
Protein Phosphatase-1 Regulates Expression of Neuregulin-1


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Sample Preparation for Mass Spectrometry Analysis
2.3. Mass Spectrometry and Data Analysis
2.4. Transient Transfections of Cell Cultures
2.5. Quantitative RT-PCR
2.6. Centrifugation in Glycerol Gradients
2.7. β-Galactosidase Activity Assay
2.8. CDK9 Kinase Activity Assay
2.9. Immunoprecipitation of RNAPII
2.10. Analysis of RNAPII Phosphorylation in Cultured Cells
2.11. Cell Growth Assays
2.12. Statistical Analysis
3. Results
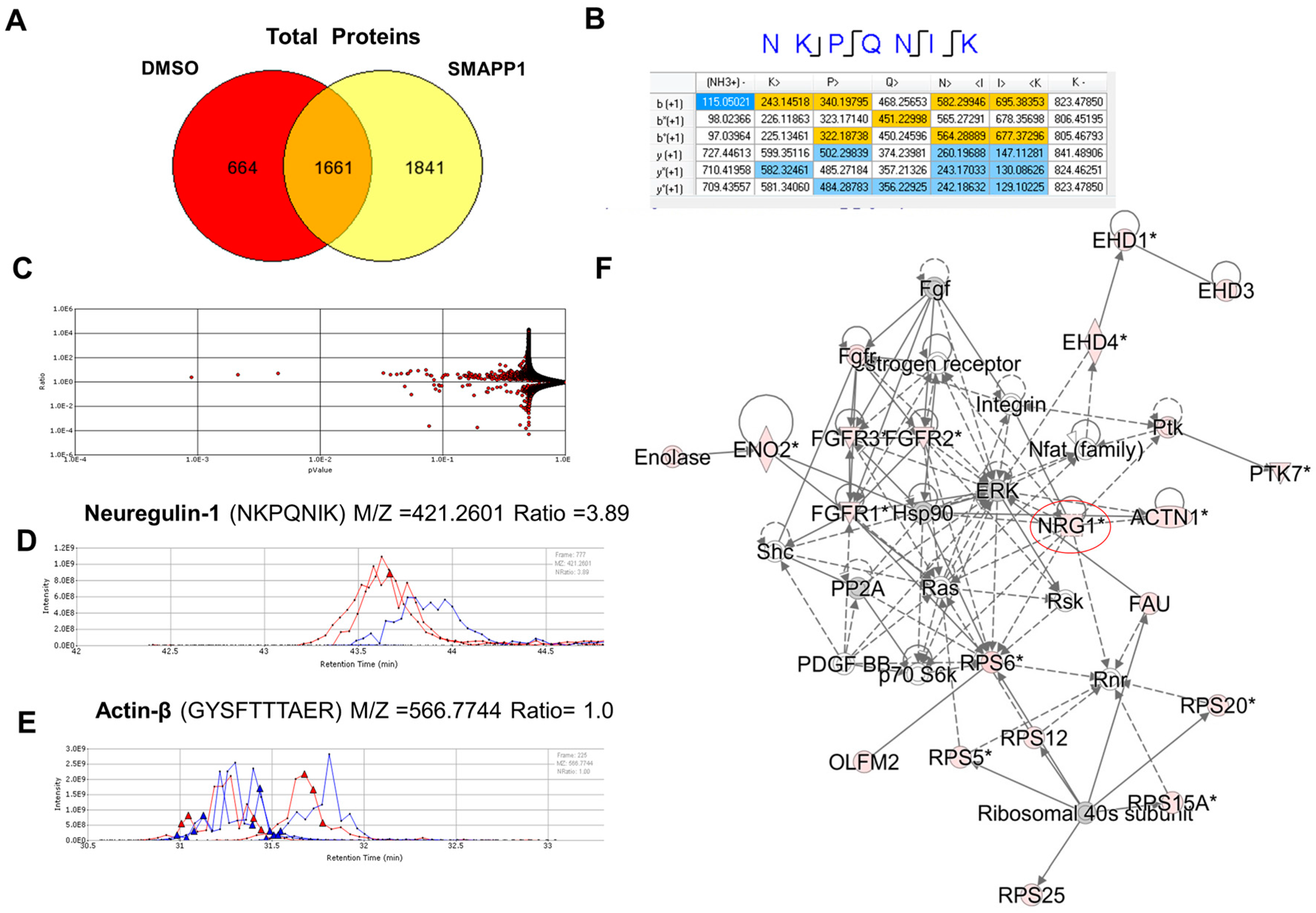
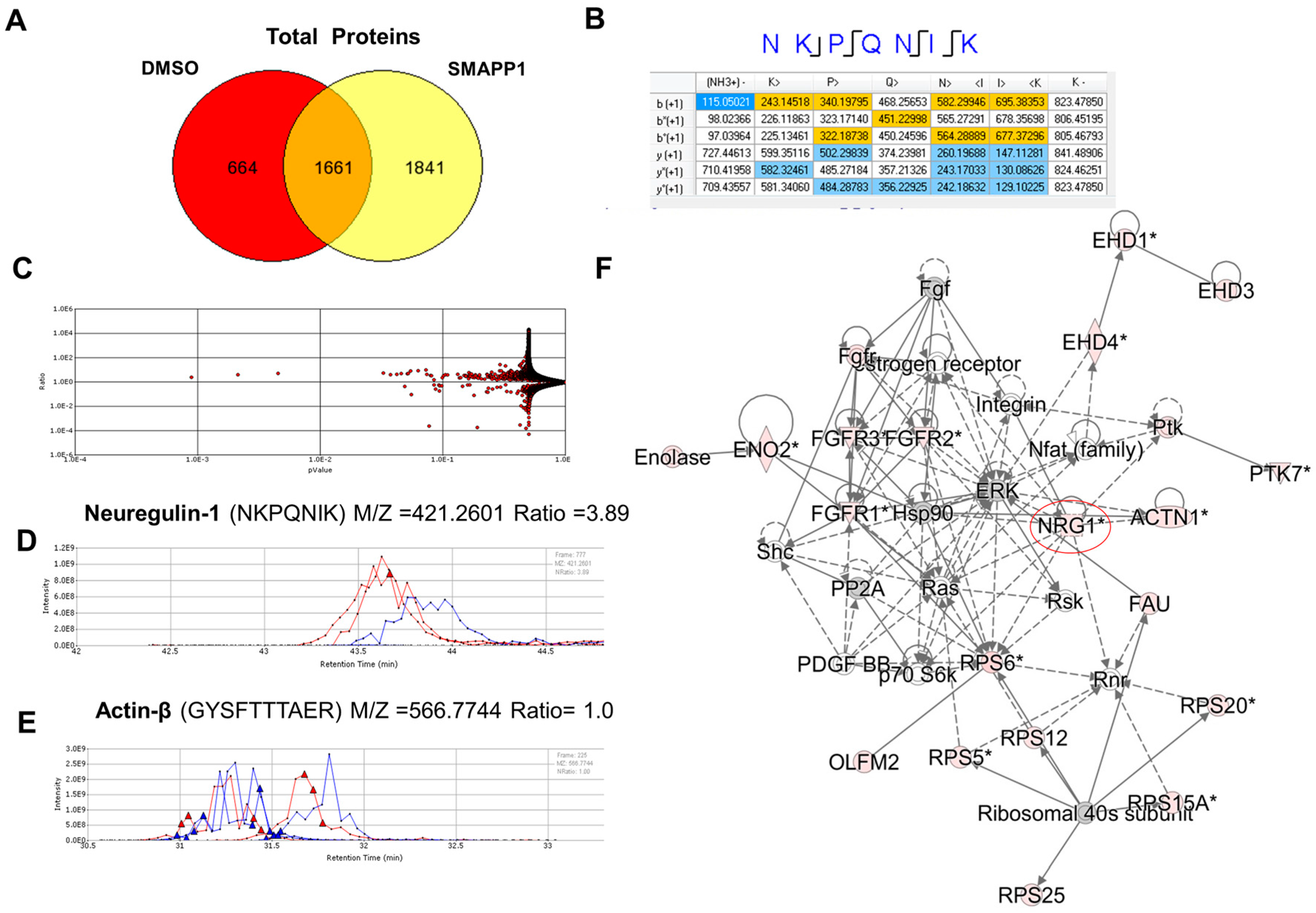
3.1. NRG-1 Expression Is Induced in Cells Treated with a Small Molecule Activator of PP1 (SMAPP1)
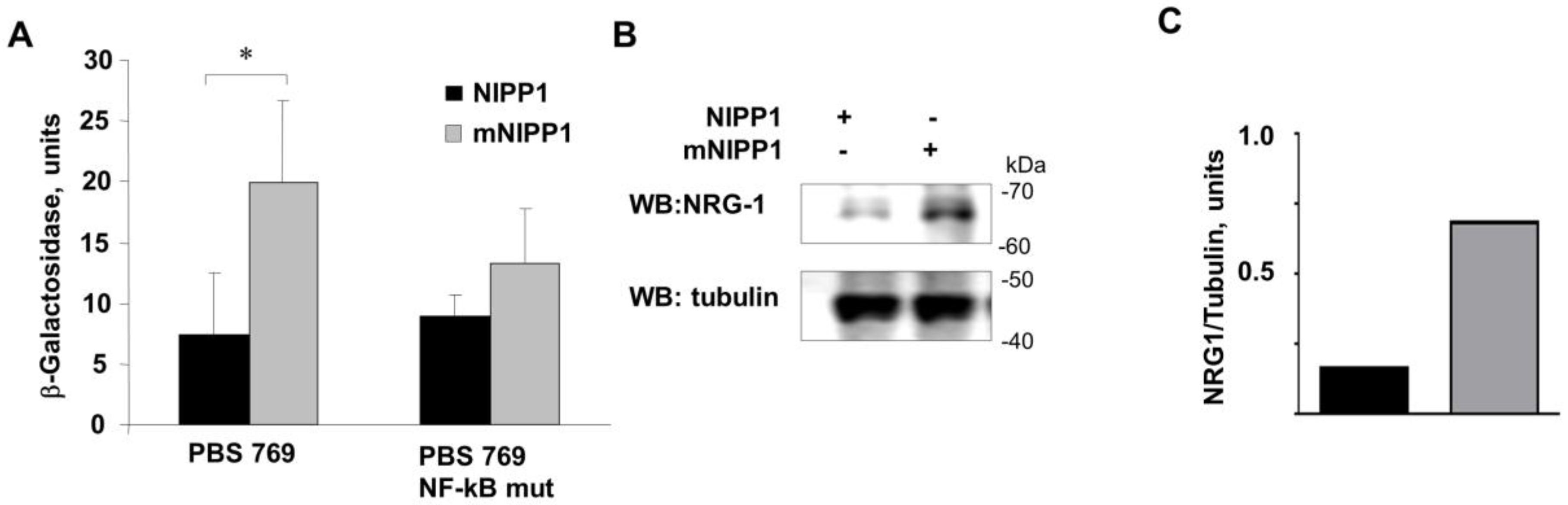
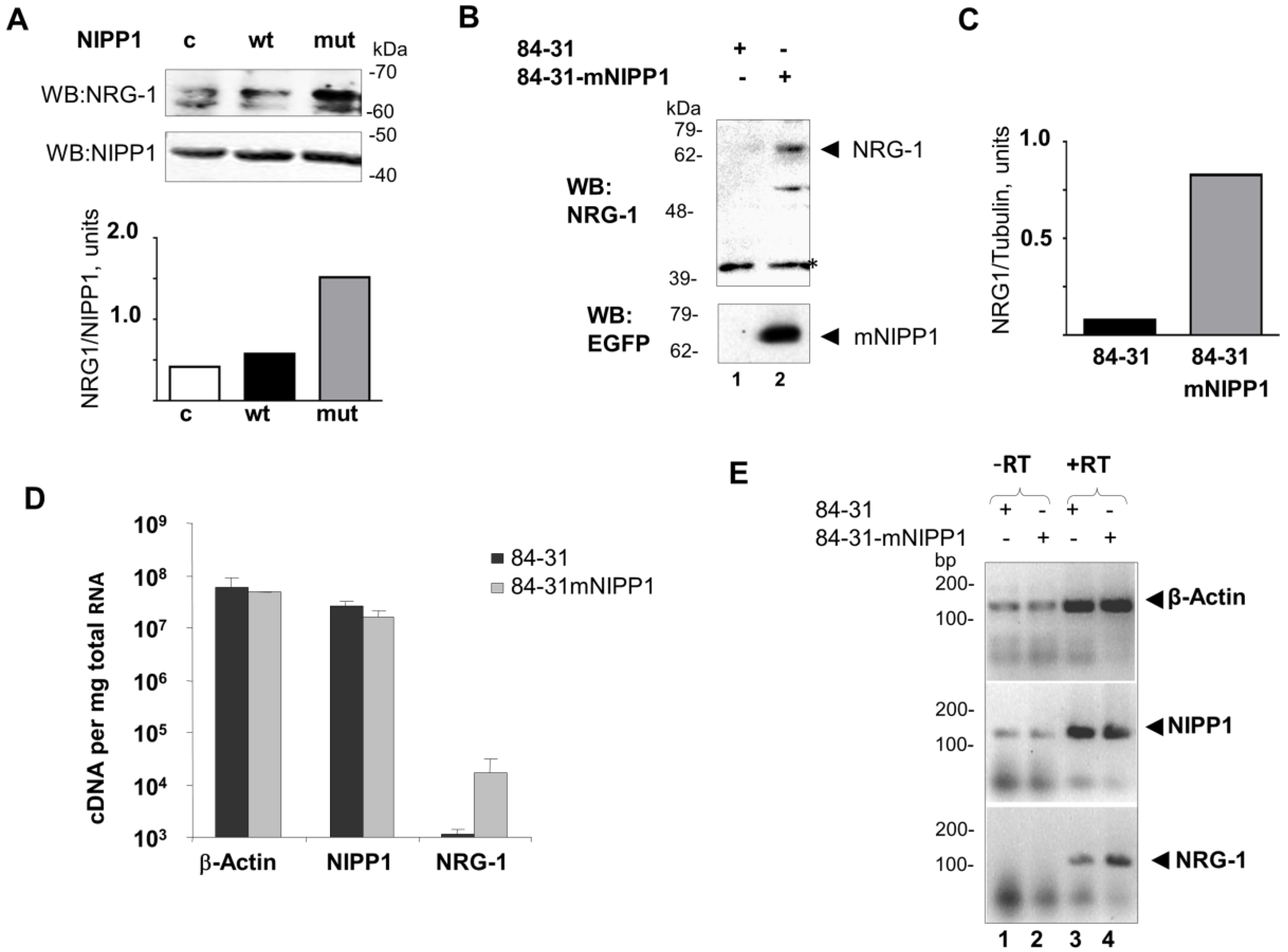
3.2. Overexpression of mNIPP1 Induces NRG-1 mRNA Expression
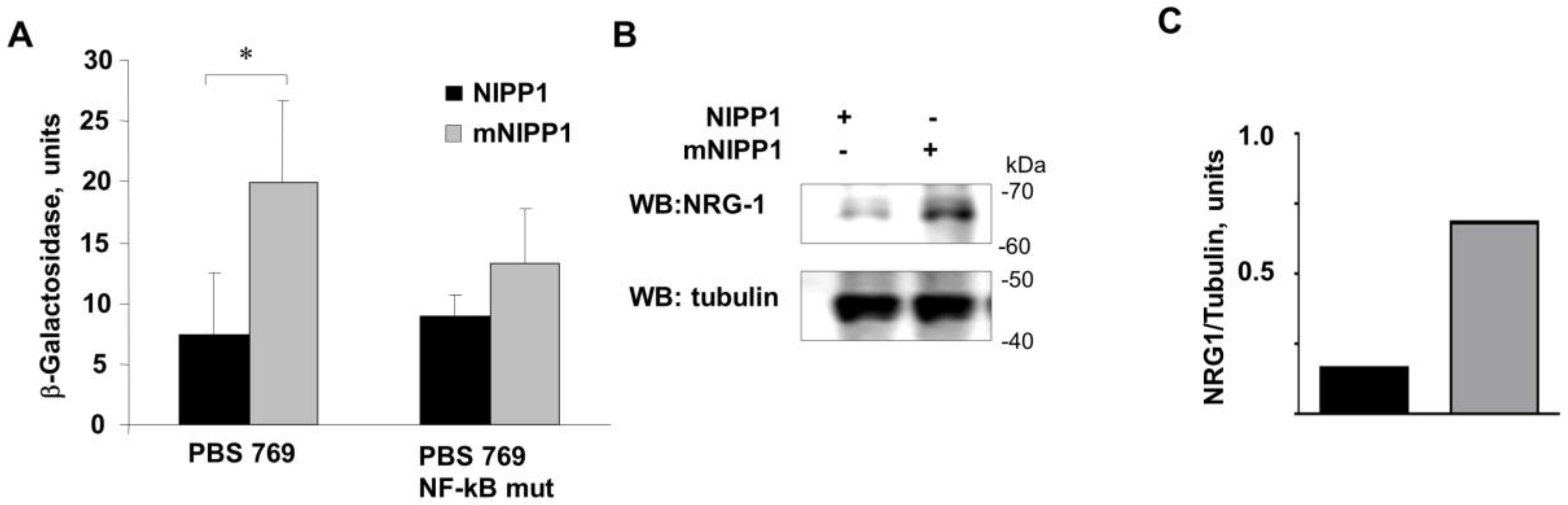
3.3. Transcription from Neuregulin-1 Promoter Was Induced by Expression of mNIPP1
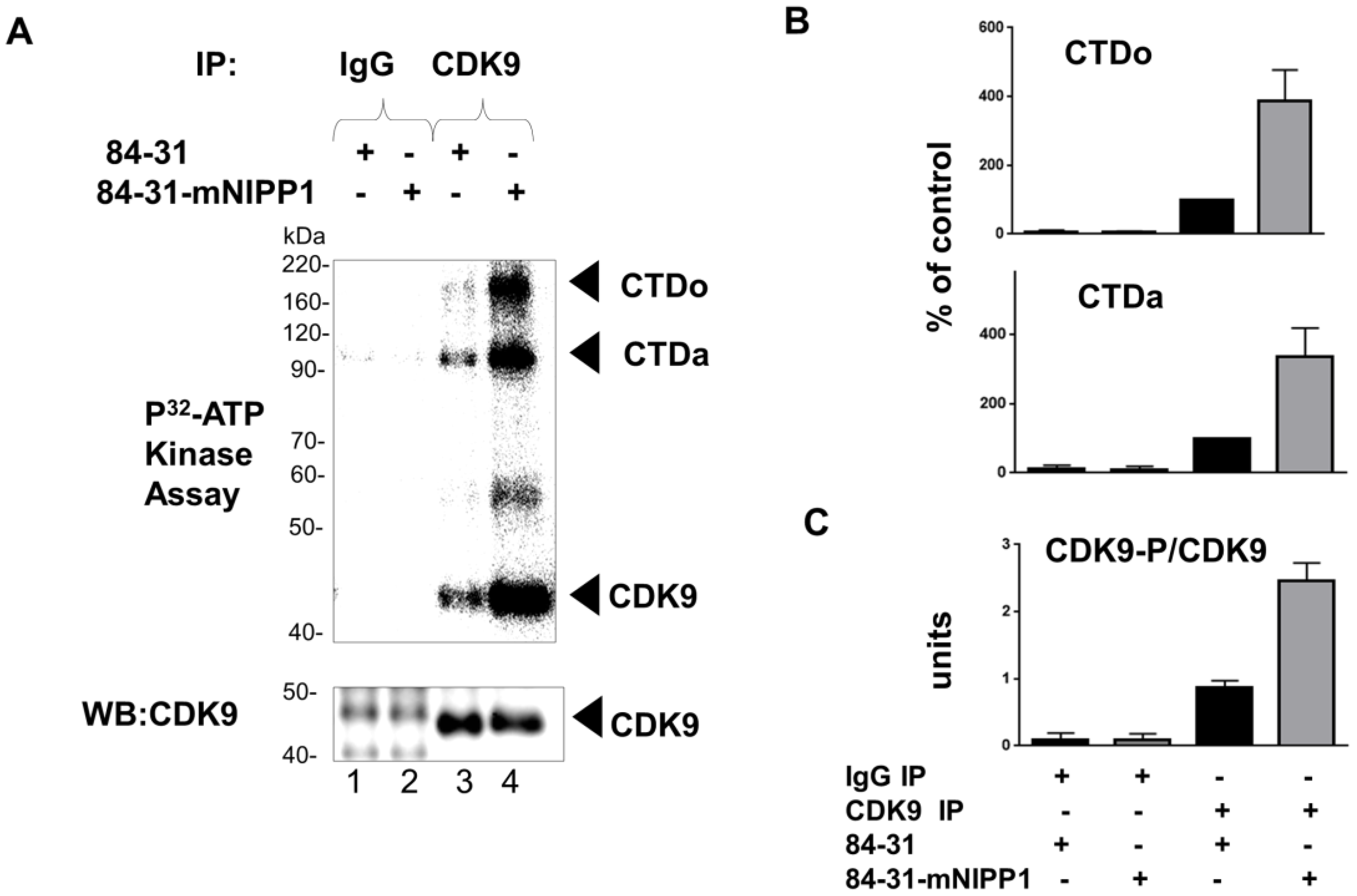
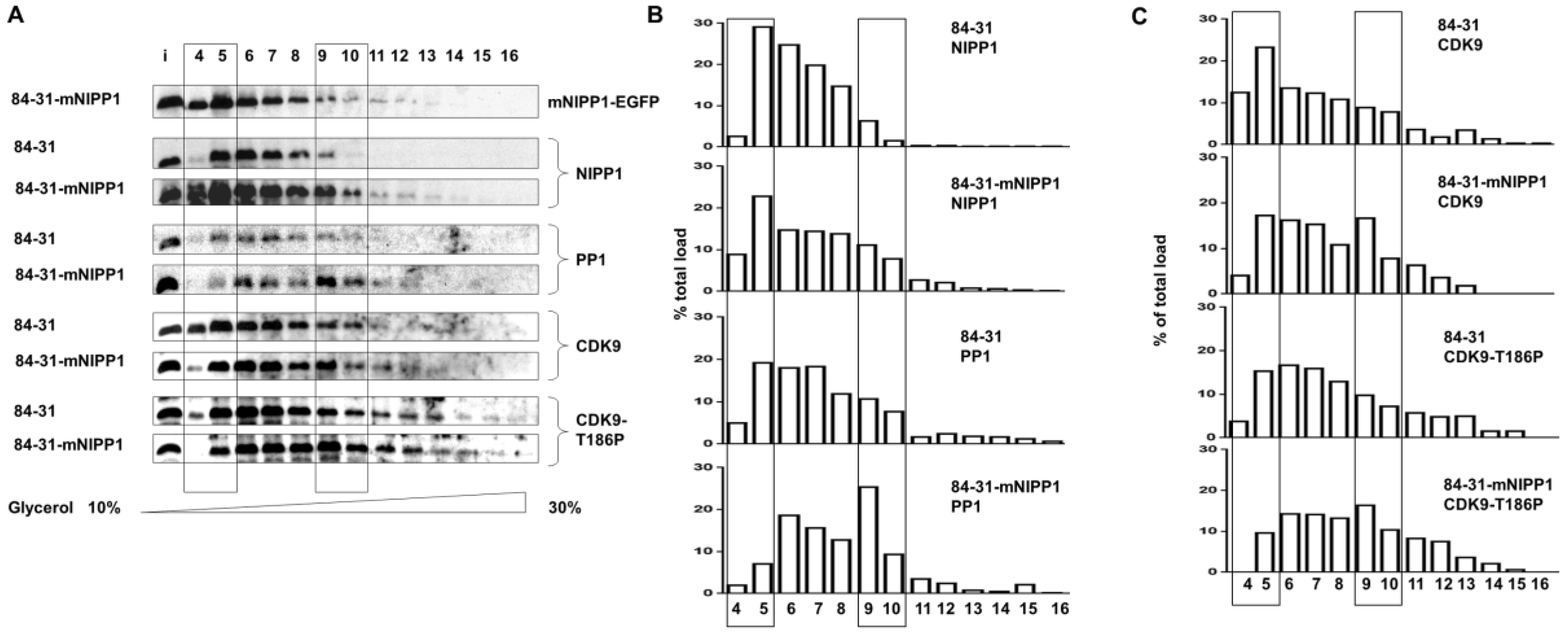
3.4. Expression of Mutant NIPP1 Redistributed PP1 and Increased Thr-186 Phosphorylation of CDK9
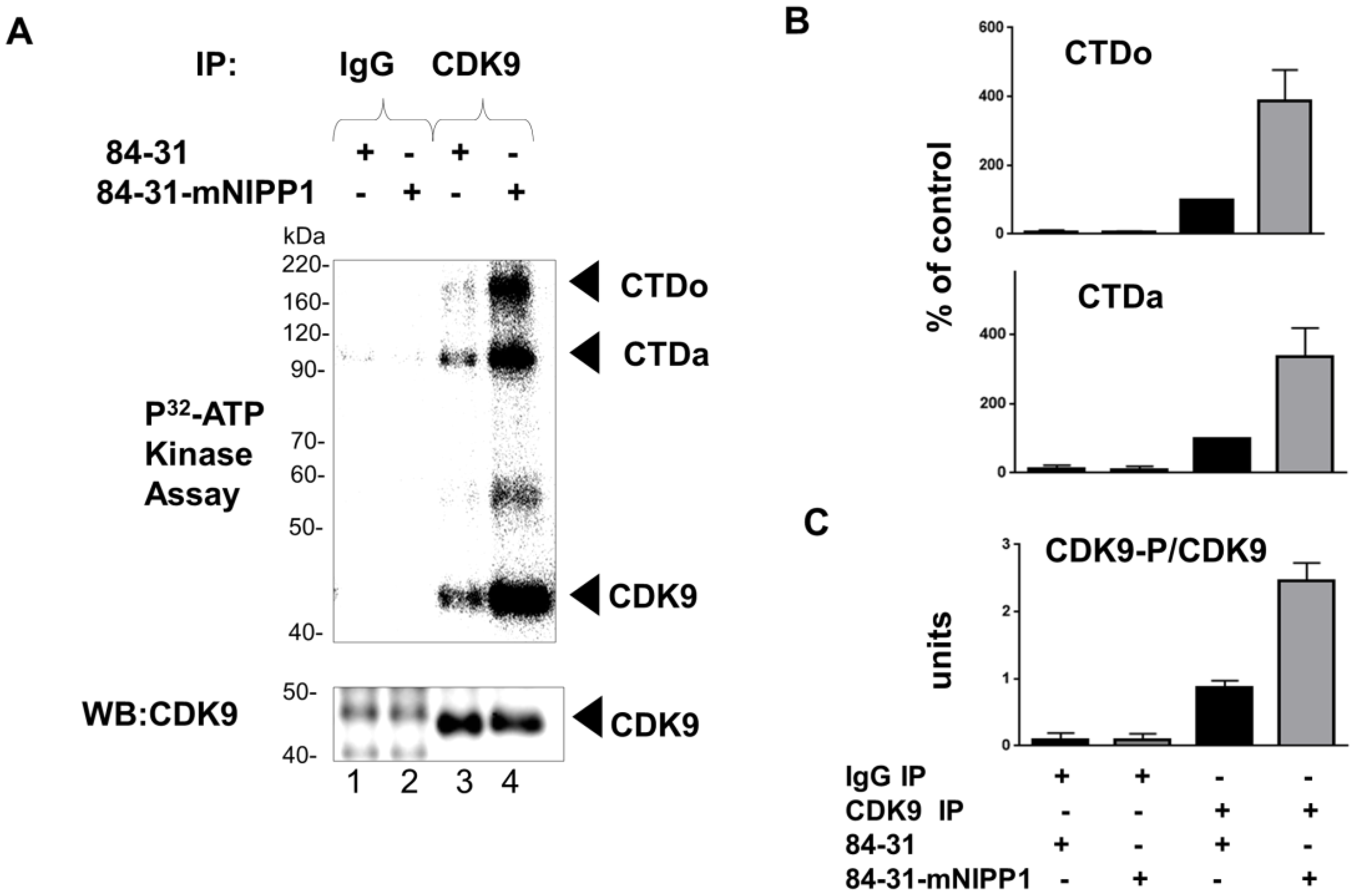
3.5. Expression of Mutant NIPP1 Induces Cellular Activity of CDK9
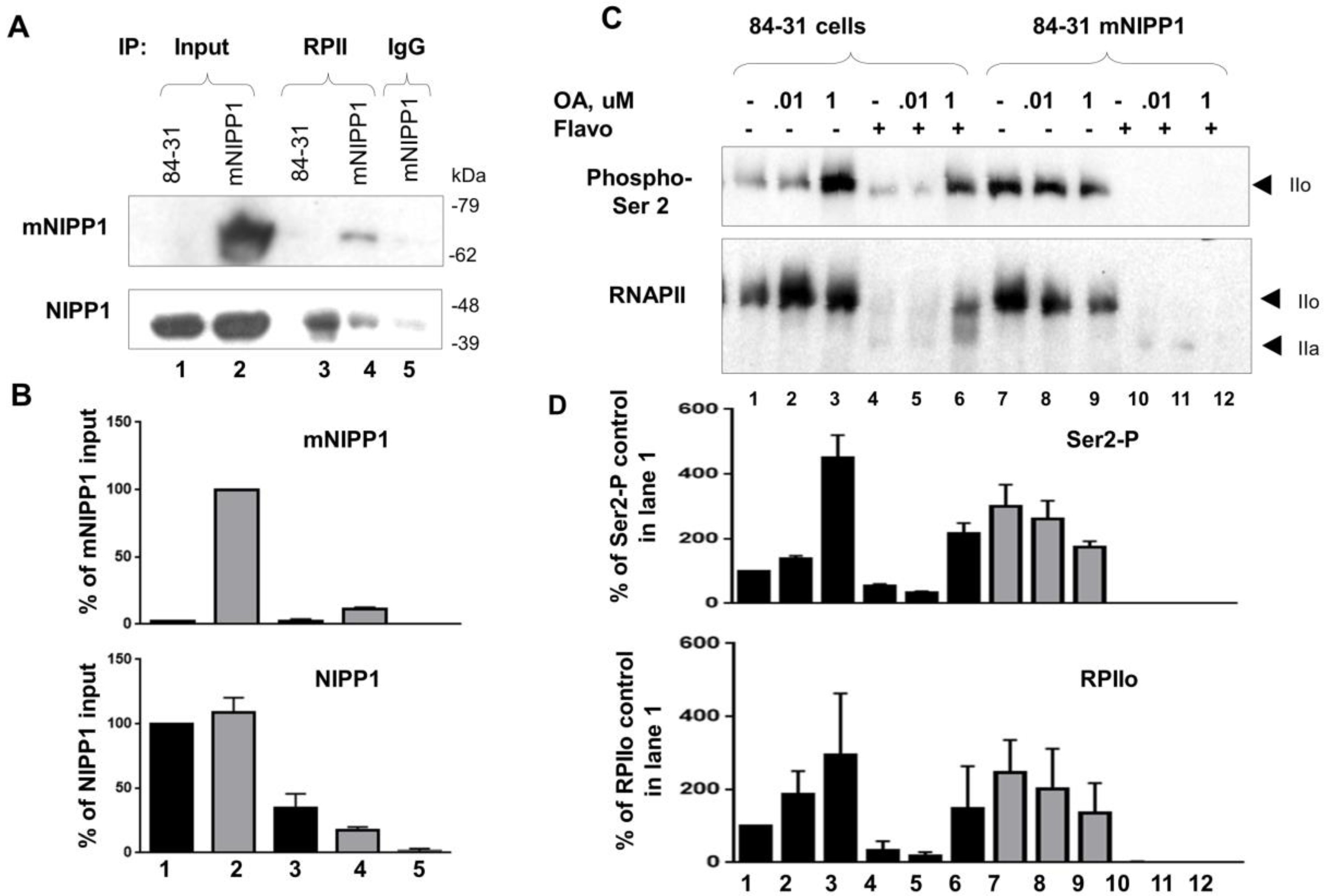
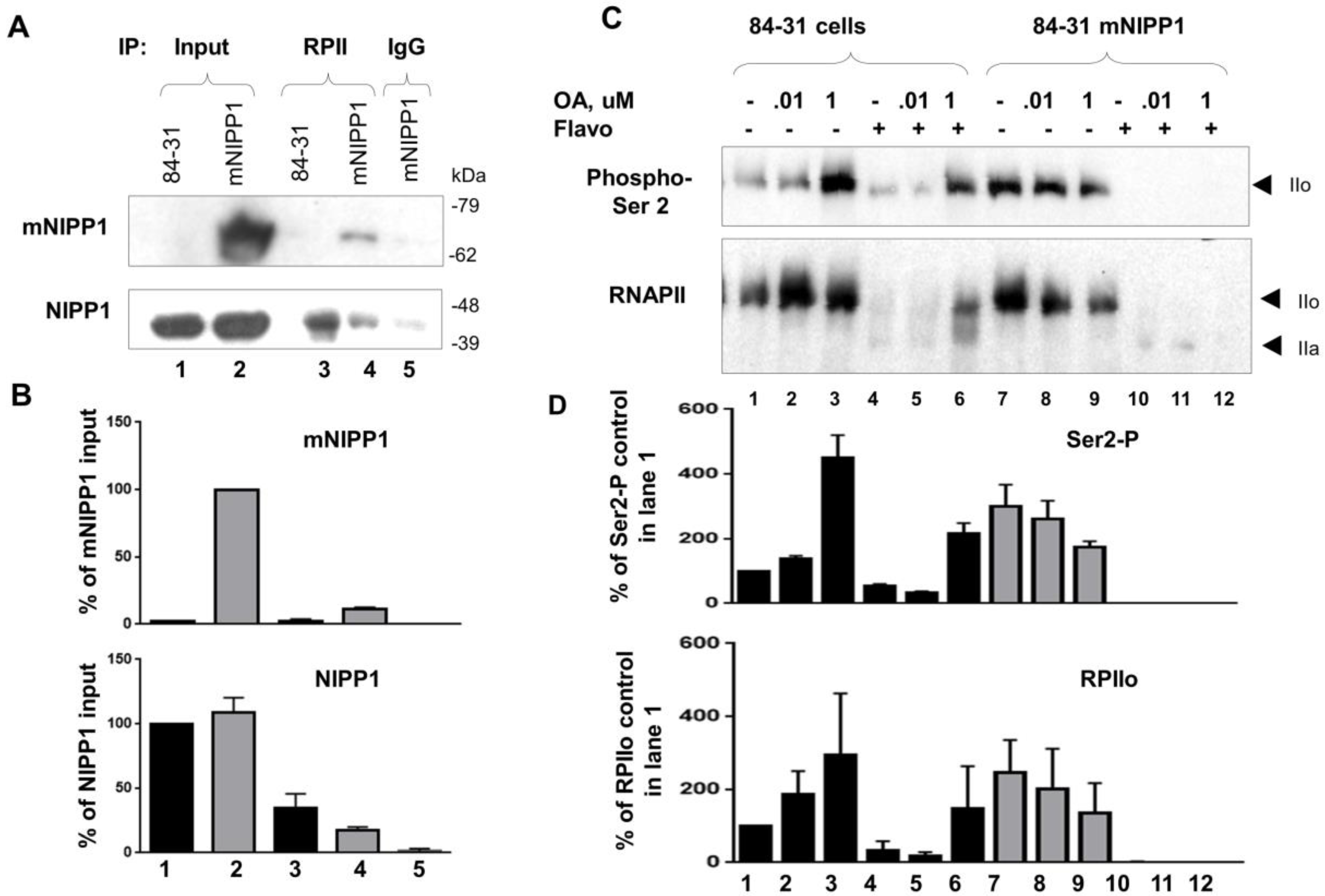
3.6. Mutant NIPP1 Associates with RNAPII
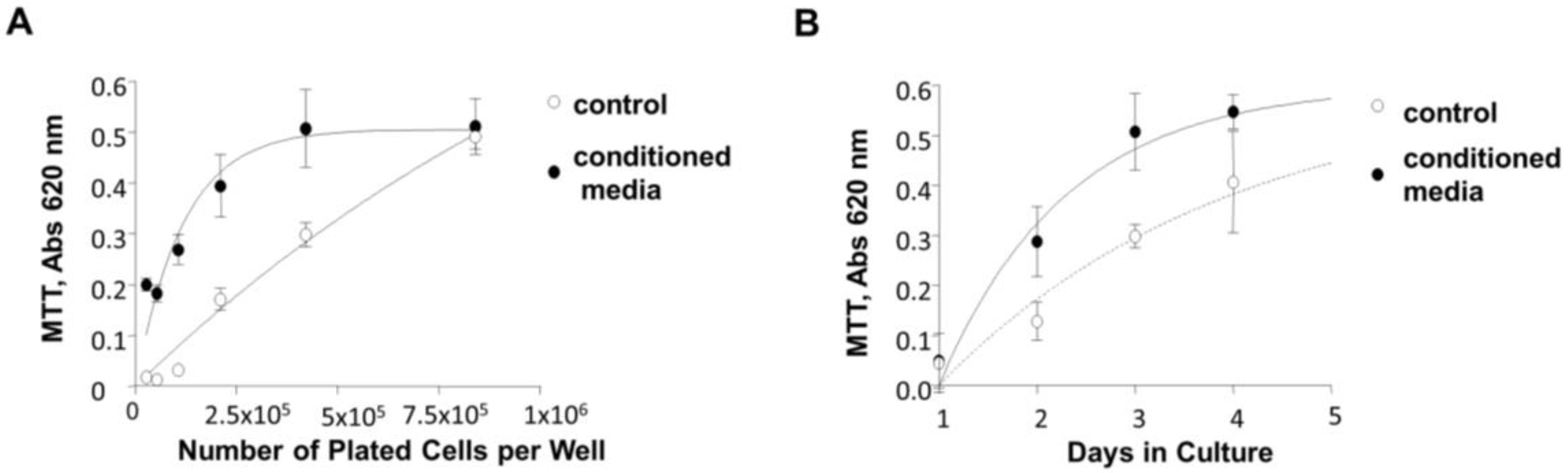
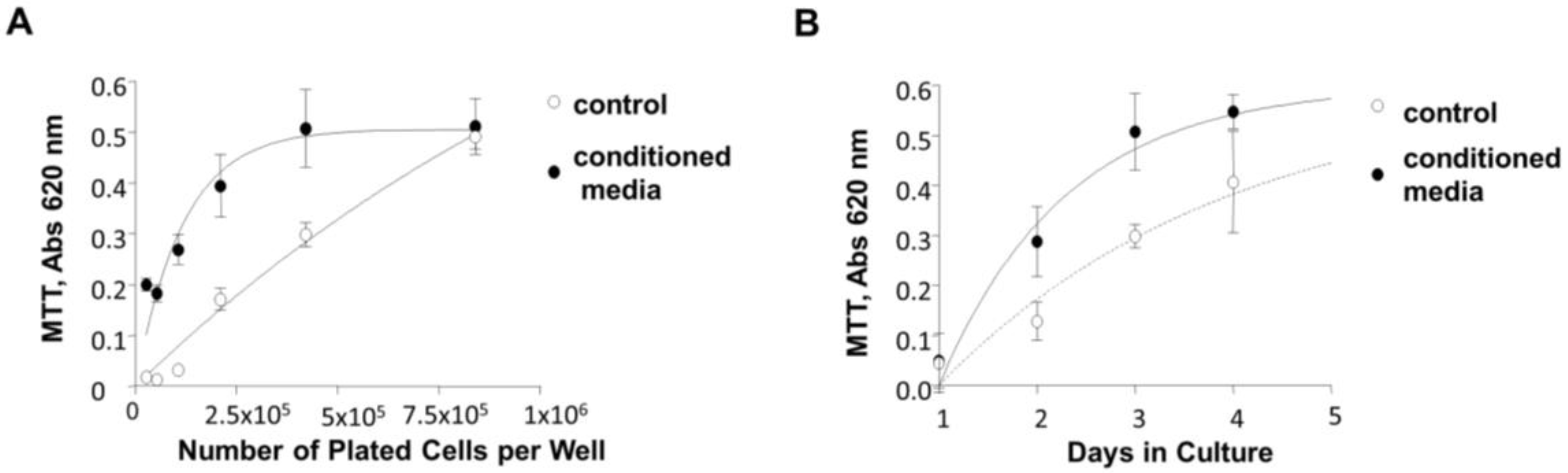
3.7. Induction of 84-31 Cell Growth in Media Conditioned with 84-31-mNIPP1 Cells
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bollen, M.; Peti, W.; Ragusa, M.J.; Beullens, M. The extended PP1 toolkit: Designed to create specificity. Trends Biochem. Sci. 2010, 35, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Verheyen, T.; Gornemann, J.; Verbinnen, I.; Boens, S.; Beullens, M.; Van Eynde, A.; Bollen, M. Genome-wide promoter binding profiling of protein phosphatase-1 and its major nuclear targeting subunits. Nucleic Acids Res. 2015, 43, 5771–5784. [Google Scholar] [CrossRef] [PubMed]
- Ilinykh, P.A.; Tigabu, B.; Ivanov, A.; Ammosova, T.; Obukhov, Y.; Garron, T.; Kumari, N.; Kovalskyy, D.; Platonov, M.O.; Naumchik, V.S.; et al. Role of protein phosphatase 1 in dephosphorylation of Ebola virus VP30 protein and its targeting for the inhibition of viral transcription. J. Biol. Chem. 2014, 289, 22723–22738. [Google Scholar] [CrossRef] [PubMed]
- Ammosova, T.; Yedavalli, V.R.; Niu, X.; Jerebtsova, M.; Van Eynde, A.; Beullens, M.; Bollen, M.; Jeang, K.T.; Nekhai, S. Expression of a protein phosphatase 1 inhibitor, cdNIPP1, increases CDK9 threonine 186 phosphorylation and inhibits HIV-1 transcription. J. Biol. Chem. 2011, 286, 3798–3804. [Google Scholar] [CrossRef] [PubMed]
- Ammosova, T.; Jerebtsova, M.; Beullens, M.; Lesage, B.; Jackson, A.; Kashanchi, F.; Southerland, W.; Gordeuk, V.R.; Bollen, M.; Nekhai, S. Nuclear targeting of protein phosphatase-1 by HIV-1 Tat protein. J. Biol. Chem. 2005, 280, 36364–36371. [Google Scholar] [CrossRef] [PubMed]
- Washington, K.; Ammosova, T.; Beullens, M.; Jerebtsova, M.; Kumar, A.; Bollen, M.; Nekhai, S. Protein phosphatase-1 dephosphorylates the C-terminal domain of RNA polymerase-II. J. Biol. Chem. 2002, 277, 40442–40448. [Google Scholar] [CrossRef] [PubMed]
- Ammosova, T.; Obukhov, Y.; Kotelkin, A.; Breuer, D.; Beullens, M.; Gordeuk, V.R.; Bollen, M.; Nekhai, S. Protein phosphatase-1 activates CDK9 by dephosphorylating Ser175. PLoS ONE 2011, 6, e18985. [Google Scholar] [CrossRef] [PubMed]
- Jerebtsova, M.; Klotchenko, S.A.; Artamonova, T.O.; Ammosova, T.; Washington, K.; Egorov, V.V.; Shaldzhyan, A.A.; Sergeeva, M.V.; Zatulovskiy, E.A.; Temkina, O.A.; et al. Mass spectrometry and biochemical analysis of RNA polymerase II: Targeting by protein phosphatase-1. Mol. Cell. Biochem. 2011, 347, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.; van Eynde, A.; Beullens, M.; Roy, N.; Thiel, G.; Stalmans, W.; Bollen, M. The protein phosphatase-1 (PP1) regulator, nuclear inhibitor of PP1 (NIPP1), interacts with the polycomb group protein, embryonic ectoderm development (EED), and functions as a transcriptional repressor. J. Biol. Chem. 2003, 278, 30677–30685. [Google Scholar] [CrossRef] [PubMed]
- Viré, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; Van Eynde, A.; Bernard, D.; Vanderwinden, J.M.; et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Van Dessel, N.; Boens, S.; Lesage, B.; Winkler, C.; Gornemann, J.; Van Eynde, A.; Bollen, M. Protein phosphatase PP1-NIPP1 activates mesenchymal genes in HeLa cells. FEBS Lett. 2015, 589, 1314–1321. [Google Scholar] [CrossRef] [PubMed]
- Ammosova, T.; Platonov, M.; Ivanov, A.; Kont, Y.S.; Kumari, N.; Kehn-Hall, K.; Jerebtsova, M.; Kulkarni, A.A.; Üren, A.; Kovalskyy, D.; et al. 1E7-03, a low MW compound targeting host protein phosphatase-1, inhibits HIV-1 transcription. Br. J. Pharmacol. 2014, 171, 5059–5075. [Google Scholar] [PubMed]
- Tyagi, M.; Iordanskiy, S.; Ammosova, T.; Kumari, N.; Smith, K.; Breuer, D.; Ilatovskiy, A.V.; Kont, Y.S.; Ivanov, A.; Üren, A.; et al. Reactivation of latent HIV-1 provirus via targeting protein phosphatase-1. Retrovirology 2015. [Google Scholar] [CrossRef] [PubMed]
- Vasti, C.; Hertig, C.M. Neuregulin-1/erbB activities with focus on the susceptibility of the heart to anthracyclines. World J. Cardiol. 2014, 6, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Falls, D.L. Neuregulins: Functions, forms, and signaling strategies. Exp. Cell Res. 2003, 284, 14–30. [Google Scholar] [CrossRef]
- Flågeng, M.H.; Larionov, A.; Geisler, J.; Knappskog, S.; Prestvik, W.S.; Bjørkøy, G.; Lilleng, P.K.; Dixon, J.M.; Miller, W.R.; Lønning, P.E.; et al. Treatment with aromatase inhibitors stimulates the expression of Epidermal growth factor receptor-1 and Neuregulin 1 in ER positive/HER-2/neu non-amplified primary breast cancers. J. Steroid Biochem. Mol. Biol. 2016, in press. [Google Scholar]
- Cai, M.X.; Shi, X.C.; Chen, T.; Tan, Z.N.; Lin, Q.Q.; Du, S.J.; Tian, Z.J. Exercise training activates neuregulin 1/ErbB signaling and promotes cardiac repair in a rat myocardial infarction model. Life Sci. 2016, 149, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Dang, R.; Guo, Y.; Cai, H.; Yang, R.; Liang, D.; Lv, C.; Jiang, P. Effects of prolonged antipsychotic administration on neuregulin-1/ErbB signaling in rat prefrontal cortex and myocardium: Implications for the therapeutic action and cardiac adverse effect. J. Toxicol. Sci. 2016, 41, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Dang, R.; Guo, Y.; Zhu, Y.; Yang, R.; Cai, H.; Jiang, P. Chronic administration of calcitriol enhanced neuregulin-1/ErbB signaling in rat myocardium. Die Pharm. 2016, 71, 192–195. [Google Scholar]
- Frensing, T.; Kaltschmidt, C.; Schmitt-John, T. Characterization of a neuregulin-1 gene promoter: Positive regulation of type I isoforms by NF-kappaB. Biochim. Biophys. Acta 2008, 1779, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Ammosova, T.; Jerebtsova, M.; Beullens, M.; Voloshin, Y.; Ray, P.E.; Kumar, A.; Bollen, M.; Nekhai, S. Nuclear protein phosphatase-1 regulates HIV-1 transcription. J. Biol. Chem. 2003, 278, 32189–32194. [Google Scholar] [CrossRef] [PubMed]
- Beullens, M.; Vulsteke, V.; Van Eynde, A.; Jagiello, I.; Stalmans, W.; Bollen, M. The C-terminus of NIPP1 (nuclear inhibitor of protein phosphatase-1) contains a novel binding site for protein phosphatase-1 that is controlled by tyrosine phosphorylation and RNA binding. Biochem J. 2000, 352, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Lesage, B.; Beullens, M.; Nuytten, M.; Van Eynde, A.; Keppens, S.; Himpens, B.; Bollen, M. Interactor-mediated nuclear translocation and retention of protein phosphatase-1. J. Biol. Chem. 2004, 279, 55978–55984. [Google Scholar] [CrossRef] [PubMed]
- Fisher, K.J.; Gao, G.P.; Weitzman, M.D.; DeMatteo, R.; Burda, J.F.; Wilson, J.M. Transduction with recombinant adeno-associated virus for gene therapy is limited by leading-strand synthesis. J. Virol. 1996, 70, 520–532. [Google Scholar] [PubMed]
- Ammosova, T.; Platonov, M.; Yedavalli, V.R.; Obukhov, Y.; Gordeuk, V.R.; Jeang, K.T.; Kovalskyy, D.; Nekhai, S. Small molecules targeted to a non-catalytic “RVxF” binding site of protein phosphatase-1 inhibit HIV-1. PLoS ONE 2012, 7, e39481. [Google Scholar] [CrossRef] [PubMed]
- Peterlin, B.M.; Price, D.H. Controlling the elongation phase of transcription with P-TEFb. Mol. Cell 2006, 23, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Nekhai, S.; Petukhov, M.; Breuer, D. Regulation of CDK9 activity by phosphorylation and dephosphorylation. BioMed Res. Int. 2014. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Price, J.P.; Byers, S.A.; Cheng, D.; Peng, J.; Price, D.H. Analysis of the large inactive P-TEFb complex indicates that it contains one 7SK molecule, a dimer of HEXIM1 or HEXIM2, and two P-TEFb molecules containing Cdk9 phosphorylated at threonine 186. J. Biol. Chem. 2005, 280, 28819–28826. [Google Scholar] [CrossRef] [PubMed]
- Ammosova, T.; Washington, K.; Debebe, Z.; Brady, J.; Nekhai, S. Dephosphorylation of CDK9 by protein phosphatase 2A and protein phosphatase-1 in Tat-activated HIV-1 transcription. Retrovirology 2005. [Google Scholar] [CrossRef] [PubMed]
- Chao, S.H.; Price, D.H. Flavopiridol inactivates P-TEFb and blocks most RNA polymerase II transcription in vivo. J. Biol. Chem. 2001, 276, 31793–31799. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kern, J.A. Neuregulin-1 activates the JAK-STAT pathway and regulates lung epithelial cell proliferation. Am. J. Respir. Cell Mol. Biol. 2002, 27, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ford, B.D.; Mann, M.A.; Fischbach, G.D. Neuregulin-1 increases the proliferation of neuronal progenitors from embryonic neural stem cells. Dev. Biol. 2005, 283, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Nekhai, S.; Howard University, Washington, DC, USA. Unpublished work. 2016.
- Stefansson, H.; Steinthorsdottir, V.; Thorgeirsson, T.E.; Gulcher, J.R.; Stefansson, K. Neuregulin 1 and schizophrenia. Ann. Med. 2004, 36, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Lupu, R.; Cardillo, M.; Cho, C.; Harris, L.; Hijazi, M.; Perez, C.; Rosenberg, K.; Yang, D.; Tang, C. The significance of heregulin in breast cancer tumor progression and drug resistance. Breast Cancer Res. Treat. 1996, 38, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Rupert, C.E.; Coulombe, K.L. The roles of neuregulin-1 in cardiac development, homeostasis, and disease. Biomark. Insights 2015, 10, 1–9. [Google Scholar] [PubMed]
- Zhang, Y.; Liao, M.; Dufau, M.L. Unlocking repression of the human luteinizing hormone receptor gene by trichostatin A-induced cell-specific phosphatase release. J. Biol. Chem. 2008, 283, 24039–24046. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.T.; Kiss, T.; Michels, A.A.; Bensaude, O. 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes. Nature 2001, 414, 322–325. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhu, Q.; Luo, K.; Zhou, Q. The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature 2001, 414, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Yik, J.H.; Chen, R.; Nishimura, R.; Jennings, J.L.; Link, A.J.; Zhou, Q. Inhibition of P-TEFb (CDK9/Cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA. Mol. Cell 2003, 12, 971–982. [Google Scholar] [CrossRef]
- Michels, A.A.; Nguyen, V.T.; Fraldi, A.; Labas, V.; Edwards, M.; Bonnet, F.; Lania, L.; Bensaude, O. MAQ1 and 7SK RNA interact with CDK9/cyclin T complexes in a transcription-dependent manner. Mol. Cell. Biol. 2003, 23, 4859–4869. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Yang, Z.; Zhou, Q. Phosphorylated positive transcription elongation factor b (P-TEFb) is tagged for inhibition through association with 7SK snRNA. J. Biol. Chem. 2004, 279, 4153–4160. [Google Scholar] [CrossRef] [PubMed]
- Larochelle, S.; Chen, J.; Knights, R.; Pandur, J.; Morcillo, P.; Erdjument-Bromage, H.; Tempst, P.; Suter, B.; Fisher, R.P. T-loop phosphorylation stabilizes the CDK7-cyclin H-MAT1 complex in vivo and regulates its CTD kinase activity. Embo J. 2001, 20, 3749–3759. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Liu, M.; Li, H.; Xue, Y.; Ramey, W.N.; He, N.; Ai, N.; Luo, H.; Zhu, Y.; Zhou, N.; et al. PP2B and PP1alpha cooperatively disrupt 7SK snRNP to release P-TEFb for transcription in response to Ca2+ signaling. Genes Dev. 2008, 22, 1356–1368. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.P. The CDK Network: Linking Cycles of Cell Division and Gene Expression. Genes Cancer 2012, 3, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Yeo, M.; Lee, S.K.; Lee, B.; Ruiz, E.C.; Pfaff, S.L.; Gill, G.N. Small CTD phosphatases function in silencing neuronal gene expression. Science 2005, 307, 596–600. [Google Scholar] [CrossRef] [PubMed]
- Vaquero, J.; Gutierrez, M.; Villalobo, A.; Rey, J.A. Molecular analysis of the erbB gene family calmodulin-binding and calmodulin-like domains in astrocytic gliomas. Int. J. Oncol. 2004, 25, 1489–1494. [Google Scholar]
- Ozaki, M.; Itoh, K.; Miyakawa, Y.; Kishida, H.; Hashikawa, T. Protein processing and releases of neuregulin-1 are regulated in an activity-dependent manner. J. Neurochem. 2004, 91, 176–188. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ammosova, T.; Washington, K.; Rotimi, J.; Kumari, N.; Smith, K.A.; Niu, X.; Jerebtsova, M.; Nekhai, S. Protein Phosphatase-1 Regulates Expression of Neuregulin-1. Biology 2016, 5, 49. https://doi.org/10.3390/biology5040049
Ammosova T, Washington K, Rotimi J, Kumari N, Smith KA, Niu X, Jerebtsova M, Nekhai S. Protein Phosphatase-1 Regulates Expression of Neuregulin-1. Biology. 2016; 5(4):49. https://doi.org/10.3390/biology5040049
Chicago/Turabian StyleAmmosova, Tatiana, Kareem Washington, Jamie Rotimi, Namita Kumari, Kahli A. Smith, Xiaomei Niu, Marina Jerebtsova, and Sergei Nekhai. 2016. "Protein Phosphatase-1 Regulates Expression of Neuregulin-1" Biology 5, no. 4: 49. https://doi.org/10.3390/biology5040049
APA StyleAmmosova, T., Washington, K., Rotimi, J., Kumari, N., Smith, K. A., Niu, X., Jerebtsova, M., & Nekhai, S. (2016). Protein Phosphatase-1 Regulates Expression of Neuregulin-1. Biology, 5(4), 49. https://doi.org/10.3390/biology5040049