; Knight, E.M.; Venkataraman, K.; Fong, N.L.; Xie, B.; Gao, Y.; Palsson, B.Ø. Genetic Basis of growth adaptation of Escherichia coli after deletion of pgi, a major metabolic gene. PLoS Genet. 2010, 6, e1001186. [Google Scholar] [CrossRef] [PubMed]
- Conrad, T.M.; Frazier, M.; Joyce, A.R.; Cho, B.-K.; Knight, E.M.; Lewis, N.E.; Landick, R.; Palsson, B.Ø. RNA polymerase mutants found through adaptive evolution reprogram Escherichia coli for optimal growth in minimal media. Proc. Natl. Acad. Sci. 2010, 107, 20500–20505. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carroll, S.M.; Chubiz, L.M.; Agashe, D.; Marx, C.J. Parallel and Divergent Evolutionary Solutions for the Optimization of an Engineered Central Metabolism in Methylobacterium extorquens AM1. Microorganisms 2015, 3, 152-174. https://doi.org/10.3390/microorganisms3020152
Carroll SM, Chubiz LM, Agashe D, Marx CJ. Parallel and Divergent Evolutionary Solutions for the Optimization of an Engineered Central Metabolism in Methylobacterium extorquens AM1. Microorganisms. 2015; 3(2):152-174. https://doi.org/10.3390/microorganisms3020152
Chicago/Turabian StyleCarroll, Sean Michael, Lon M. Chubiz, Deepa Agashe, and Christopher J. Marx. 2015. "Parallel and Divergent Evolutionary Solutions for the Optimization of an Engineered Central Metabolism in Methylobacterium extorquens AM1" Microorganisms 3, no. 2: 152-174. https://doi.org/10.3390/microorganisms3020152
APA StyleCarroll, S. M., Chubiz, L. M., Agashe, D., & Marx, C. J. (2015). Parallel and Divergent Evolutionary Solutions for the Optimization of an Engineered Central Metabolism in Methylobacterium extorquens AM1. Microorganisms, 3(2), 152-174. https://doi.org/10.3390/microorganisms3020152