Massively Parallel Sequencing of Genes Implicated in Heritable Cardiac Disorders: A Strategy for a Small Diagnostic Laboratory

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples
2.2. DNA Extraction
2.3. Custom Cardiac Panel Design (SureSelectQXT Target Enrichment System)
2.4 Library Preparation and Sequencing
2.4.1. SureSelectQXT Target Enrichment System
2.4.2. TruSight® Cardio Sequencing System
2.5. Bidirectional Sanger-Based Sequencing
2.6. Bioinformatic Processing
2.6.1. SureSelectQXT Target Enrichment System
2.6.2. TruSight Cardio Sequencing System
2.7. Validation Process
2.7.1. Read-Depth Analysis
2.7.2. Statistical Analysis
2.7.3. Inter/Intra-Run Repeatability Analysis
3. Results
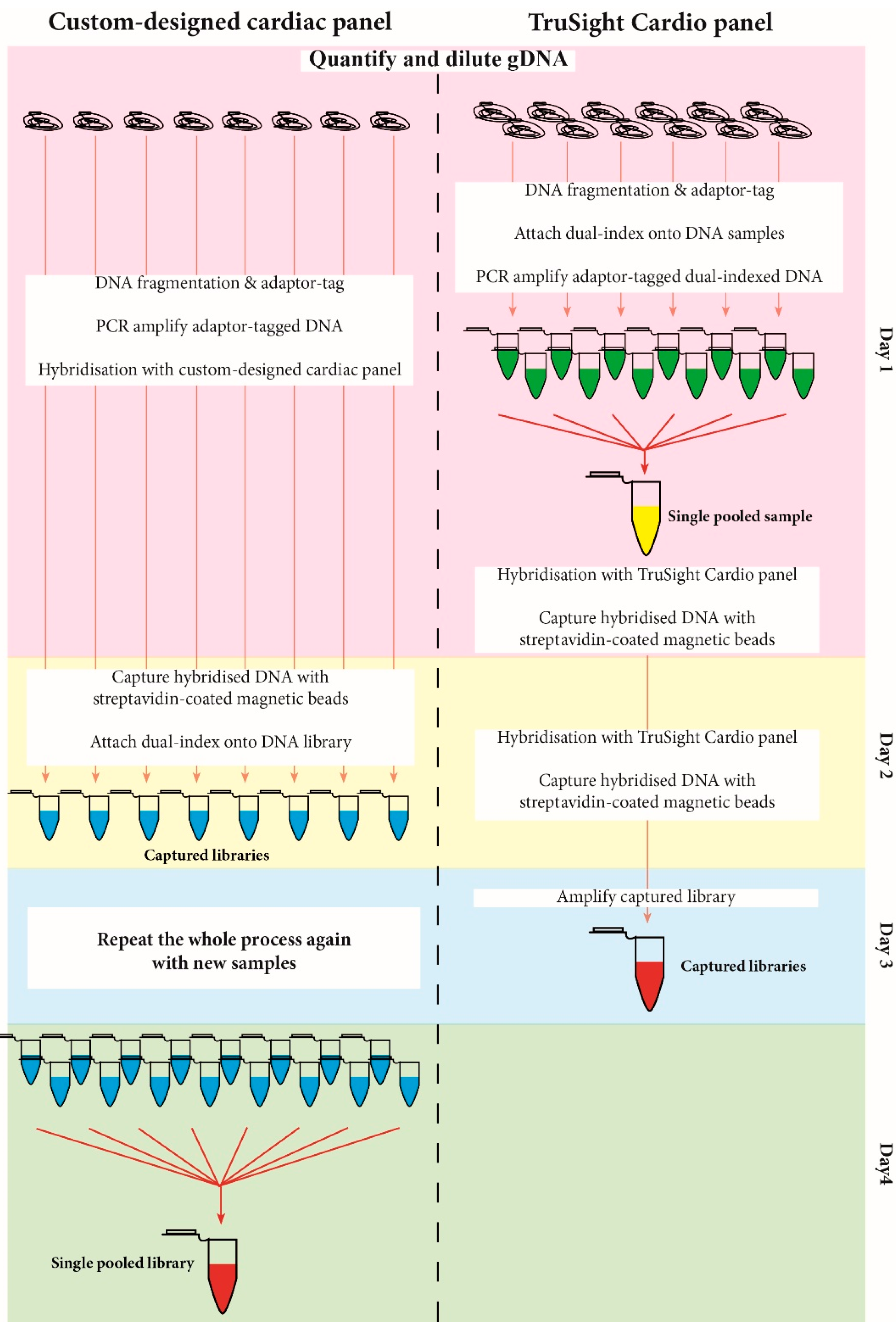
3.1. Library Preparation Workflow
3.2. Gene Read-Depth Analysis
3.2.1. Custom-Designed Cardiac Panel
3.2.2. TruSight® Cardio Panel
3.2.3. Variant Calling Concordance
3.2.4. Custom-Designed Cardiac Panel
3.2.5. TruSight® Cardio Panel
3.2.6. Inter/Intra-Run Repeatability Analysis
4. Discussion
5. Limitations
6. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Meyer, L.; Stubbs, B.; Fahrenbruch, C.; Maeda, C.; Harmon, K.; Eisenberg, M.; Drezner, J. Incidence, causes, and survival trends from cardiovascular-related sudden cardiac arrest in children and young adults 0 to 35 years of age: A 30-year review. Circulation 2012, 126, 1363–1372. [Google Scholar] [CrossRef]
- Leong, I.U.S.; Skinner, J.R.; Love, D.R. Application of massively parallel sequencing in the clinical diagnostic testing of inherited cardiac conditions. Med. Sci. 2014, 2, 98–126. [Google Scholar] [CrossRef]
- Bagnall, R.D.; Weintraub, R.G.; Ingles, J.; Duflou, J.; Yeates, L.; Lam, L.; Davis, A.M.; Thompson, T.; Connel, V.; Wallace, J.; et al. A Prospective Study of Sudden Cardiac Death among Children and Young Adults. N. Engl. J. Med. 2016, 374, 2441–2452. [Google Scholar] [CrossRef]
- Van der Werf, C.; Hofman, N.; Tan, H.L.; van Dessel, P.F.; Alders, M.; van der Wal, A.C.; van Langen, I.M.; Wilde, A.A. Diagnostic yield in sudden unexplained death and aborted cardiac arrest in the young: The experience of a tertiary referral center in The Netherlands. Heart Rhythm 2010, 7, 1383–1389. [Google Scholar] [CrossRef]
- Tester, D.J.; Medeiros-Domingo, A.; Will, M.L.; Haglund, C.M.; Ackerman, M.J. Cardiac channel molecular autopsy: Insights from 173 consecutive cases of autopsy-negative sudden unexplained death referred for postmortem genetic testing. Mayo Clin. Proc. 2012, 87, 524–539. [Google Scholar] [CrossRef]
- Lahrouchi, N.; Raju, H.; Lodder, E.M.; Papatheodorou, E.; Ware, J.S.; Papadakis, M.; Tadros, R.; Cole, D.; Skinner, J.R.; Crawford, J.; et al. Utility of Post-Mortem Genetic Testing in Cases of Sudden Arrhythmic Death Syndrome. J. Am. Coll. Cardiol. 2017, 69, 2134–2145. [Google Scholar] [CrossRef]
- Behr, E.; Wood, D.A.; Wright, M.; Syrris, P.; Sheppard, M.N.; Casey, A.; Davies, M.J.; McKenna, W. Sudden Arrhythmic Death Syndrome Steering Group. Cardiological assessment of first-degree relatives in sudden arrhythmic death syndrome. Lancet 2003, 62, 1457–1459. [Google Scholar] [CrossRef]
- Tan, H.L.; Hofman, N.; van Langen, I.M.; van der Wal, A.C.; Wilde, A.A. Sudden unexplained death: Heritability and diagnostic yield of cardiological and genetic examination in surviving relatives. Circulation 2005, 112, 207–213. [Google Scholar] [CrossRef]
- Skinner, J.R.; Duflou, J.A.; Semsarian, C. Reducing sudden death in young people in Australia and New Zealand: The TRAGADY initiative. Med. J. Aust. 2008, 189, 539–540. [Google Scholar]
- Earle, N.; Crawford, J.; Smith, W.; Hayes, I.; Shelling, A.; Hood, M.; Stiles, M.; Maxwell, F.; Heaven, D.; Love, D.R.; et al. Community detection of long QT syndrome with a clinical registry: An alternative to ECG screening programs? Heart Rhythm 2013, 10, 233–238. [Google Scholar] [CrossRef]
- Next Generation Sequencing-Oxford University Hospitals. Available online: http://www.ouh.nhs.uk/-services/referrals/genetics/genetics-laboratories/molecular-genetics-laboratory/next-generation-sequencing.aspx (accessed on June 2016).
- VCGS-Genetic Testing for Genetic Heart Conditions: Patient Information Sheet. Available online: https://www.vcgs.org.au/tests/cardiac-gene-panels (accessed on June 2016).
- Lakdawala, N.K.; Winterfield, J.R.; Funke, B.H. Arrhythmogenic disorders of genetic origin. Dilated cardiomyopathy. Circ. Arrhythm. Electrophysiol. 2013, 6, 228–237. [Google Scholar] [CrossRef]
- Crotti, L.; Marcou, C.A.; Tester, D.J.; Castelletti, S.; Giudicessi, J.R.; Torchio, M.; Medeiros-Domingo, A.; Simone, S.; Will, M.L.; Dagradi, F.; et al. Spectrum and prevalence of mutations involving BrS1- through BrS12-susceptibility genes in a cohort of unrelated patients referred for Brugada syndrome genetic testing: Implications for genetic testing. J. Am. Coll. Cardiol. 2012, 60, 1410–1418. [Google Scholar] [CrossRef]
- Burashnikov, E.; Pfeiffer, R.; Barajas-Martinez, H.; Delpon, E.; Hu, D.; Desai, M.; Borggrefe, M.; Häissaguerre, M.; Kanter, R.; Pollevick, G.D.; et al. Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death. Heart Rhythm 2010, 7, 1872–1882. [Google Scholar] [CrossRef]
- Antzelevitch, C.; Pollevick, G.D.; Cordeiro, J.M.; Casis, O.; Sanguinetti, M.C.; Aizawa, Y.; Guerchicoff, A.; Pfeiffer, R.; Oliva, A.; Wollnik, B.; et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 2007, 115, 442–449. [Google Scholar] [CrossRef]
- Perrin, M.J.; Gollob, M.H. Genetics of cardiac electrical disease. Can. J. Cardiol. 2013, 29, 89–99. [Google Scholar] [CrossRef]
- Lahat, H.; Pras, E.; Olender, T.; Avidan, N.; Ben-Asher, E.; Man, O.; Levy-Nissenbaum, E.; Khoury, A.; Lorber, A.; Goldman, B.; et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am. J. Hum. Genet. 2001, 69, 1378–1384. [Google Scholar] [CrossRef]
- Ackerman, M.J.; Priori, S.G.; Willems, S.; Berul, C.; Brugada, R.; Calkins, H.; Camm, A.J.; Ellinor, P.T.; Gollob, M.; Hamilton, R.; et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: This document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace 2011, 13, 1077–1109. [Google Scholar] [CrossRef]
- Awad, M.M.; Dalal, D.; Cho, E.; Amat-Alarcon, N.; James, C.; Tichnell, C.; Tucker, A.; Russell, S.D.; Bluemke, D.A.; Dietz, H.C.; et al. DSG2 mutations contribute to arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am. J. Hum. Genet. 2006, 49, 136–142. [Google Scholar] [CrossRef]
- Pilichou, K.; Remme, C.A.; Basso, C.; Campian, M.E.; Rizzo, S.; Barnett, P.; Scicluna, B.P.; Bauce, B.; van den Hoff, M.J.B.; de Bakker, J.M.T.; et al. Myocyte necrosis underlies progressive myocardial dystrophy in mouse dsg2-related arrhythmogenic right ventricular cardiomyopathy. J. Exp. Med. 2009, 206, 1787–1802. [Google Scholar] [CrossRef]
- Sen-Chowdhry, S.; Syrris, P.; Ward, D.; Asimaki, A.; Sevdalis, E.; McKenna, W.J. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation 2007, 115, 1710–1720. [Google Scholar] [CrossRef]
- Rampazzo, A.; Nava, A.; Malacrida, S.; Beffagna, G.; Bauce, B.; Rossi, V.; Zimbello, R.; Simionati, B.; Basso, C.; Thiene, G.; et al. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 2002, 71, 1200–1206. [Google Scholar] [CrossRef]
- Elliott, P.; Baker, R.; Pasquale, F.; Quarta, G.; Ebrahim, H.; Mehta, A.B.; Hughes, D.A.; ACES Study Group. Prevalence of Anderson-Fabry disease in patients with hypertrophic cardiomyopathy: The European Anderson-Fabry Disease survey. Heart 2011, 97, 1957–1960. [Google Scholar] [CrossRef]
- Havndrup, O.; Christiansen, M.; Stoevring, B.; Jensen, M.; Hoffman-Bang, J.; Andersen, P.S.; Hasholt, L.; Nørremølle, A.; Feldt-Rasmussen, U.; Køber, L.; et al. Fabry disease mimicking hypertrophic cardiomyopathy: Genetic screening needed for establishing the diagnosis in women. Eur. J. Heart Fail 2010, 12, 535–540. [Google Scholar] [CrossRef]
- Hedley, P.L.; Jørgensen, P.; Schlamowitz, S.; Wangari, R.; Moolman-Smook, J.; Brink, P.A.; Kanters, J.K.; Corfield, V.A.; Christiansen, M. The genetic basis of long QT and short QT syndromes: A mutation update. Hum. Mutat. 2009, 30, 1486–1511. [Google Scholar] [CrossRef]
- Houston, B.A.; Stevens, G.R. Hypertrophic cardiomyopathy: A review. Clin. Med. Insights Cardiol. 2014, 8, 53–65. [Google Scholar] [CrossRef]
- Dalal, D.; Molin, L.H.; Piccini, J.; Tichnell, C.; James, C.; Bomma, C.; Prakasa, K.; Towbin, J.A.; Marcus, F.I.; Spevak, P.J.; et al. Clinical features of arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in plakophilin-2. Circulation 2006, 113, 1641–1649. [Google Scholar] [CrossRef]
- Van Tintelen, J.P.; Entius, M.M.; Bhuiyan, Z.A.; Jongbloed, R.; Wiesfeld, A.C.; Wilde, A.A.; van der Smagt, J.; Boven, L.G.; Mannens, M.M.; van Langen, I.M.; et al. Plakophilin-2 mutations are the major determinant of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation 2006, 113, 1650–1658. [Google Scholar] [CrossRef]
- Romero, J.; Mejia-Lopez, E.; Manrique, C.; Lucariello, R. Arrhythmogenic right ventricular cardiomyopathy (ARVC/D): A systematic literature review. Clin. Med. Insights Cardiol. 2013, 7, 97–114. [Google Scholar] [CrossRef]
- Priori, S.G.; Napolitano, C.; Memmi, M.; Colombi, B.; Drago, F.; Gasparini, M.; DeSimone, L.; Coltorti, F.; Bloise, R.; Keegan, R.; et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation 2002, 106, 69–74. [Google Scholar] [CrossRef]
- Kapplinger, J.D.; Giudicessi, J.R.; Ye, D.; Tester, D.J.; Callis, T.E.; Valdivia, C.R.; Makielski, J.C.; Wilde, A.A.; Ackerman, M.J. Enhanced classification of Brugada Syndrome-associated and Long-QT Syndrome-associated genetic variants in the SCN5A-encoded Na(v)1.5 cardiac sodium channel. Circ. Cardiovasc. Genet. 2015, 8, 582–595. [Google Scholar] [CrossRef]
- Hu, D.; Barajas-Martinez, H.; Pfeiffer, R.; Dezi, F.; Pfeiffer, J.; Buch, T.; Betzenhauser, M.J.; Belardinelli, L.; Kahlig, K.M.; Rajamani, S.; et al. Mutations in SCN10A are responsible for a large fraction of cases of Brugada syndrome. J. Am. Coll. Cardiol. 2014, 64, 66–79. [Google Scholar] [CrossRef]
- Behr, E.R.; Savio-Galimberti, E.; Barc, J.; Holst, A.G.; Petropoulou, E.; Prins, B.P.; Jabbari, J.; Torchio, M.; Berthet, M.; Mizusawa, Y.; et al. Role of common and rare variants in SCN10A: Results from the Brugada syndrome QRS locus gene discovery collaborative study. Cardiovasc. Res. 2015, 106, 520–529. [Google Scholar] [CrossRef]
- Tian, T.; Liu, Y.; Zhou, X.; Song, L. Progress in the molecular genetics of hypertrophic cardiomyopathy: A mini-review. Gerontology 2013, 59, 199–205. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Dagradi, F. Management of survivors of cardiac arrest—The importance of genetic investigation. Nat. Rev. Cardiol. 2016, 13, 560–566. [Google Scholar] [CrossRef]
- Gourraud, J.B.; Barc, J.; Thollet, A.; Le Scouarnec, S.; Le Marec, H.; Schott, J.J.; Redon, R.; Probst, V. The Brugada Syndrome: A rare arrhythmia disorder with complex inheritance. Front. Cardiovasc. Med. 2016, 3, 9. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Ackerman, M.J.; George, A.L.; Wilde, A.A. Impact of genetics on the clinical management of channelopathies. J. Am. Coll. Cardiol. 2013, 62, 169–180. [Google Scholar] [CrossRef]
- Faragli, A.; Underwood, K.; Priori, S.G.; Mazzanti, A. Is there a role for genetics in the prevention of sudden cardiac death? J. Cardiovasc. Electrophysiol. 2016, 27, 1124–1132. [Google Scholar] [CrossRef]
- RCPA: Massively Parallel Sequencing Implementation Guidelines (Approval Date May 2015). Available online: https://www.rcpa.edu.au/getattachment/7d264a73-938f-45b5-912f-272872661aaa/Massively-Parallel-652 (accessed on June 2016).
- Aziz, N.; Zhao, Q.; Bry, L.; Driscoll, D.K.; Funke, B.; Gibson, J.S.; Grody, W.W.; Hegde, M.R.; Hoeltge, G.A.; Leonard, D.G.; et al. College of American Pathologists’ laboratory standards for next-generation sequencing clinical tests. Arch. Pathol. Lab. Med. 2015, 139, 481–493. [Google Scholar] [CrossRef]
- Gargis, A.S.; Kalman, L.; Berry, M.W.; Bick, D.P.; Dimmock, D.P.; Hambuch, T.; Lu, F.; Lyon, E.; Voelkerding, K.V.; Zehnbauer, B.A.; et al. Assuring the quality of next-generation sequencing in clinical laboratory practice. Nat. Biotechnol. 2012, 30, 1033–1036. [Google Scholar] [CrossRef]
- Ellard, S.; Linday, H.; Camm, N.; Watson, C.; Abbs, S.; Wallis, Y.; Mattocks, C.; Taulor, G.R.; Charlton, R. ACGS: Practice guidelines for Targeted Next Generation Sequencing Analysis and Interpretation. Available online: http://www.acgs.uk.com/media/774807/bpg_for_targeted_next_generation_sequencing_may_2014_final.pdf (accessed on June 2016).
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- De Leeneer, K.; De Schrijver, J.; Clement, L.; Baetens, M.; Lefever, S.; De Keulenaer, S.; Van Criekinge, W.; Deforce, D.; Van Nieuwerburgh, F.; Bekaert, S.; et al. Practical tools to implement massive parallel pyrosequencing of PCR products in next generation molecular diagnostics. PLoS ONE 2011, 6, e25531. [Google Scholar] [CrossRef]
- Mardis, E.R. Next-generation DNA sequencing methods. Annu. Rev. Genomics Hum. Genet. 2008, 9, 387–402. [Google Scholar] [CrossRef]
- Rehm, H.L.; Bale, S.J.; Bayrak-Toydemir, P.; Berg, J.S.; Brown, K.K.; Deignan, J.L.; Friez, M.J.; Funke, B.H.; Hegde, M.R.; Lyon, E.; et al. ACMG clinical laboratory standards for next-generation sequencing. Genet. Med. 2013, 15, 733–747. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Mattocks, C.J.; Morris, M.A.; Matthijs, G.; Swinnen, E.; Corveleyn, A.; Dequeker, E.; Müller, C.R.; Pratt, V.; Wallace, A. A standardized framework for the validation and verification of clinical molecular genetic tests. Eur. J. Hum. Genet. 2010, 18, 1276–1288. [Google Scholar] [CrossRef]
- Shearer, A.E.; Hildebrand, M.S.; Ravi, H.; Joshi, S.; Guiffre, A.C.; Novak, B.; Happe, S.; LeProust, E.M.; Smith, R.J.H. Pre-capture multiplexing improves efficiency and cost-effectiveness of targeted genomic enrichment. BMC Genom. 2012, 13, 618. [Google Scholar] [CrossRef]
- Ackerman, M.J. Genetic purgatory and the cardiac channelopathies: Exposing the variants of uncertain/unknown significance issue. Heart Rhythm 2015, 12, 2325–2331. [Google Scholar] [CrossRef]
- Leong, I.U.; Stuckey, A.; Lai, D.; Skinner, J.R.; Love, D.R. Assessment of the predictive accuracy of five in silico prediction tools, alone or in combination, and two metaservers to classify long QT syndrome gene mutations. BMC Med. Genet. 2015, 16, 34. [Google Scholar] [CrossRef]


{kind=link}
| Gene | Transcript Accession # | LRG Reference # | ARVC | BrS | CPVT | DCM | HCM | LQTS | SQTS |
|---|---|---|---|---|---|---|---|---|---|
| BAG3 | NM_004281.3 | LRG_742t1 | |||||||
| 2–4% [13] | |||||||||
| CACNA1C | NM_000719.6 | LRG_334t1 | BrS 3 | SQT 4 | |||||
| 2–12% [14,15,16] | Limited data [17] | ||||||||
| CACNB2 | NM_201596.2 | LRG_381t1 | BrS 4 | SQT 5 | |||||
| (pending approval) | 2–12% [14,15,16] | Limited data [17] | |||||||
| CASQ2 | NM_001232.3 | LRG_404t1 | CPVT 2 | ||||||
| Rare [18] | |||||||||
| DSC2 | NM_004949.3 | LRG_400t1 | ARVC 11 | ||||||
| 2–7% [19] | |||||||||
| DSG2 | NM_001943.3 | LRG_397t1 | ARVC 10 | ||||||
| 7–26% [20,21,22] | |||||||||
| DSP | NM_004415.2 | LRG_423t1 | ARVC 8 | ||||||
| 6–16% [23] | 1–3% [13] | ||||||||
| GLA | NM_000169.2 | LRG_672t1 | |||||||
| 0.5–3% [24,25] | |||||||||
| KCNA5 | NM_002234.2 | ||||||||
| KCNE1 | NM_000219.3 | LRG_290t1 | LQT 5 | ||||||
| <1% [26] | |||||||||
| KCNE2 | NM_172201.1 | LRG_291t1 | LQT 6 | ||||||
| <1% [26] | |||||||||
| KCNH2 | NM_000238.3 | LRG_288t1 | LQT 2 | SQT 1 | |||||
| 35–45% [26] | Limited data [17] | ||||||||
| KCNJ2 | NM_000891.2 | LRG_328t1 | LQT 7 | SQT 3 | |||||
| <1% [26] | Limited data [17] | ||||||||
| KCNQ1 | NM_000218.2 | LRG_287t1 | LQT 1 | SQT 2 | |||||
| 40–55% [26] | Limited data [17] | ||||||||
| LMNA | NM_005572.3 | LRG_254t1 | |||||||
| 4–8% [13] | |||||||||
| MYBPC3 | NM_000256.3 | LRG_386t1 | |||||||
| >20% [27] | |||||||||
| MYH6 | NM_002471. | LRG_389t1 | |||||||
| Rare [27] | |||||||||
| MYH7 | NM_000257.2 | LRG_384t1 | |||||||
| 4–8% [13] | >30% [27] | ||||||||
| MYL2 | NM_000432.3 | LRG_393t1 | |||||||
| <5% [27] | |||||||||
| PKP2 | NM_004572.3 | LRG_398t1 | ARVC 9 | ||||||
| [28,29] | |||||||||
| RBM20 | NM_001134363.1 | LRG_382t1 | |||||||
| 3–6% [13] | |||||||||
| RYR2 | NM_001035.2 | LRG_402t1 | ARVC 2 | CPVT 1 | |||||
| (pending approval) | Rare [30] | 50% [31] | |||||||
| SCN1B | NM_001037.4 | LRG_420t1 | BrS 5 | ||||||
| (pending approval) | <1% [14] | ||||||||
| SCN5A | NM_198056.2 | LRG_289t1 | BrS 1 | LQT 3 | |||||
| 2–30% [32] | 1–2% [13] | 2–8% [26] | |||||||
| SCN10A | NM_006514.3 | BrS 21 | |||||||
| [33,34] | |||||||||
| TNNI3 | NM_000363.4 | LRG_432t1 | |||||||
| >5% [27] | |||||||||
| TNNT2 | NM_000364.2 | LRG_431t1 | |||||||
| (pending approval) | 3–6% [13] | >20% [27] | |||||||
| TPM1 | NM_001018005.1 | LRG_387t1 | |||||||
| (pending approval) | 2–4% [13] | >5% [27] | |||||||
| TTN | NM_001267550.1 | LRG_391t1 | |||||||
| 15–25% [13] | Rare [35] | ||||||||
| % of diagnosis covered | 45–70% [36] | 20–25% [37] | 50–70% | 20–25% [36] | 60% [36] | 75–80% | NA | ||
| [36,37,38,39] | [36,37,38,39] |
| Custom-Designed Cardiac Panel | TruSight Cardio Panel | |||
|---|---|---|---|---|
| Iteration 1 | Iteration 2 | Iteration 3 | ||
| Number of probes | 9556 | 14,038 | 15,123 | N/A |
| Capture size (kbp) | 293.727 | 307.071 | 344.742 | 572.00 |
| Number of individual samples | 16 | 24 | 36 | 31 |
| Number of inter/intra-run controls | 0 | 8 | 12 | 4 |
| Total number of samples | 16 | 32 | 48 | 35 |
| Sanger-Based Results | ||||
|---|---|---|---|---|
| Variant Detected | Variant not Detected | |||
| MPS results | Variant detected | TP | FP | PPV |
| [TP/(TP + FP)] × 100% | ||||
| Variant not detected | FN | TN | ||
| Sensitivity | Specificity | |||
| [TP/(TP + FN)] × 100% | [TN/(FP + TN)] × 100% | |||
| Gene | Missense | Deletion | Insertion/Duplication | Intronic | Total |
|---|---|---|---|---|---|
| BAG3 | |||||
| CACNA1C | |||||
| CACNB2 | |||||
| CASQ2 | 2 | 3 | 5 | ||
| DSC2 | 1 | 2 | 3 | ||
| DSG2 | 1 | 6 | 7 | ||
| DSP | 1 | 10 | 11 | ||
| GLA | 2 | 2 | |||
| KCNA5 | |||||
| KCNE1 | 3 | 3 | |||
| KCNE2 | |||||
| KCNH2 | 4 | 3 | 10 | 17 | |
| KCNJ2 | 4 | 4 | |||
| KCNQ1 | 4 | 6 | 1 | 7 | 18 |
| LMNA | 1 | 4 | 5 | ||
| MYBPC3 | 4 | 4 | 2 | 12 | 22 |
| MYH6 | |||||
| MYH7 | 1 | 1 | 18 | 20 | |
| MYL2 | 2 | 1 | 3 | ||
| PKP2 | 5 | 5 | |||
| RBM20 | |||||
| RYR2 | 8 | 11 | 19 | ||
| SCN1B | |||||
| SCN5A | 1 | 1 | 2 | ||
| SCN10A | 1 | 5 | 1 | 12 | 19 |
| TNNI3 | 3 | 2 | 5 | ||
| TNNT2 | 2 | 2 | 7 | 11 | |
| TPM1 | 4 | 4 | |||
| TTN | 19 | 96 | 115 | ||
| Total | 18 | 54 | 10 | 218 | 300 |
| Iteration | Total # Variants Expected | # Missense Variants | # Deletion Variants | # Insertion/Duplication Variants | # Intronic Variants |
|---|---|---|---|---|---|
| 1 | 171 | 132 | 9 | 1 | 29 |
| 2 | 420 | 323 | 27 | 4 | 66 |
| 3 | 587 | 469 | 38 | 10 | 70 |
| Total | 1178 | 924 | 74 | 15 | 165 |
| TruSight | 584 | 460 | 36 | 10 | 78 |
| Gene | Iteration 1 | Iteration 2 | Iteration 3 | Total | TruSight |
|---|---|---|---|---|---|
| BAG3 | |||||
| CACNA1C | |||||
| CACNB2 | |||||
| CASQ2 | 4 | 6 | 10 | 6 | |
| DSC2 | 6 | 6 | 4 | ||
| DSG2 | 15 | 15 | 9 | ||
| DSP | 29 | 29 | 26 | ||
| GLA | 2 | 2 | 2 | ||
| KCNA5 | |||||
| KCNE1 | 10 | 10 | 16 | 36 | 11 |
| KCNE2 | |||||
| KCNH2 | 39 | 28 | 63 | 130 | 39 |
| KCNJ2 | 6 | 6 | |||
| KCNQ1 | 15 | 13 | 14 | 42 | 16 |
| LMNA | 5 | 5 | 10 | 10 | |
| MYBPC3 | 8 | 36 | 56 | 100 | 43 |
| MYH6 | |||||
| MYH7 | 9 | 44 | 93 | 146 | 56 |
| MYL2 | 4 | 3 | 7 | 5 | |
| PKP2 | 5 | 5 | 3 | ||
| RBM20 | |||||
| RYR2 | 25 | 31 | 1 | 57 | 31 |
| SCN1B | 2 | 2 | 4 | 4 | |
| SCN5A | 48 | 61 | 78 | 187 | 72 |
| SCN10A | |||||
| TNNI3 | 4 | 23 | 45 | 72 | 26 |
| TNNT2 | 4 | 27 | 44 | 75 | 29 |
| TPM1 | 3 | 15 | 27 | 45 | 15 |
| TTN | 117 | 77 | 194 | 176 | |
| Total | 171 | 420 | 587 | 1178 | 583 |
| Iteration 1 | Iteration 2 | Iteration 3 | ||||
|---|---|---|---|---|---|---|
| SC | JSI | SC | JSI | SC | JSI | |
| Expected TP | 171 | 420 | 587 | |||
| TP detected | 170 | 171 | 420 | 419 | 587 | 587 |
| FP detected | 1 | 5 | 2 | 2 | 2 | 2 |
| Sensitivity | 99.42% | 100% | 100% | 99.76% | 100% | 100% |
| (96.78–99.99%) | (96.78–99.99%) | (99.13–100.00%) | (98.69–99.99%) | (99.37–100.00%) | (99.37–100.00%) | |
| Specificity | 99.42% | 97.16% | 99.53% | 99.53% | 99.66% | 99.66% |
| (97.26–100.00%) | (93.46–99.07%) | (98.30–99.94%) | (98.30–99.94%) | (98.78–99.96%) | (98.78–99.96%) | |
| Positive predictive value | 99.42% | 97.16% | 99.53% | 99.52% | 99.66% | 99.66% |
| (96.78–99.99%) | (93.50–99.07%) | (98.30–99.94%) | (98.30–99.94%) | (98.78–99.96%) | (98.78–99.96%) | |
| TruSight Cardio Panel | Custom-Designed Cardiac Panel | |||
|---|---|---|---|---|
| VariantStudio | JSI | SureCall | JSI | |
| Expected TP | 584 | 584 | ||
| TP detected | 472 | 580 | 583 | 581 |
| FP detected | 2 | 5 | 4 | 6 |
| Sensitivity | 80.82% | 99.14% | 99.83% | 99.49% |
| (77.39–83.94%) | (98.26–99.81%) | (99.05–100.00%) | (98.51–99.89% | |
| Specificity | 99.58% | 99.15% | 99.32% | 98.98% |
| (98.48–99.95%) | (98.02–99.72%) | (98.26–99.81%) | (97.78–99.62%) | |
| Positive predictive value | 99.58% | 99.15% | 99.32% | 98.97% |
| (98.48–99.95%) | (98.02–99.72%) | (99.05–100.00%) | (97.78–99.62%) | |
| Design | ||
|---|---|---|
| Custom-Cardiac Panel | TruSight® Cardio Panel | |
| Number of cardiac genes targeted | 29 | 174 |
| (SCN10A is not part of this panel) | ||
| Workflow | ||
| Number samples processed | 8 samples per working batch | 12 samples for working batch |
| 16 samples for a full MiSeq run | 12 samples for a full MiSeq run | |
| Time for library preparation (including incubation time) | 2 days for 8 samples (could be scaled to 16 samples for the same time) | 3 days for 12 samples |
| Capture workflow type | Post-capture pooling | Pre-capture pooling |
| Labour intensity | More labour intensive than pre-capture pooling | Less labour intensive than post-capture pooling |
| Control over quality of sample libraries | More control over each sample library | Less control over sample libraries |
| Coverage | ||
| Regions with poor read-depth (<30×) | MYL2 exon 1 | GLA exon 4 |
| TTN exon 10, 47, 178 and 189 | KCNH2 exon 1 and 2 | |
| KCNQ1 exon 5 and 15 | ||
| MYBPC3 exon 3, 11 and 26 | ||
| SCN1B exon 1 | ||
| SCN5A exon 3 | ||
| TNNI3 exon 1 | ||
| TNNT2 exon 11 and 15 | ||
| Total # regions: 5 | Total # regions: 13 | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leong, I.U.S.; Stuckey, A.; Belluoccio, D.; Fan, V.; Skinner, J.R.; Prosser, D.O.; Love, D.R. Massively Parallel Sequencing of Genes Implicated in Heritable Cardiac Disorders: A Strategy for a Small Diagnostic Laboratory. Med. Sci. 2017, 5, 22. https://doi.org/10.3390/medsci5040022
Leong IUS, Stuckey A, Belluoccio D, Fan V, Skinner JR, Prosser DO, Love DR. Massively Parallel Sequencing of Genes Implicated in Heritable Cardiac Disorders: A Strategy for a Small Diagnostic Laboratory. Medical Sciences. 2017; 5(4):22. https://doi.org/10.3390/medsci5040022
Chicago/Turabian StyleLeong, Ivone U. S., Alexander Stuckey, Daniele Belluoccio, Vicky Fan, Jonathan R. Skinner, Debra O. Prosser, and Donald. R. Love. 2017. "Massively Parallel Sequencing of Genes Implicated in Heritable Cardiac Disorders: A Strategy for a Small Diagnostic Laboratory" Medical Sciences 5, no. 4: 22. https://doi.org/10.3390/medsci5040022
APA StyleLeong, I. U. S., Stuckey, A., Belluoccio, D., Fan, V., Skinner, J. R., Prosser, D. O., & Love, D. R. (2017). Massively Parallel Sequencing of Genes Implicated in Heritable Cardiac Disorders: A Strategy for a Small Diagnostic Laboratory. Medical Sciences, 5(4), 22. https://doi.org/10.3390/medsci5040022