Characterization of Plastidial and Nuclear SSR Markers for Understanding Invasion Histories and Genetic Diversity of Schinus molle L.

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
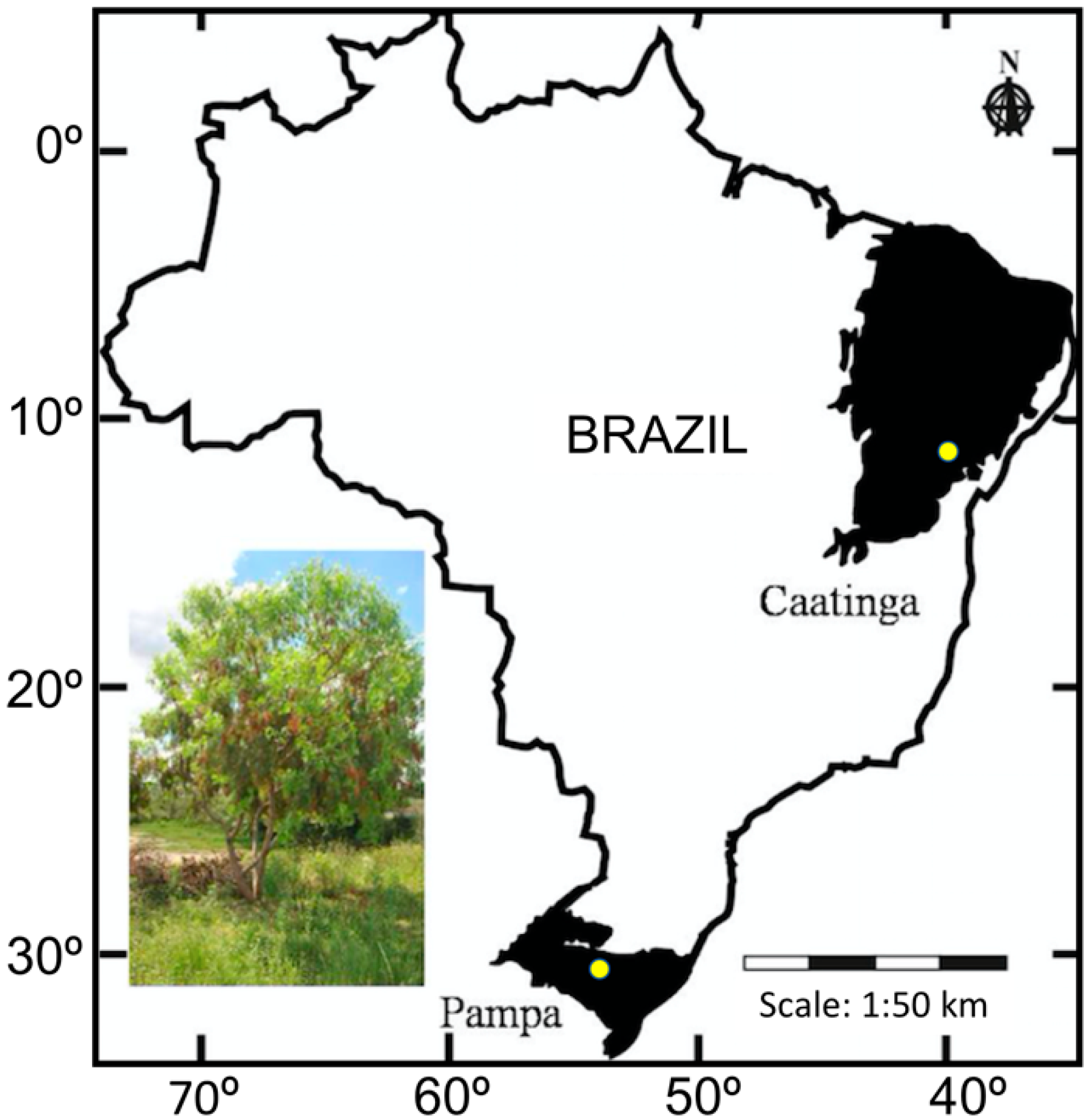
2.1. Sample Strategies and DNA Extraction
2.2. NGS Sequencing and de novo Assembly
2.3. Discovery and Characterization of SSR Markers
3. Results
3.1. Sequencing Output and SSR Discovery
3.2. Characterization of SSR Markers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Milton, S.J.; Wilson, J.R.U.; Richardson, D.M.; Seymour, C.L.; Dean, W.R.J.; Iponga, D.M.; Procheş, Ş. Invasive Alien Plants Infiltrate Bird-Mediated Shrub Nucleation Processes in Arid Savanna. J. Ecol. 2007, 95, 648–661. [Google Scholar] [CrossRef]
- Iponga, D.M. Seed Set of the Invasive Tree Schinus molle (Anacardiaceae) in Semi-Arid Savanna, South Africa: The Role of Pollinators and Selfing. J. Arid Environ. 2010, 74, 414–416. [Google Scholar] [CrossRef]
- Dikshit, A.; Naqvi, A.; Husain, A. Schinus molle: A New Source of Natural Fungitoxicant. Appl. Environ. Microbiol. 1986, 51, 1085–1088. [Google Scholar] [PubMed]
- Marongiu, B.; Porcedda, A.P.S.; Casu, R.; Pierucci, P. Chemical Composition of the Oil and Supercritical CO2 Extract of Schinus molle L. Flavour Fragr. J. 2004, 19, 554–558. [Google Scholar] [CrossRef]
- Goldstein, D.J.; Coleman, R.C. Schinus molle L. (Anacardiaceae) Chicha Production in the Central Andes. Econ. Bot. 2004, 58, 523–529. [Google Scholar] [CrossRef]
- Howard, L.F.; Minnich, R.A. The Introduction and Naturalization of Schinus molle (Pepper Tree) in Riverside, California. Landsc. Urban Plan. 1989, 18, 77–95. [Google Scholar] [CrossRef]
- Danin, A. The Inclusion of Adventive Plants in the Second Edition of Flora Palaestina. Willdenowia 2000, 30, 305–314. [Google Scholar] [CrossRef]
- Asner, G.P.; Jones, M.O.; Martin, R.E.; Knapp, D.E.; Hughes, R.F. Remote Sensing of Native and Invasive Species in Hawaiian Forests. Remote Sens. Environ. 2008, 112, 1912–1926. [Google Scholar] [CrossRef]
- Iponga, D.M.; Milton, S.J.; Richardson, D.M. Reproductive Potential and Seedling Establishment of the Invasive Alien Tree Schinus molle (Anacardiaceae) in South Africa. Austral Ecol. 2009, 34, 678–687. [Google Scholar] [CrossRef]
- Ramírez-Albores, J.E.; Bustamante, R.O.; Badano, E.I. Improved Predictions of the Geographic Distribution of Invasive Plants Using Climatic Niche Models. PLoS ONE 2016, 11, e0156029. [Google Scholar] [CrossRef] [PubMed]
- Stinca, A.; Chianese, G.; D’Auria, G.; Del Guacchio, E.; Fascetti, S.; Perrino, E.V.; Rosati, L.; Salerno, G.; Santangelo, A. New Alien Vascular Species for the Flora of Southern Italy. Webbia 2017, 72, 295–301. [Google Scholar] [CrossRef]
- Lemos, R.P.M.; D’Oliveira-Matielo, C.B.; Rodrigues, C.R.; Roesch, L.F.W.; Stefenon, V.M. Modeling distribution of Schinus molle L. in the Brazilian Pampa: Insights on vegetation dynamics and conservation of the biome. Ann. For. Res. 2014, 57, 205–214. [Google Scholar] [CrossRef]
- Ward, S.M.; Gaskin, J.F.; Wilson, L.M. Ecological Genetics of Plant Invasion: What Do We Know? Invasive Plant Sci. Manag. 2008, 1, 98–109. [Google Scholar] [CrossRef]
- Lemos, R.P.M.; D’Oliveira, C.B.; Stefenon, V.M. Genetic Structure and Internal Gene Flow in Populations of Schinus molle (Anacardiaceae) in the Brazilian Pampa. Tree Genet. Genomes 2015, 11, 75. [Google Scholar] [CrossRef]
- Nagel, J.C.; Ceconi, D.E.; Poletto, I.; Stefenon, V.M. Historical Gene Flow within and among Populations of Luehea divaricata in the Brazilian Pampa. Genetica 2015, 143, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Stefenon, V.; Nagel, J.; Poletto, I. Evidences of Genetic Bottleneck and Fitness Decline in Luehea divaricata Populations from Southern Brazil. Silva Fenn. 2016, 50, 1566. [Google Scholar] [CrossRef]
- Taheri, S.; Lee Abdullah, T.; Yusop, M.; Hanafi, M.; Sahebi, M.; Azizi, P.; Shamshiri, R. Mining and Development of Novel SSR Markers Using Next Generation Sequencing (NGS) Data in Plants. Molecules 2018, 23, 399. [Google Scholar] [CrossRef] [PubMed]
- Meekins, J.F.; Ballard, J.H.E.; McCarthy, B.C. Genetic Variation and Molecular Biogeography of a North American Invasive Plant Species (Alliaria petiolata, Brassicaceae). Int. J. Plant Sci. 2001, 162, 161–169. [Google Scholar] [CrossRef]
- Doyle, J.; Doyle, J. A Rapid DNA Isolation Procedure for Small Quantities of Fresh Leaf Tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Chikhi, R.; Medvedev, P. Informed and Automated K-Mer Size Selection for Genome Assembly. Bioinformatics 2014, 30, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de Novo Short Read Assembly Using de Bruijn Graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- da Maia, L.C.; Palmieri, D.A.; de Souza, V.Q.; Kopp, M.M.; de Carvalho, F.I.F.; Costa de Oliveira, A. SSR Locator: Tool for Simple Sequence Repeat Discovery Integrated with Primer Design and PCR Simulation. Int. J. Plant Genom. 2008, 2008, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New Capabilities and Interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Wang, L.; Xu, K.; Kou, C.; Zhang, Y.; Wei, G.; He, J.; Wang, Y.; Zhao, L. Information Theory-Based Algorithm for in Silico Prediction of PCR Products with Whole Genomic Sequences as Templates. BMC Bioinform. 2005, 6, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peakall, R.; Smouse, P.E. Genalex 6: Genetic Analysis in Excel. Population Genetic Software for Teaching and Research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic Analysis in Excel. Population Genetic Software for Teaching and Research—An Update. Bioinform. Oxf. Engl. 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Raymond, M.; Rousset, F. GENEPOP (Version 1.2): Population Genetics Software for Exact Tests and Ecumenicism. J. Hered. 1995, 86, 248–249. [Google Scholar] [CrossRef]
- Rousset, F. Genepop’007: A Complete Re-Implementation of the Genepop Software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Pivetta, M. Da quaresmeira ao jerivá: Tamanho do genoma de 100 árvores brasileiras varia até 20 vezes. Pesquisa FAPESP 2005, 115, 42–44. [Google Scholar]
- Owusu, S.A.; Staton, M.; Jennings, T.N.; Schlarbaum, S.; Coggeshall, M.V.; Romero-Severson, J.; Carlson, J.E.; Gailing, O. Development of Genomic Microsatellites in Gleditsia triacanthos (Fabaceae) Using Illumina Sequencing1. Appl. Plant Sci. 2013, 1, 1300050. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhang, R.; Staton, M.; Schlarbaum, S.E.; Coggeshall, M.V.; Romero-Severson, J.; Carlson, J.E.; Liang, H.; Xu, Y.; Drautz-Moses, D.I.; et al. Development of Genic and Genomic Microsatellites in Gleditsia triacanthos L. (Fabaceae) Using Illumina Sequencing. Ann. For. Res. 2017. [Google Scholar] [CrossRef]
- Staton, M.; Best, T.; Khodwekar, S.; Owusu, S.; Xu, T.; Xu, Y.; Jennings, T.; Cronn, R.; Arumuganathan, A.K.; Coggeshall, M.; et al. Preliminary Genomic Characterization of Ten Hardwood Tree Species from Multiplexed Low Coverage Whole Genome Sequencing. PLoS ONE 2015, 10, e0145031. [Google Scholar] [CrossRef] [PubMed]
- Rasmusen, D.A.; Noor, M.A.F. What can you do with 0.1× genome coverage? A case study based on a genome survey of the scuttle fly Megaselia scalaris (Phoridae). BMC Genom. 2009, 10, 382. [Google Scholar] [CrossRef] [PubMed]
- Petit, R.J.; Duminil, J.; Fineschi, S.; Hampe, A.; Salvini, D.; Vendramin, G.G. Comparative organization of chloroplast, mitochondrial and nuclear diversity in plant populations. Mol. Ecol. 2005, 14, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Franca-Rocha, W.; Silva, A.D.; Nolasco, M.C.; Lobão, J.; Britto, D.; Chaves, J.M.; Rocha, C.D. Levantamento Da Cobertura Vegetal e Do Uso Do Solo Do Bioma Caatinga. In Proceedings of the Ananis XIII Simpósio Brasileiro Sensoriamento Remoto INPE, Florianópolis, Brazil, 21–26 April 2007; pp. 2629–2636. [Google Scholar]
- de Oliveira, G.; Araújo, M.B.; Rangel, T.F.; Alagador, D.; Diniz-Filho, J.A.F. Conserving the Brazilian Semiarid (Caatinga) Biome under Climate Change. Biodivers. Conserv. 2012, 21, 2913–2926. [Google Scholar] [CrossRef]
- Roesch, L.F.W.; Vieira, F.C.B.; Pereira, V.A.; Schünemann, A.L.; Teixeira, I.F.; Senna, A.J.T.; Stefenon, V.M. The Brazilian Pampa: A Fragile Biome. Diversity 2009, 1, 182–190. [Google Scholar] [CrossRef]
- Morris, A.B.; Shaw, J. Markers in time and space: A review of the last decade of plant phylogeographic approaches. Mol. Ecol. 2018, 27, 2317–2333. [Google Scholar] [CrossRef] [PubMed]
- Mensous, M.; Van de Paer, C.; Manzi, S.; Bouchez, O.; Baâli-Cherif, D.; Besnard, G. Diversity and evolution of plastomes in Saharan mimosoids: potential use for phylogenetic and population genetic studies. Tree Genet. Genomes 2017, 13, 48. [Google Scholar] [CrossRef]


{kind=link}
| Locus | Primer Sequence (5′→3′) a | Rep Motif | Prod. Size | Ta (°C) | Genome Region b | GenBank ID | Bit-Score | Ident. |
|---|---|---|---|---|---|---|---|---|
| Smolle03 | AAGTTTTATTTTCCCAGAAT AATCATAGGTTCTTCTCTCC | (TTC)3 | 169 | 51 | ptSSR | MH536214 | 1050 | 99% |
| Smolle04 | CTCCTAGGGATAAGAGACAT GAATAATTGTTGGAGACTCA | (AG)4 | 206 | 49 | ptSSR | MH536215 | 941 | 99% |
| Smolle05 | CGTAGACCAAATGATACAAT TTATTTCTCATCAAACGAAT | (AGA)3 | 261 | 47 | ptSSR | MH536216 | 941 | 99% |
| Smolle06 | GGTCCATGAATCTAAGAAAT TTGAAATGAAATCTTTAGGA | (TC)4 | 140 | 47 | ptSSR | MH536217 | 1014 | 100% |
| Smolle07 | GAGTTGAAAATAAGCGTAGA TCTGGCTACTAAGATGTTTC | (CTG)3 | 117 | 51 | ptSSR | MH536218 | 1022 | 99% |
| Smolle08 | ATTTGTTATCTCATGTTTGC ACACATTGTCTAACCAAATC | (GA)4 | 275 | 49 | ptSSR | MH536219 | 970 | 99% |
| Smolle09 | CCCATTAACATTTTAGAAGA GCTAAAGTTGCAAAAATAAG | (GTT)3 | 188 | 47 | ptSSR | MH536220 | 625 | 98% |
| Smolle10 | TAGTTCATCCTATTGGCTC GAAACGAATTTTCATTTTTA | (TAA)3 | 234 | 47 | ptSSR | MH536221 | 492 | 95% |
| Smolle11 | AGAGGAGTAGTTATGAACCC TCACTATATTTATTCCTTTTTCT | (GAA)3 | 140 | 51 | ptSSR | MH536222 | 837 | 99% |
| Smolle12 | CCACTAGAGATCAGAAATTG AATTGAGACGGTATTTTGTA | (AAG)3 | 128 | 47 | ptSSR | MH536223 | 580 | 99% |
| Smolle13 | CTGTGTTTTTGGTAACAGTC GGTGGGTAGGTAGAGAATAC | (CT)4 | 148 | 55 | ptSSR | MH536224 | 1369 | 99% |
| Smolle14 | AGTTTCTTTTTACACATCCA AAGAAGATCCATTTTGAGTT | (AT)4 | 161 | 47 | ptSSR | MH536225 | 523 | 96% |
| Smolle15 | GTACAAATAAGAATCCCCTT AGATCTTGTAGCACTTACCA | (TTC)3 | 268 | 51 | ptSSR | MH536226 | 1387 | 99% |
| Smolle16 | GCAGATTCATCTAATTATGG AAGTTATAAGTTGTGAAGCG | (TCT)3 | 193 | 49 | ptSSR | MH536227 | 588 | 98% |
| Smolle19 | GTCAACTAAGGGGATAAGAT ATCCAATATCAATAAACCAA | (AT)6 | 159 | 47 | ptSSR | MH536230 | 1061 | 97% |
| Smolle22 | CTATAGTGGCTAGGGTGAG AATACCTTCCTCTGTCATCT | (AG)4 | 172 | 51 | ptSSR | MH536232 | 483 | 94% |
| Smolle28 | ATTTGTGCTCAATTTTCATA ACACATTGTCTAACCAAATC | (GA)4 | 124 | 49 | ptSSR | MH536237 | 494 | 96% |
| Smolle17 | AGAATTCTCTACCATTCTCC ACAAAAATCACATGAAAATC | (TTC)3 | 203 | 47 | nSSR | MH536228 | - | - |
| Smolle18 | TCTAGTACCAGAATCTTTGC AGGAGGTAAATCCAACTATC | (CT)5 | 127 | 51 | nSSR | MH536229 | - | - |
| Smolle21 | GTGTTTCGTTAAGACAAAAG GTGAGAAACGAATAAAGAAA | (TG)5 | 113 | 47 | nSSR | MH536231 | - | - |
| Smolle23 | GAAGATAAGTTCATACCCCT TTCATTAATTGGCTCTAATC | (CTT)3 | 202 | 47 | nSSR | MH536233 | - | - |
| Smolle24 | AGATTTCCCGAACTATTATT TGTTCAAGGAATAAAGGTAA | (TGA)3 | 226 | 47 | nSSR | MH536234 | - | - |
| Smolle25 | TGCACCTTATATGAAAGACT ACCATCACTACAGCTCATAC | (GAA)3 | 145 | 53 | nSSR | MH536235 | - | - |
| Smolle27 | AGTCAATGAAGTTTTCACAG TGAGAACTCAAGATGCTATT | (CT)4 | 224 | 49 | nSSR | MH536236 | - | - |
| N | A | Ae | HO | HE | FIS | |
|---|---|---|---|---|---|---|
| Pampa | 23.62 | 5.54 | 3.56 | 0.64 | 0.68 | 0.08 |
| Caatinga | 24.75 | 7.33 | 5.15 | 0.62 | 0.72 | 0.20 |
| Overall | 48.37 | 10.08 | 6.03 | 0.64 | 0.81 | 0.21 |
| ptSSRs | nSSRs | |||
|---|---|---|---|---|
| Caatinga | Pampa | Caatinga | Pampa | |
| N | 25.17 | 24.82 | 23.71 | 20.71 |
| A | 6.70 | 5.70 | 8.85 | 5.14 |
| Ae | 4.67 | 3.55 | 6.32 | 3.57 |
| Ho | 0.57 | 0.67 | 0.75 | 0.56 |
| He | 0.68 | 0.68 | 0.82 | 0.66 |
| FIS | 0.25 | 0.03 | 0.10 | 0.22 |
| Pairs of Loci | p-Values | ||
|---|---|---|---|
| Caatinga | Pampa | ||
| Smolle03 | Smolle10 | 0.02184 | 0.02184 |
| Smolle08 | Smolle12 | 0.00356 | 0.00356 |
| Smolle05 | Smolle19 | 0.00111 | 0.00111 |
| Smolle14 | Smolle19 | 0.00000 | 0.00000 |
| Smolle07 | Smolle21 | 0.03328 | 0.03328 |
| Smolle19 | Smolle21 | 0.02626 | 0.02626 |
| Smolle22 | Smolle17 | 0.03454 | 0.03454 |
| Smolle07 | Smolle24 | 0.01180 | 0.01180 |
| Smolle03 | Smolle25 | 0.00057 | 0.00057 |
| Smolle11 | Smolle25 | 0.04062 | 0.04062 |
| Smolle19 | Smolle27 | 0.00000 | 0.00000 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lemos, R.P.M.; Matielo, C.B.D.; Beise, D.C.; Da Rosa, V.G.; Sarzi, D.S.; Roesch, L.F.W.; Stefenon, V.M. Characterization of Plastidial and Nuclear SSR Markers for Understanding Invasion Histories and Genetic Diversity of Schinus molle L. Biology 2018, 7, 43. https://doi.org/10.3390/biology7030043
Lemos RPM, Matielo CBD, Beise DC, Da Rosa VG, Sarzi DS, Roesch LFW, Stefenon VM. Characterization of Plastidial and Nuclear SSR Markers for Understanding Invasion Histories and Genetic Diversity of Schinus molle L. Biology. 2018; 7(3):43. https://doi.org/10.3390/biology7030043
Chicago/Turabian StyleLemos, Rafael Plá Matielo, Cristiane Barbosa D’Oliveira Matielo, Dalvan Carlos Beise, Vanessa Gonçalves Da Rosa, Deise Schröder Sarzi, Luiz Fernando Würdig Roesch, and Valdir Marcos Stefenon. 2018. "Characterization of Plastidial and Nuclear SSR Markers for Understanding Invasion Histories and Genetic Diversity of Schinus molle L." Biology 7, no. 3: 43. https://doi.org/10.3390/biology7030043