A Structure-Based Classification and Analysis of Protein Domain Family Binding Sites and Their Interactions

Abstract
:1. Introduction
2. Methods
2.1. The KBDOCK Database
2.2. Structural Annotation of DFBSs
3. Results
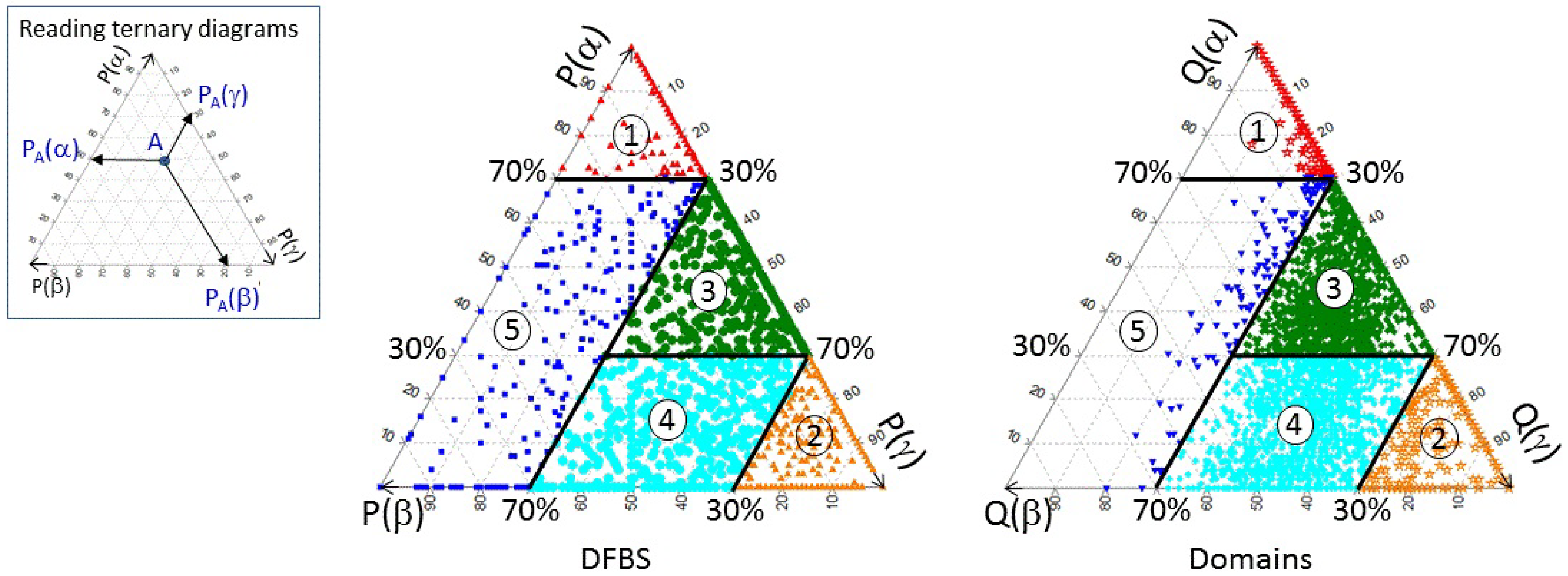
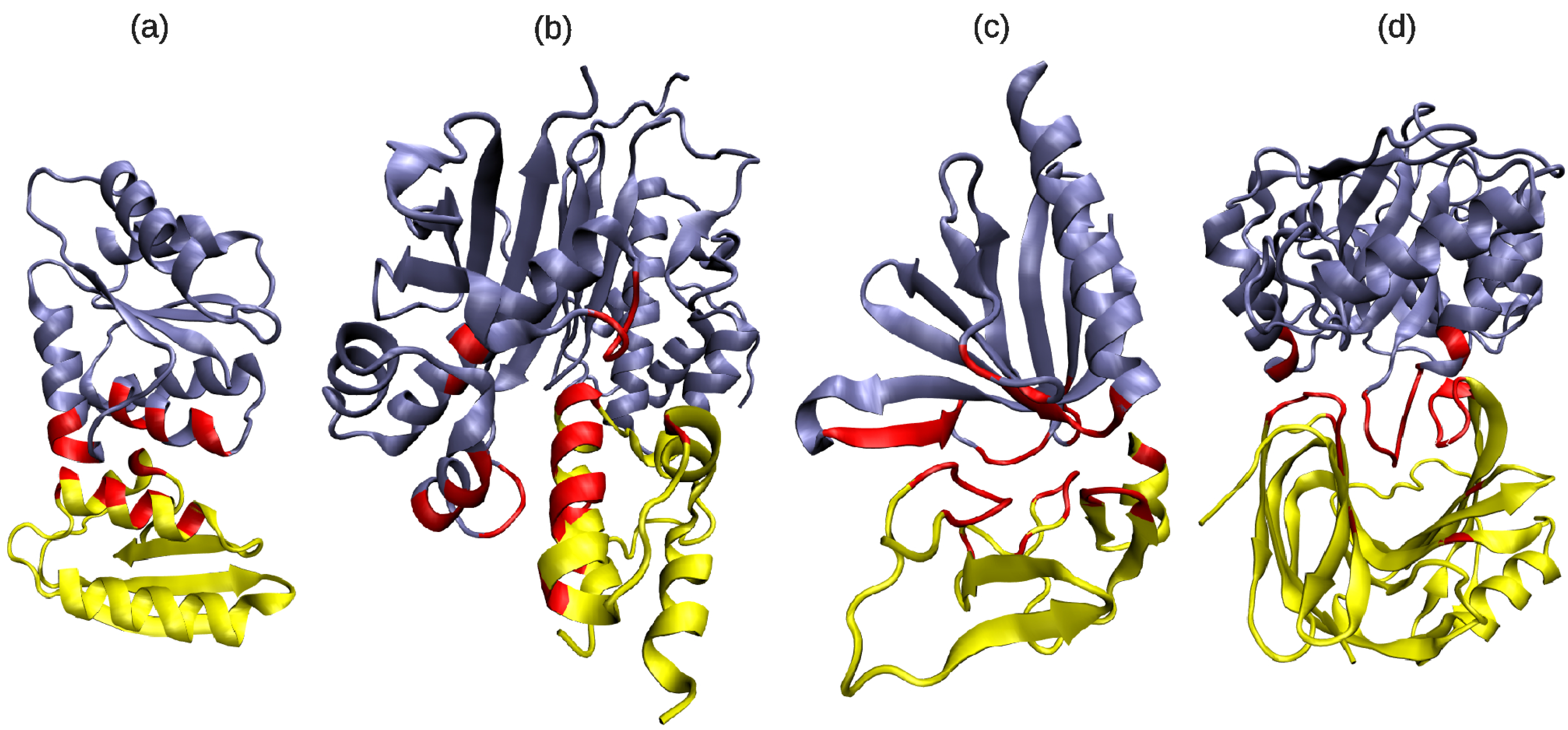
3.1. Classifying and Analysing DFBSs


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Definition | Class No. | No. DFBSs | No. Domains |
|---|---|---|---|
| P(α) ≥ 70% | 1 | 835 | 504 |
| P(γ) ≥ 70% | 2 | 996 | 249 |
| 30% ≤ P(γ) < 70% AND 30% ≤ P(α) < 70% | 3 | 1117 | 1805 |
| 30% ≤ P(γ) < 70% AND P(α) < 30% | 4 | 784 | 1319 |
| P(γ) < 30% AND P(α) < 70% | 5 | 269 | 124 |
| Class No. | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| Class Name | α-rich | γ-rich | α + γ | β + γ | α + β + γ |
| DFBS | |||||
| P(α) | 86.6 ± 10.7 | 7.3 ± 9.8 | 48.5 ± 11.5 | 9.3 ± 10.0 | 29.3 ± 24.7 |
| P(β) | 0.5 ± 2.8 | 5.5 ± 8.7 | 3.9 ± 7.6 | 39.1 ± 15.1 | 53.1 ± 27.5 |
| P(γ) | 12.9 ± 10.5 | 87.2 ± 11.2 | 47.6 ± 10.9 | 51.7 ± 10.8 | 17.6 ± 9.5 |
| Domain Surface | |||||
| Q(α) | 79.7 ± 7.6 | 9.6 ± 9.9 | 46.7 ± 11.2 | 14.7 ± 9.6 | 53.1 ± 12.8 |
| Q(β) | 0.2 ± 0.9 | 0.9 ± 8.0 | 9.7 ± 9.0 | 33.0 ± 12.0 | 21.0 ± 12.5 |
| Q(γ) | 20.1 ± 7.5 | 82.4 ± 10.4 | 43.6 ± 8.3 | 52.3 ± 8.6 | 26.0 ± 2.9 |
3.2. Are Binding Site Surfaces Special?
| Domain Surface | DFBS Class | ||||
|---|---|---|---|---|---|
| α-rich | γ-rich | α + γ | β + γ | α + β + γ | |
| α-rich | 72.4 | 4.2 | 23.0 | 0.0 | 0.4 |
| γ-rich | 1.6 | 80.3 | 7.2 | 10.4 | 0.4 |
| α + γ | 22.0 | 19.4 | 42.7 | 9.8 | 6.1 |
| β + γ | 2.7 | 31.8 | 13.7 | 43.1 | 8.7 |
| α + β + γ | 26.6 | 4.8 | 25.0 | 10.5 | 33.1 |
| All domains | 20.9 | 24.9 | 27.9 | 19.6 | 6.7 |

3.3. Do DFIs have SSE Pairing Preferences?
| α-rich | γ-rich | α + γ | β + γ | α + β + γ | |
|---|---|---|---|---|---|
| α-rich | 227 (124) | 177 (320) | 334 (359) | 137 (252) | 40 (87) |
| 7.4% | 5.7% | 10.8% | 4.4% | 1.3% | |
| γ-rich | 251 (191) | 327 (429) | 356 (301) | 84 (103) | |
| 8.1% | 10.6% | 11.5% | 2.7% | ||
| α + γ | 333 (240) | 72 (337) | 307 (116) | ||
| 10.8% | 2.3% | 10.0% | |||
| β + γ | 312 (118) | 97 (81) | |||
| 10.1% | 3.1% | ||||
| α + β + γ | 30 (14) | ||||
| 1.0% |
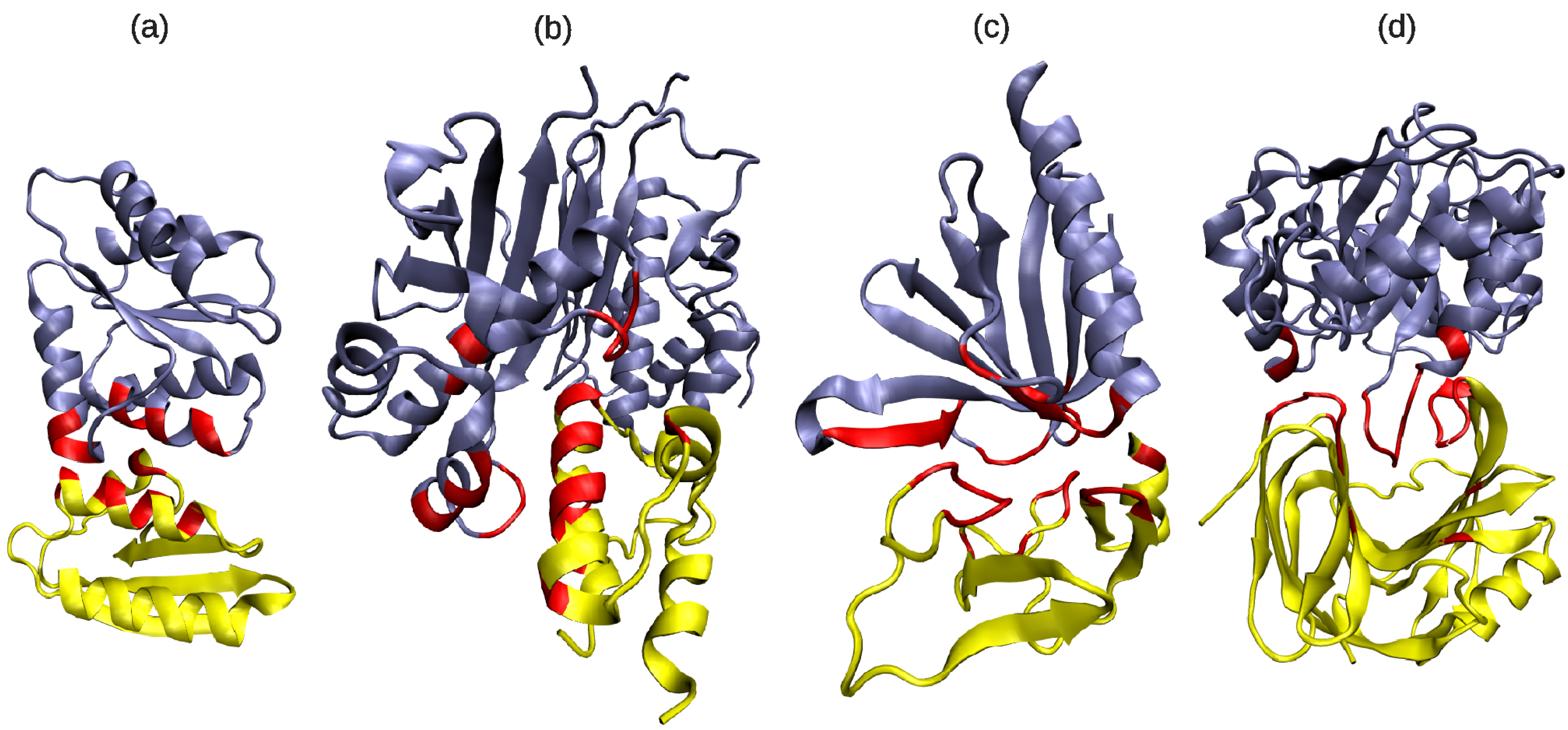
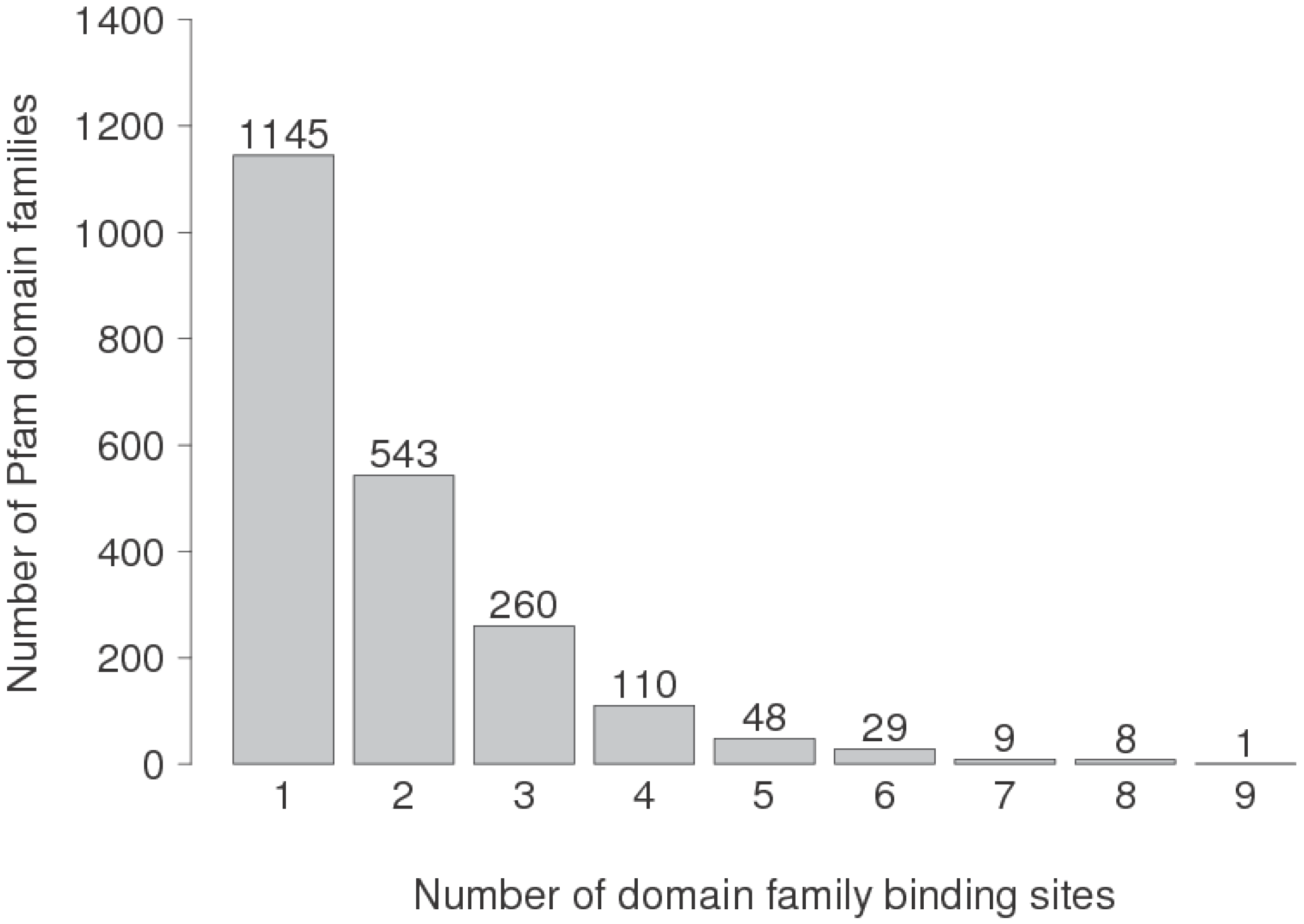
3.4. Are Multi-Partner Binding Sites Special?
| DFBSs | No. Partners | α-rich | γ-rich | α + γ | β + γ | α + β + γ |
|---|---|---|---|---|---|---|
| 3245 | 1 | 21.6 | 24.4 | 28.0 | 18.8 | 7.2 |
| 499 | 2 | 18.4 | 28.7 | 28.5 | 19.8 | 4.6 |
| 119 | 3 | 21.8 | 28.6 | 22.7 | 21.9 | 5.0 |
| 138 | >3 | 10.9 | 19.6 | 29.7 | 36.2 | 3.6 |
| 4001 | Any | 20.9 | 24.9 | 27.9 | 19.6 | 6.7 |
| DFBSs | No. Partners | Average SAS (Å2) | No. Residues |
|---|---|---|---|
| 3245 | 1 | 678 ± 496 | 12 ± 9 |
| 499 | 2 | 579 ± 429 | 10 ± 8 |
| 119 | 3 | 554 ± 482 | 10 ± 9 |
| 138 | ≥4 | 578 ± 334 | 11 ± 7 |
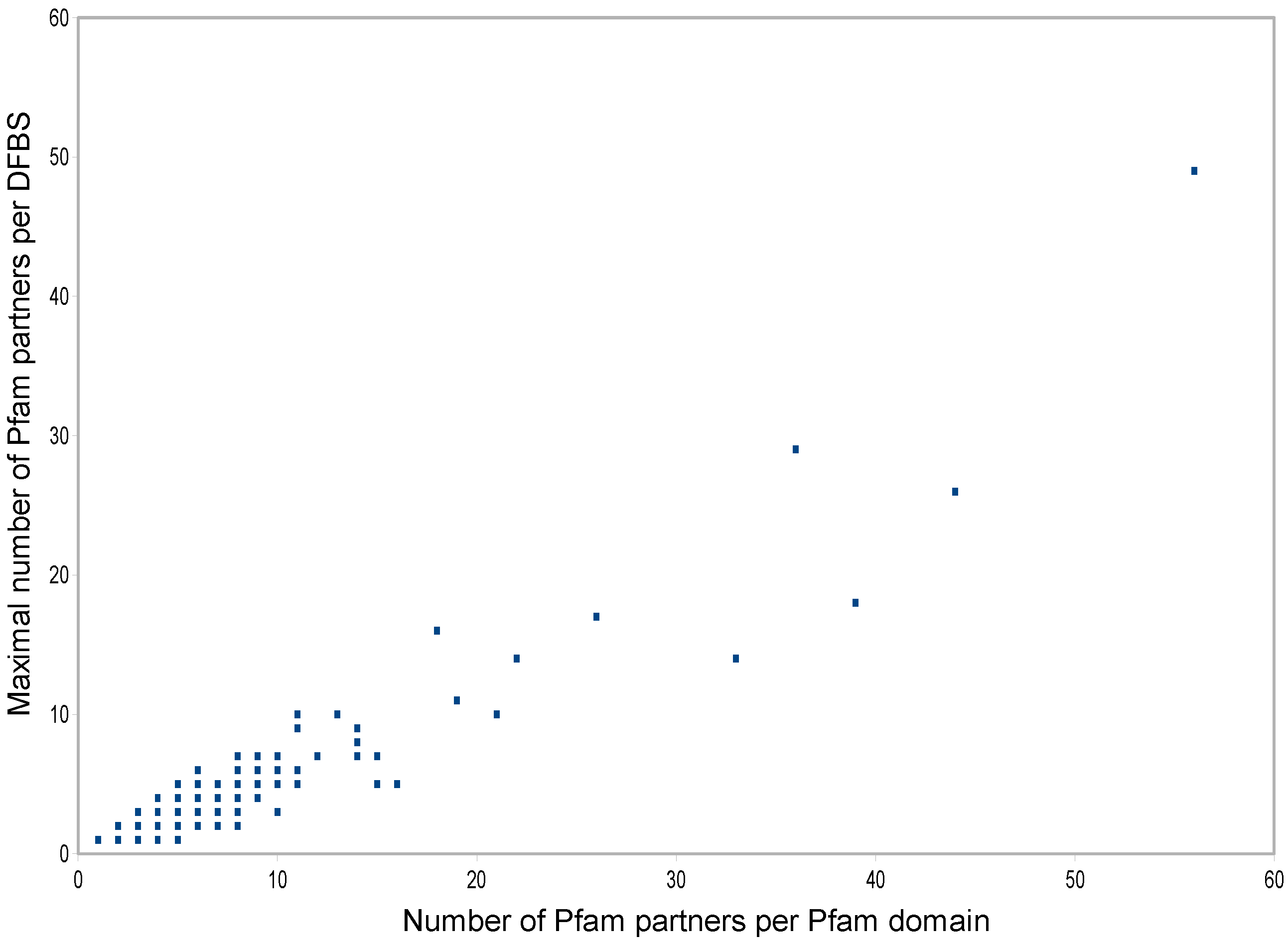
3.5. Distribution of DFBSs on Multi-Partner Pfam Domains

| Pfam Domain | DFBSs | Partners per DFBS | Total Partners | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Ubiquitin-conjugating enzyme | 6 | 2 | 4 | 11 | 5 | 3 | 1 | 19 | |||
| Ubiquitin | 6 | 3 | 29 | 3 | 1 | 1 | 6 | 36 | |||
| Ras | 7 | 49 | 3 | 3 | 1 | 3 | 1 | 1 | 56 | ||
| Trypsin | 7 | 26 | 7 | 13 | 7 | 4 | 4 | 2 | 44 | ||
| Immunoglobulin C1-set | 7 | 5 | 5 | 14 | 8 | 2 | 1 | 8 | 33 | ||
| Protein kinase domain | 8 | 16 | 18 | 6 | 1 | 1 | 1 | 3 | 1 | 39 | |
| Class I histocompatibility antigen | 8 | 1 | 5 | 5 | 2 | 1 | 1 | 1 | 1 | 10 | |
| RNA polymerase Rpb1 | 8 | 1 | 3 | 2 | 1 | 2 | 1 | 1 | 1 | 10 | |
| Actin | 9 | 3 | 10 | 4 | 2 | 2 | 1 | 1 | 1 | 1 | 21 |

4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ezkurdia, I.; Bartoli, L.; Fariselli, P.; Casadio, R.; Valencia, A.; Tress, M.L. Progress and challenges in predicting protein-protein interaction sites. Brief. Bioinform. 2009, 10, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M. The Protein Data Bank: A historical perspective. Acta Crystallogr. 2008, A38, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Mistry, J.; Tate, J.; Coggill, P.; Heger, A.; Pollington, J.E.; Gavin, O.L.; Gunasekaran, P.; Ceric, G.; Forslund, K.; et al. The Pfam protein families database. Nucleic Acids Res. 2010, 38, D211–D222. [Google Scholar] [CrossRef] [PubMed]
- Murzin, A.G.; Brenner, S.E.; Hubbard, T.; Chothia, C. SCOP—A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995, 247, 536–540. [Google Scholar] [CrossRef]
- Cuff, A.L.; Sillitoe, I.; Lewis, T.; Redfern, O.C.; Garratt, R.; Thornton, J.; Orengo, C.A. The CATH classification revisited-architectures reviewed and new ways to characterize structural divergence in superfamilies. Nucleic Acids Res. 2009, 37, D310–D314. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.; Kääriänen, S.; Rosentröm, P.; Schenkel, A. Seaching protein structure databases with DaliLite v.3. Bioinformatics 2008, 24, 2780–2781. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, B.A.; Panchenko, A.R.; Bryant, S.H. Finding biologically relevant protein domain interactions: Conserved binding mode analysis. Protein Sci. 2006, 15, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Keskin, O.; Nussinov, R. Similar binding Sites and different partners: Implications to shared proteins in cellular pathways. Structure 2007, 15, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Ghoorah, A.W.; Devignes, M.D.; Smail-Tabbone, M.; Ritchie, D.W. Spatial clustering of protein binding sites for template based protein docking. Bioinformatics 2011, 27, 2820–2827. [Google Scholar] [CrossRef] [PubMed]
- De Vries, S.J.; Bonvin, A.M.J.J. How proteins get in touch: Interface prediction in the study of biomolecular complexes. Curr. Protein Pept. Sci. 2008, 9, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Keskin, O.; Gursoy, A.; Ma, B.; Nussinov, R. Principles of protein-protein interactions: What are the preferred ways for proteins to interact? Chem. Rev. 2008, 108, 1225–1244. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Recio, J. Prediction of protein binding sites and hot spots. Wiley Interdiscip. Rev.-Comput. Mol. Sci. 2011, 1, 680–698. [Google Scholar] [CrossRef]
- Janin, J.; Chothia, C. The structure of protein-protein recognition sites. J. Biol. Chem. 1990, 265, 16027–16030. [Google Scholar] [PubMed]
- Jones, S.; Thornton, J.M. Principles of protein-protein interactions. Proc. Natl. Acad. Sci. USA 1996, 93, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Lo Conte, L.; Chothia, C.; Janin, J. The atomic structure of protein-protein recognition sites. J. Mol. Biol. 1999, 285, 2177–2198. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Tsai, C.J.; Nussinov, R. Hydrogen bonds and salt bridges across protein-protein interfaces. Protein Eng. 1997, 10, 999–1012. [Google Scholar] [CrossRef] [PubMed]
- Bogan, A.A.; Thorn, K.S. Anatomy of hot spots in protein interfaces. J. Mol. Biol. 1998, 280, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.Y.; Elkayam, T.; Wolfson, H.; Nussinov, R. Protein-protein interactions: Structurally conserved residues distinguish between binding sites and exposed protein surfaces. Proc. Natl. Acad. Sci. USA 2003, 100, 5772–5777. [Google Scholar] [CrossRef] [PubMed]
- Caffrey, D.R.; Somaroo, S.; Hughes, J.D.; Mintseris, J.; Huang, E.S. Are protein-protein interfaces more conserved in sequence than the rest of the protein surface? Protein Sci. 2004, 13, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Guharoy, M.; Chakrabarti, P. Secondary structure based analysis and classification of biological interfaces: Identification of binding motifs in protein-protein interactions. Bioinformatics 2007, 23, 1909–1918. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure—Pattern-Recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Patil, A.; Kinoshita, K.; Nakamura, H. Hub promiscuity in protein-protein interaction networks. Int. J. Mol. Sci. 2010, 11, 1930–1943. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.J.; Ma, B.; Nussinov, R. Protein-protein interaction networks: How can a hub protein bind so many different partners? Trends Biochem. Sci. 2009, 34, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Albert, R. Scale-free networks in cell biology. J. Cell Biol. 2005, 118, 4947–4957. [Google Scholar] [CrossRef] [PubMed]
- Nacher, J.; Hayashida, M.; Akutsu, T. Frequency of scale-free distribution in protein-protein interaction networks based on random selection of interacting domain pairs. BioSystems 2009, 95, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Ghoorah, A.W.; Devignes, M.D.; Smail-Tabbone, M.; Ritchie, D.W. KBDOCK 2013: A spatial classification of 3D protein domain family interactions. Nucleic Acids Res. 2014, D42, 389–295. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, D42, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Iype, L.; Jain, S.; Fagan, P.; Marvin, J.; et al. The protein data bank. Acta Crystallogr. Sect. D-Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef]
- Holm, L.; Sander, C. Removing near-neighbour redundancy from large protein sequence collections. Bioinformatics 1998, 14, 423–429. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghoorah, A.W.; Devignes, M.-D.; Alborzi, S.Z.; Smaïl-Tabbone, M.; Ritchie, D.W. A Structure-Based Classification and Analysis of Protein Domain Family Binding Sites and Their Interactions. Biology 2015, 4, 327-343. https://doi.org/10.3390/biology4020327
Ghoorah AW, Devignes M-D, Alborzi SZ, Smaïl-Tabbone M, Ritchie DW. A Structure-Based Classification and Analysis of Protein Domain Family Binding Sites and Their Interactions. Biology. 2015; 4(2):327-343. https://doi.org/10.3390/biology4020327
Chicago/Turabian StyleGhoorah, Anisah W., Marie-Dominique Devignes, Seyed Ziaeddin Alborzi, Malika Smaïl-Tabbone, and David W. Ritchie. 2015. "A Structure-Based Classification and Analysis of Protein Domain Family Binding Sites and Their Interactions" Biology 4, no. 2: 327-343. https://doi.org/10.3390/biology4020327
APA StyleGhoorah, A. W., Devignes, M.-D., Alborzi, S. Z., Smaïl-Tabbone, M., & Ritchie, D. W. (2015). A Structure-Based Classification and Analysis of Protein Domain Family Binding Sites and Their Interactions. Biology, 4(2), 327-343. https://doi.org/10.3390/biology4020327