Design of Catalytically Amplified Sensors for Small Molecules

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
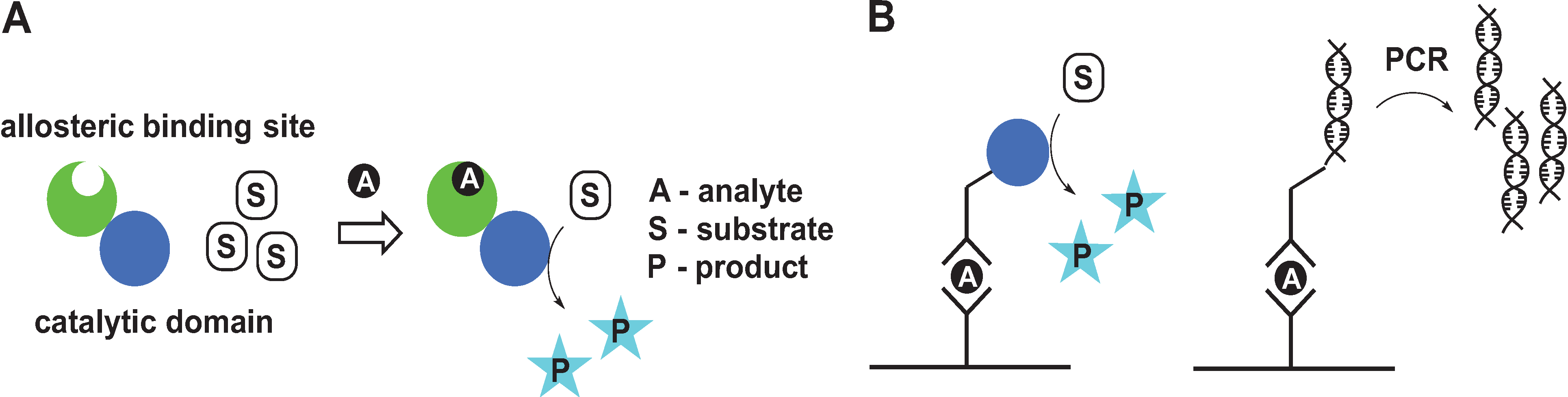
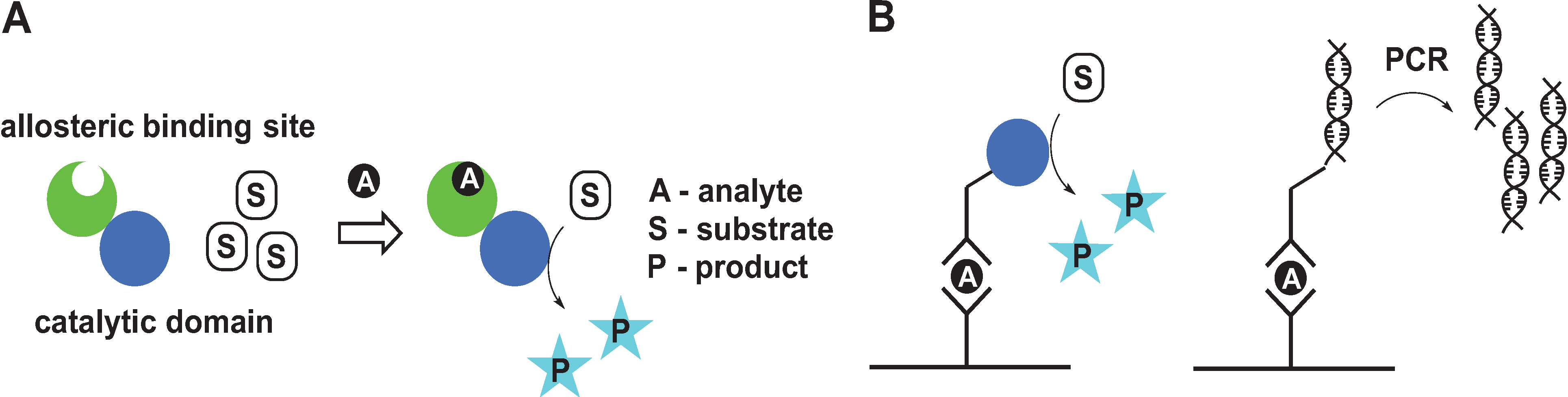
2. Signal Amplification by Protein-Based Switches
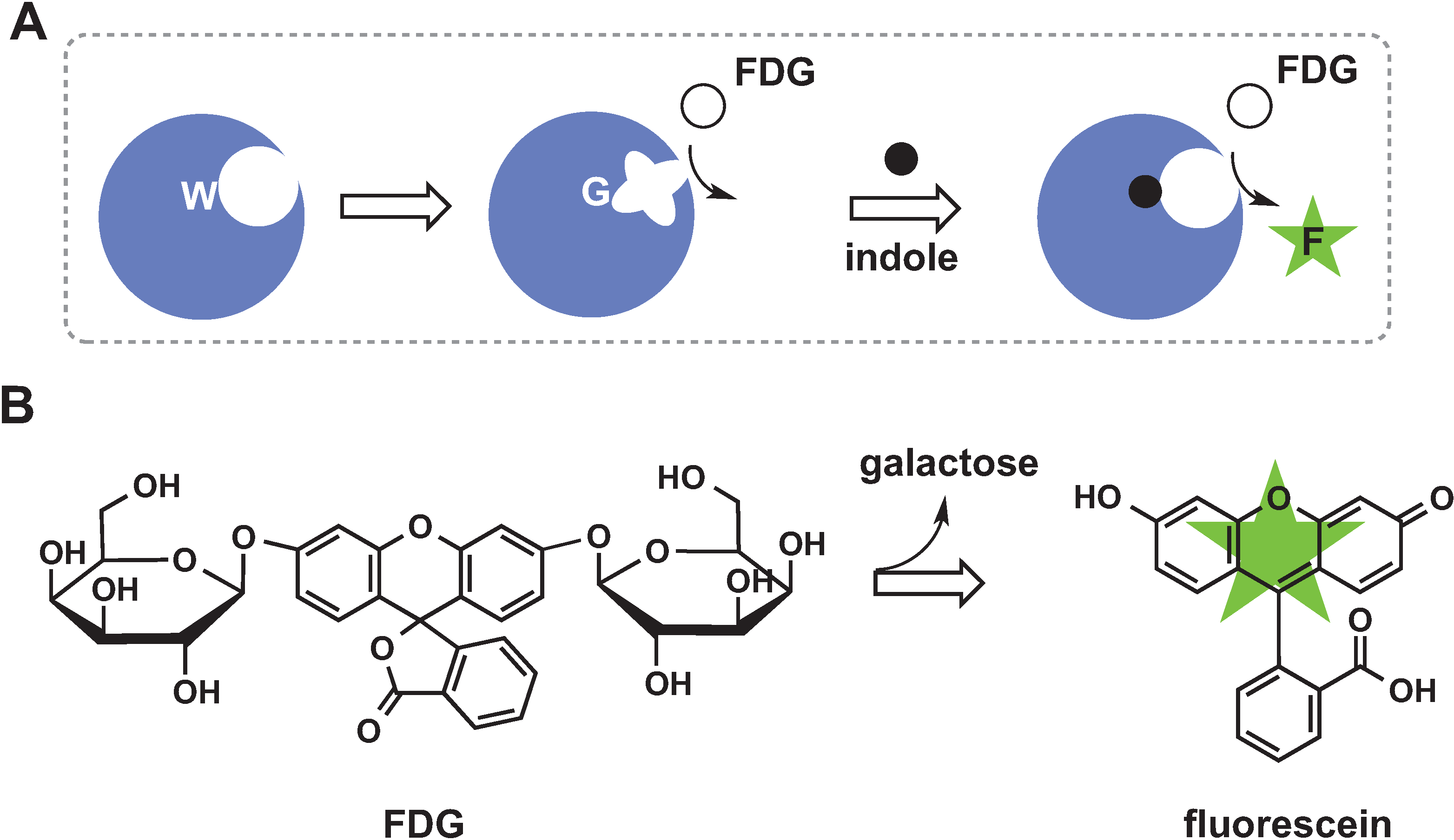
2.1. Control of Enzyme Assembly
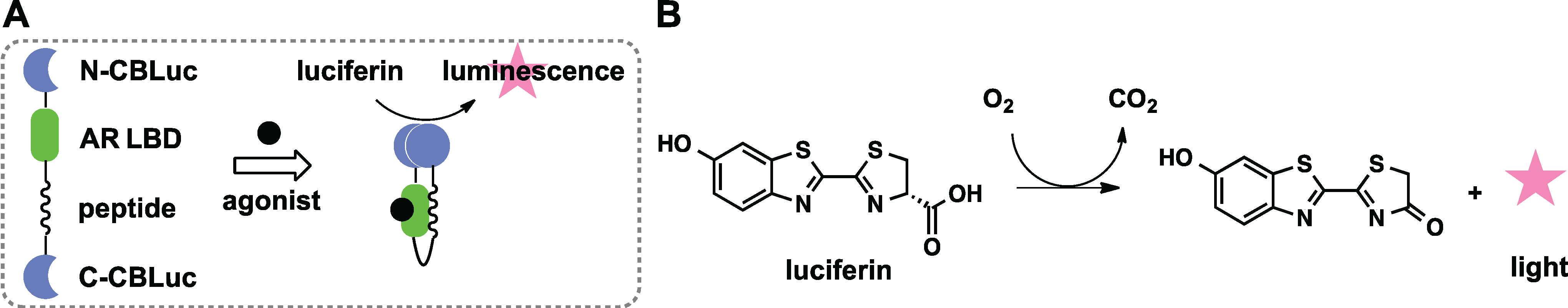
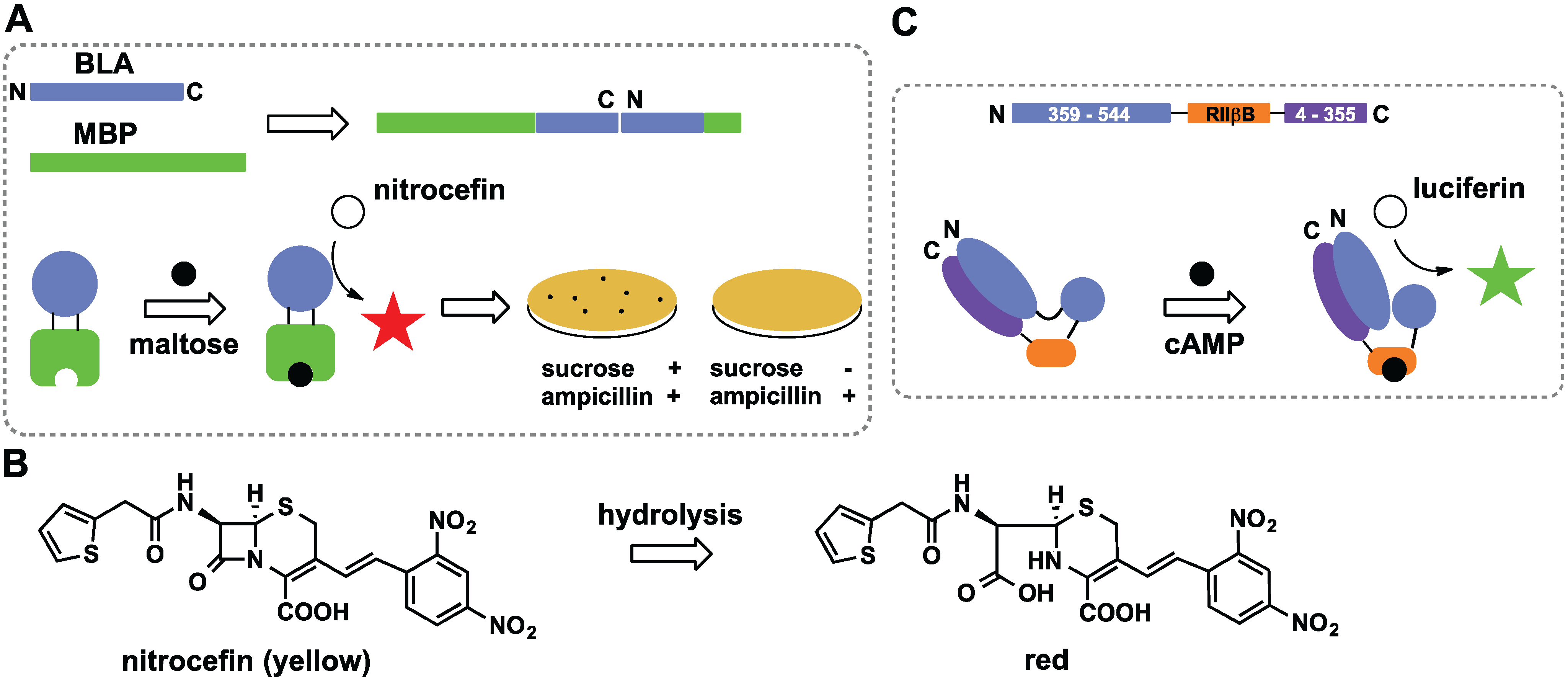
2.2. Domain Insertion
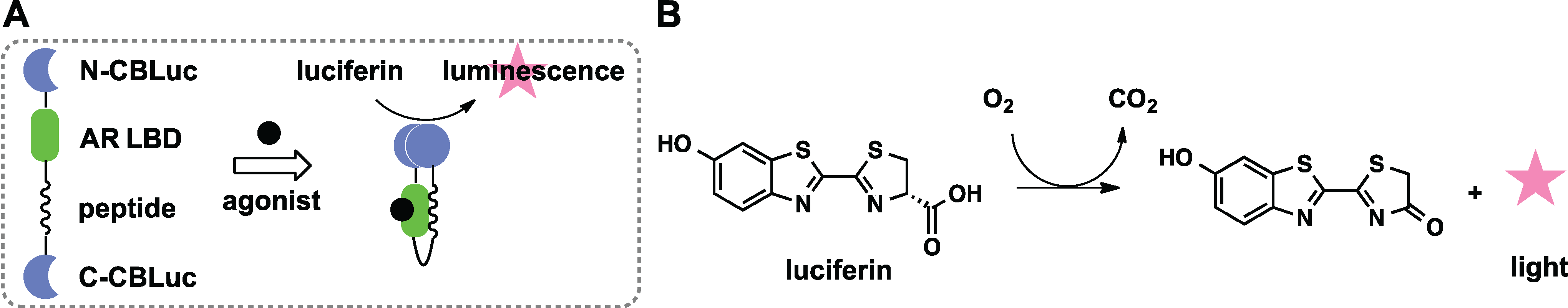
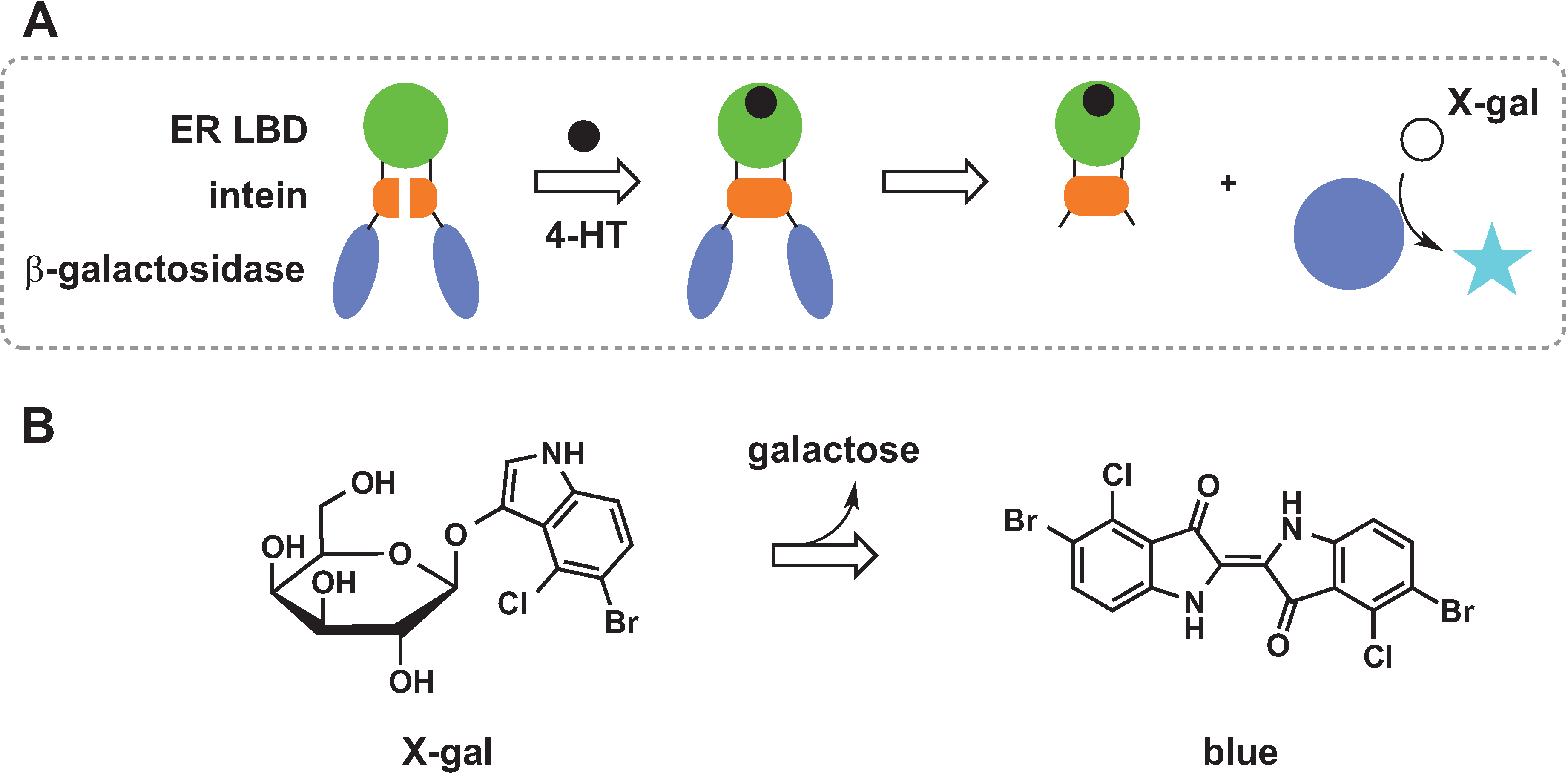
2.3. Protein Splicing

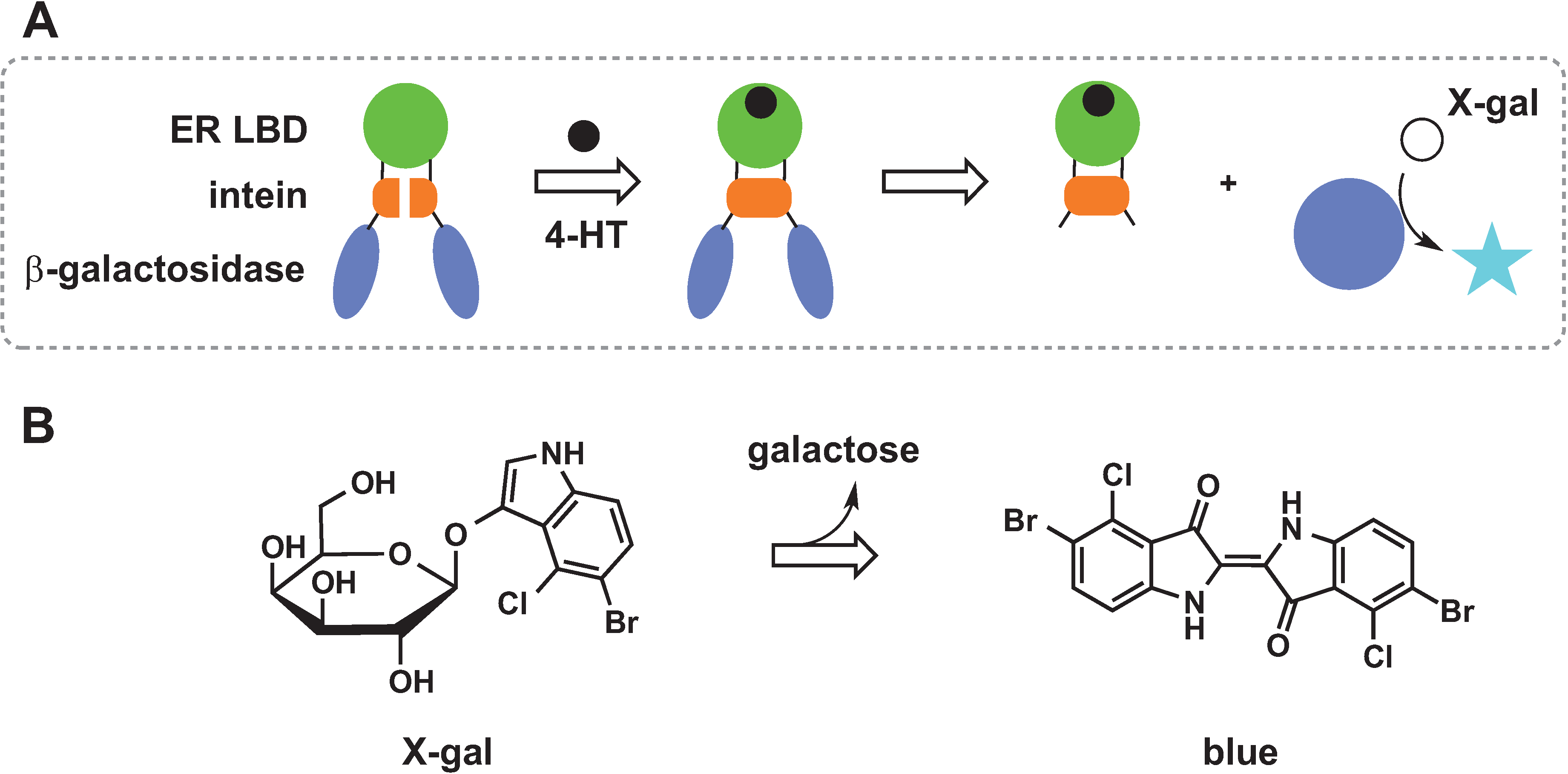
2.4. Chemical Rescue
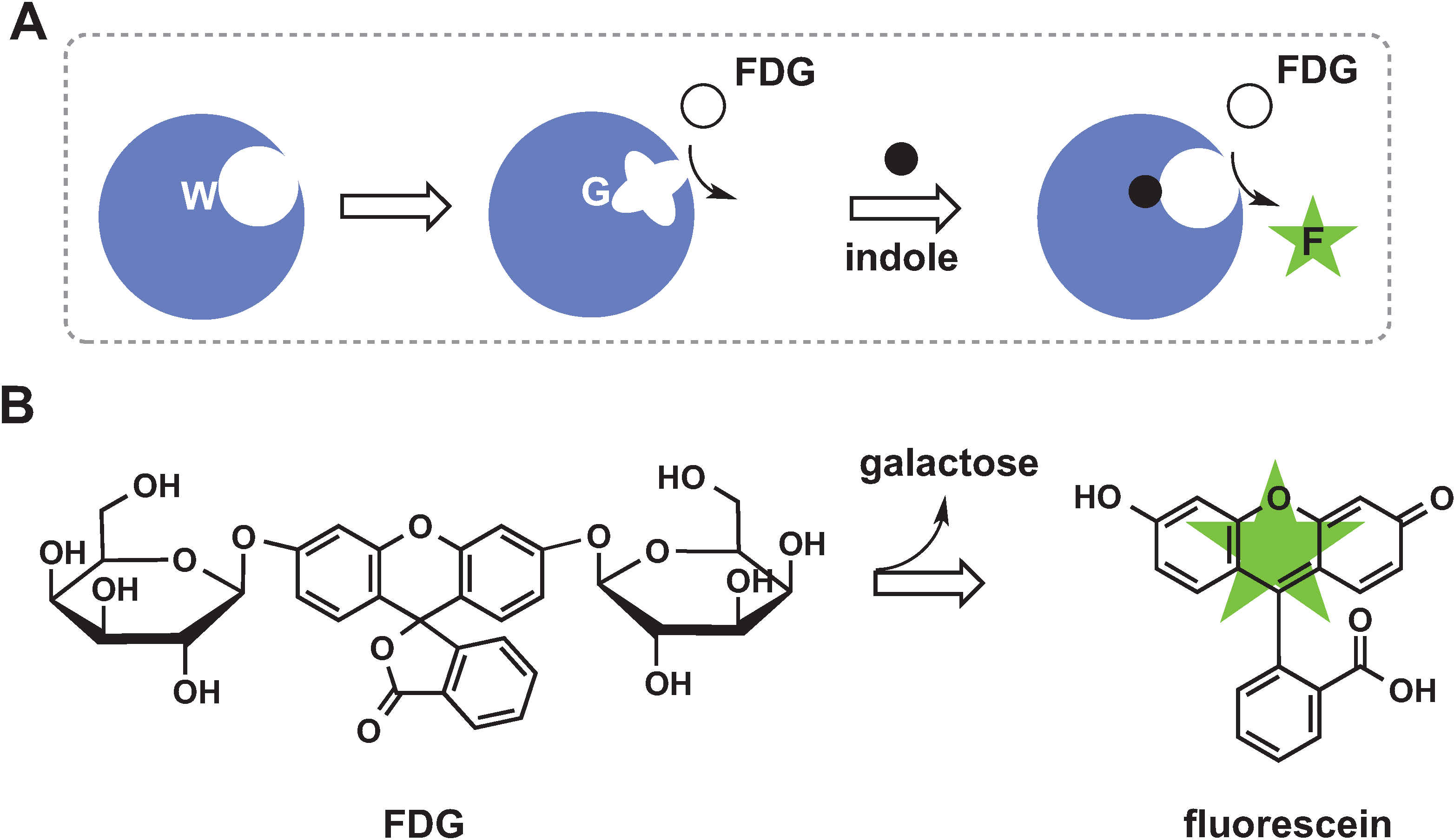
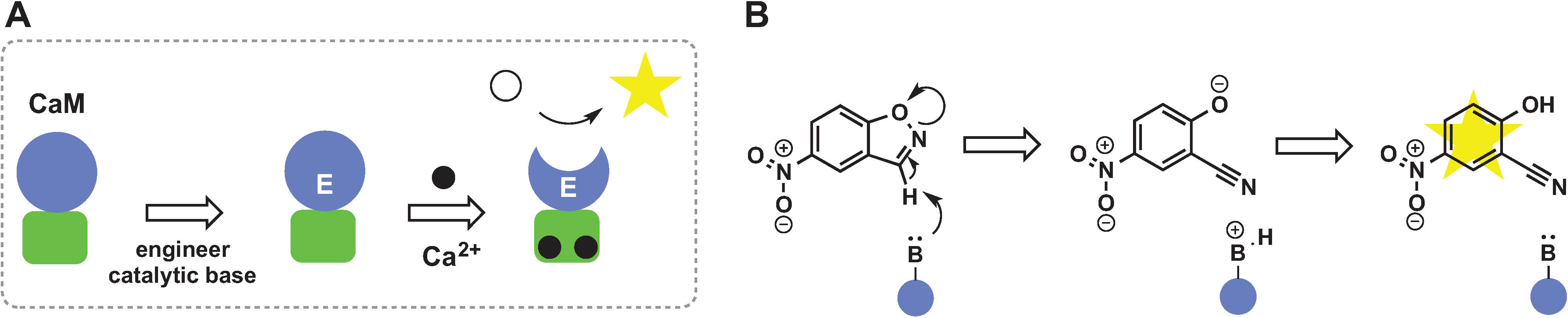
2.5. De Novo Design of Catalytic Function
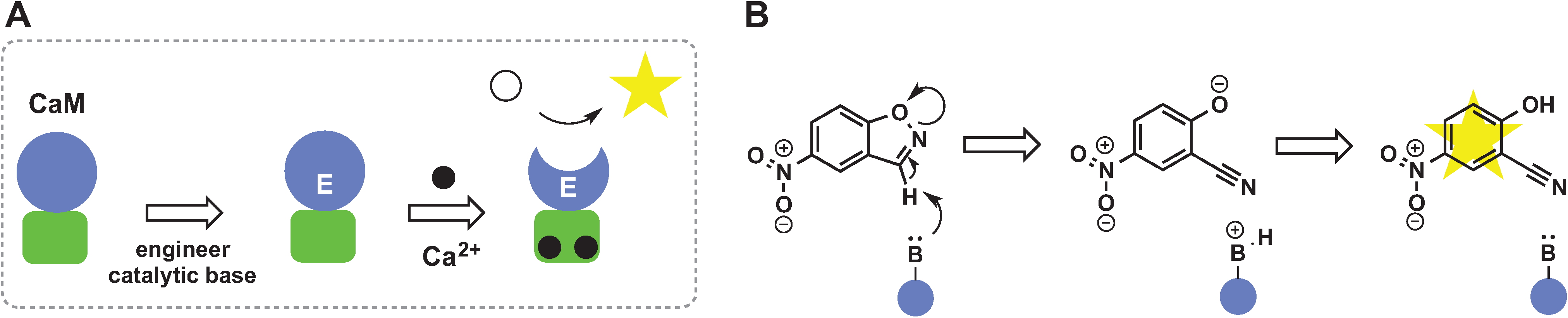
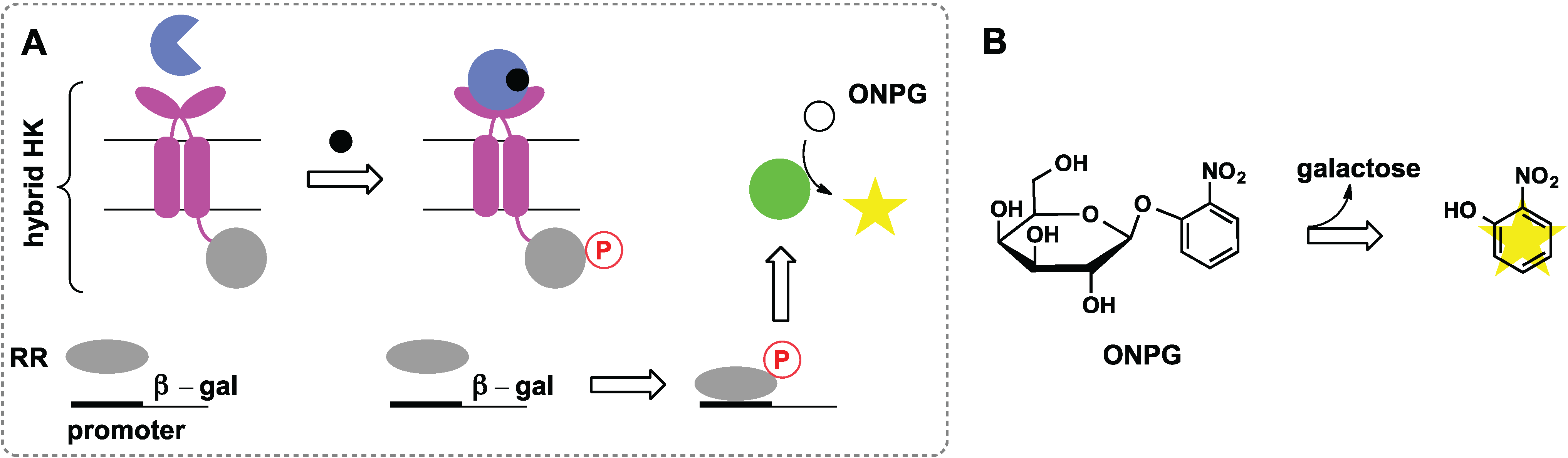
2.6. Switches Based on Two-Component Systems

3. Signal Amplification by Organometallic Catalysts
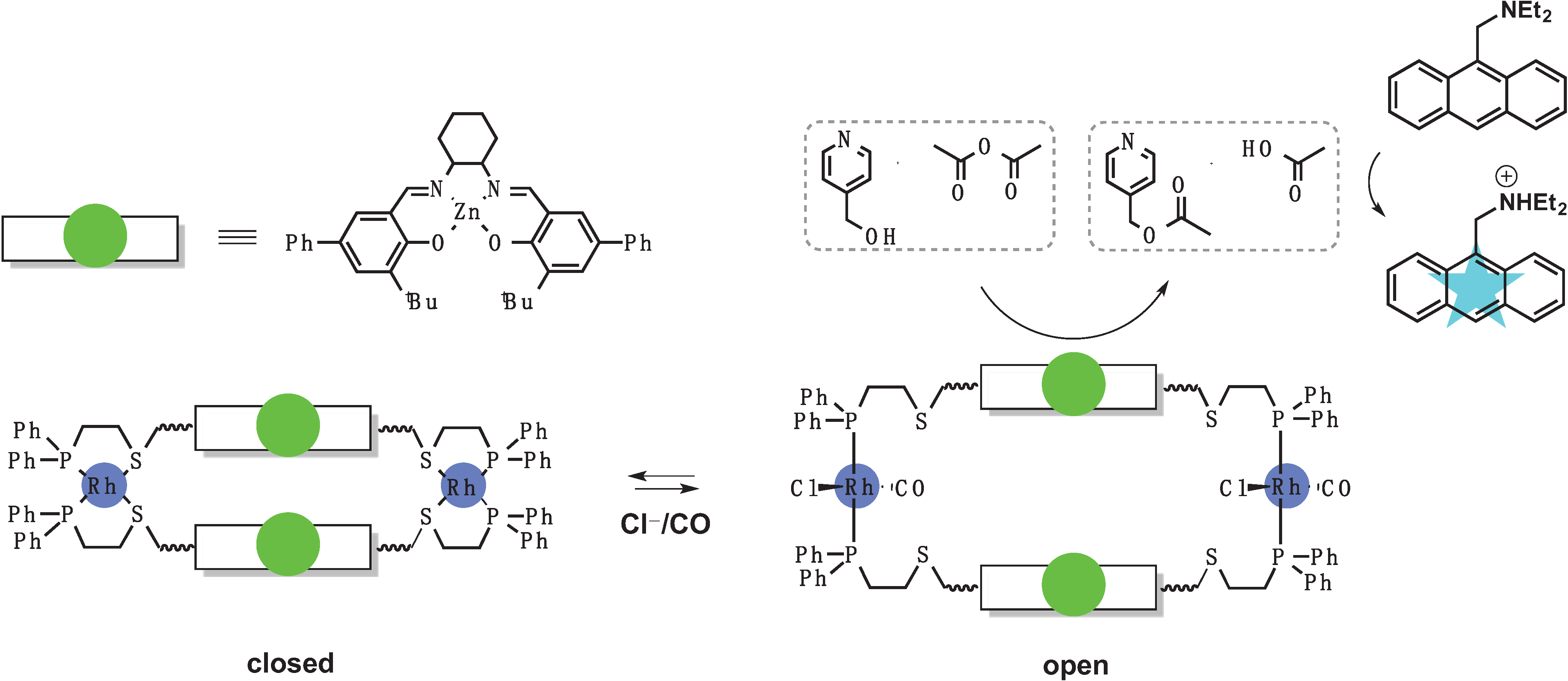
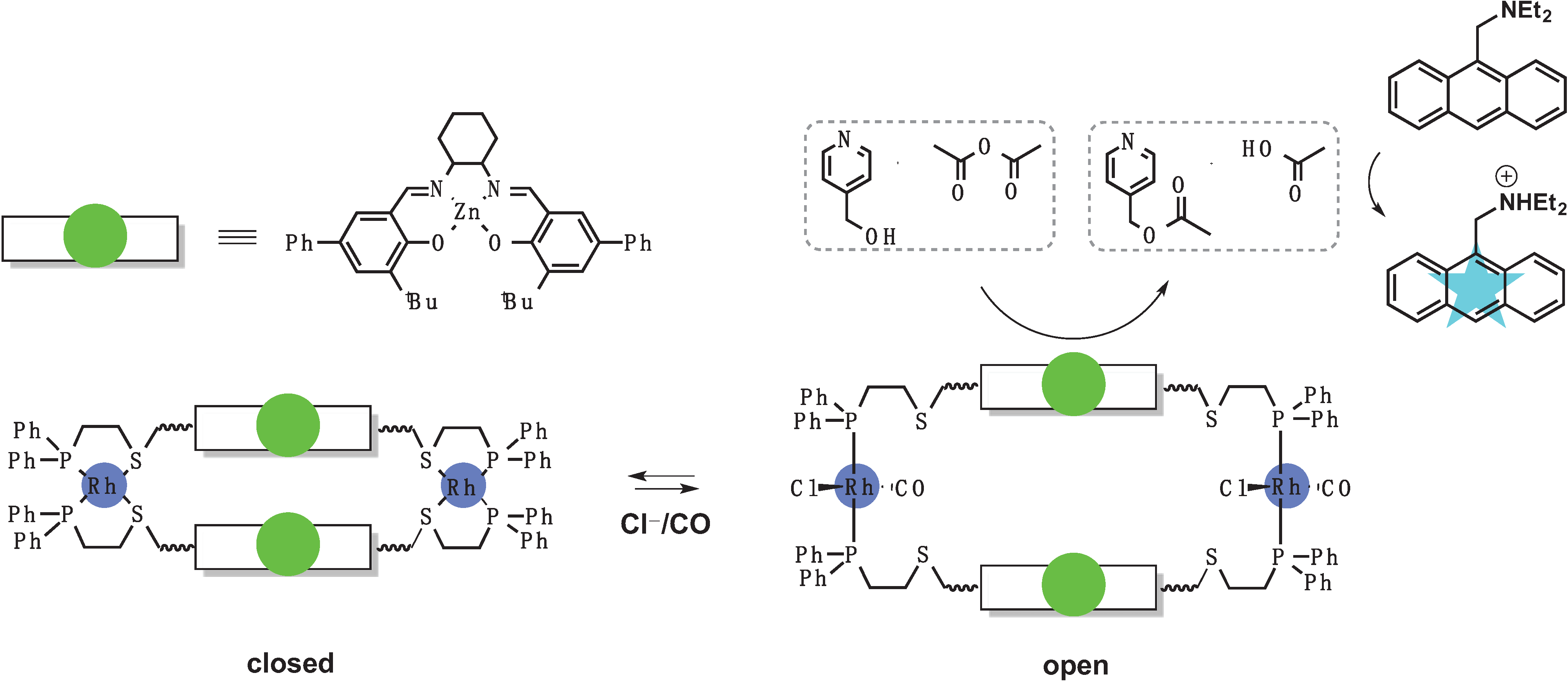
3.1. Switches Based on a Weak-Link Approach
3.2. Metal Sensors Based on Variable Affinity to Ligands
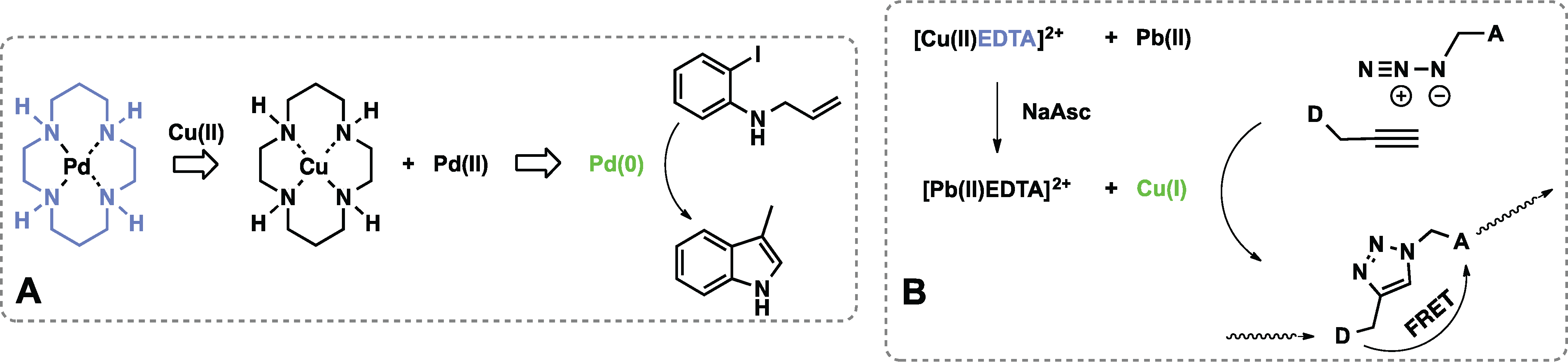
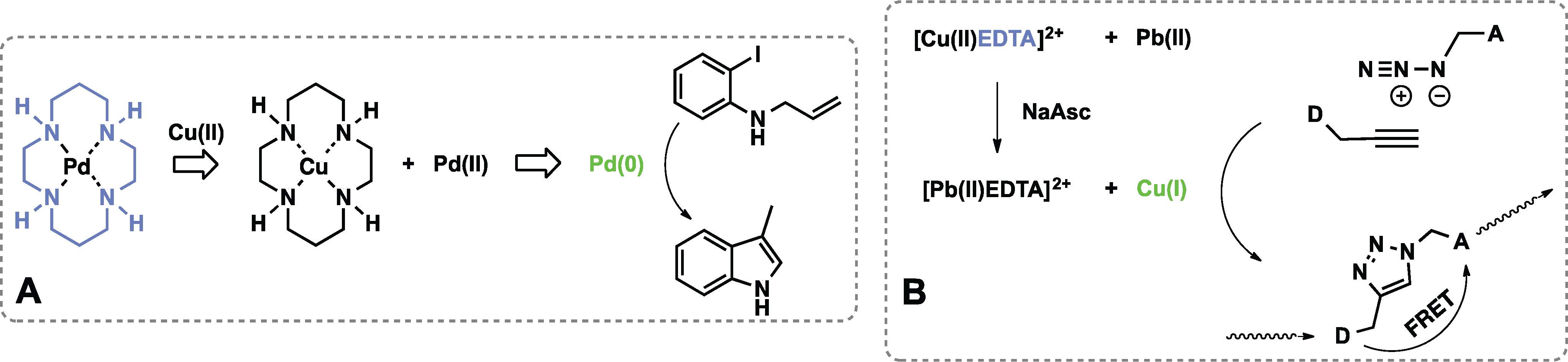
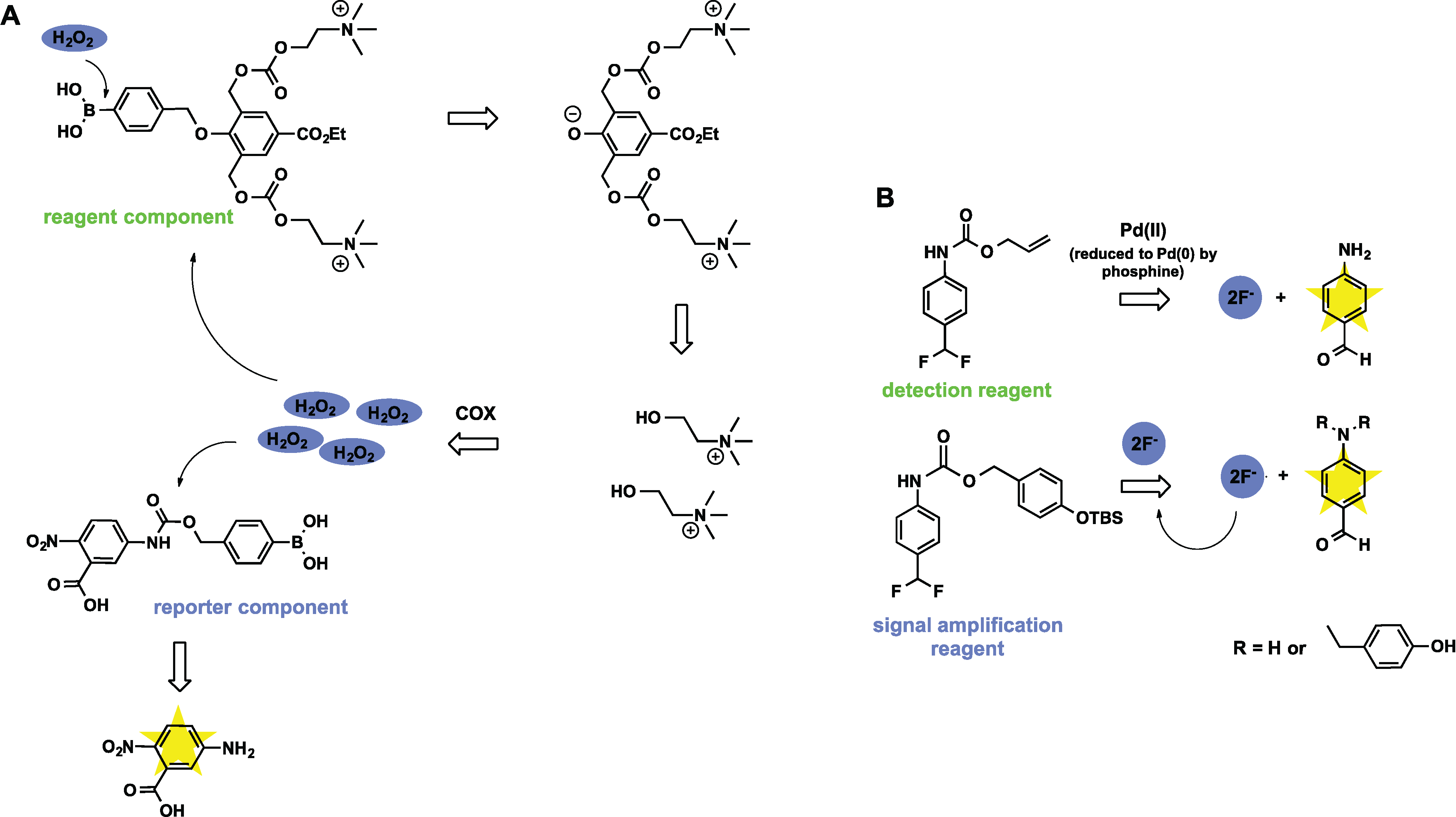
3.3. Sensors Based on Auto Amplification Mechanism
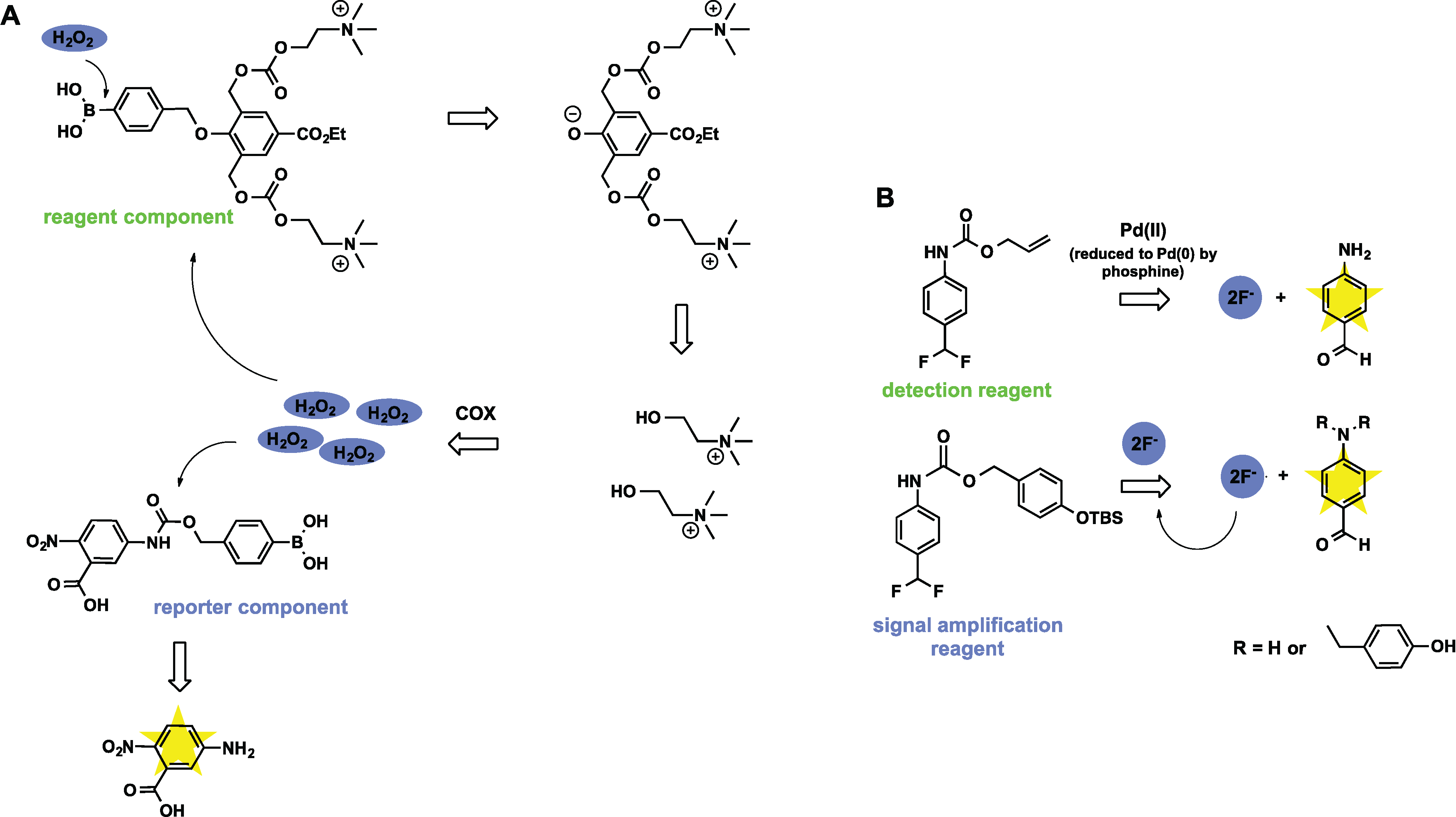
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stratton, M.M.; McClendon, S.; Eliezer, D.; Loh, S.N. Structural characterization of two alternate conformations in a calbindin D9k-based molecular switch. Biochemistry 2011, 50, 5583–5589. [Google Scholar] [CrossRef]
- Miyawaki, A.; Llopis, J.; Heim, R.; McCaffery, J.M.; Adams, J.A.; Ikura, M.; Tsien, R.Y. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature 1997, 388, 882–887. [Google Scholar] [CrossRef]
- Nagai, T.; Sawano, A.; Park, E.S.; Miyawaki, A. Circularly permuted green fluorescent proteins engineered to sense Ca2+. Proc. Natl. Acad. Sci. USA 2001, 98, 3197–3202. [Google Scholar] [CrossRef]
- Baird, G.S.; Zacharias, D.A.; Tsien, R.Y. Circular permutation and receptor insertion within green fluorescent proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 11241–11246. [Google Scholar]
- De Lorimier, R.M.; Smith, J.J.; Dwyer, M.A.; Looger, L.L.; Sali, K.M.; Paavola, C.D.; Rizk, S.S.; Sadigov, S.; Conrad, D.W.; Loew, L.; et al. Construction of a fluorescent biosensor family. Protein Sci. 2002, 11, 2655–2675. [Google Scholar]
- Yang, C.J.; Jockusch, S.; Vicens, M.; Turro, N.J.; Tan, W. Light-switching excimer probes for rapid protein monitoring in complex biological fluids. Proc. Natl. Acad. Sci. USA 2005, 102, 17278–17283. [Google Scholar] [CrossRef]
- Fehr, M.; Rfommer, W.B.; Lalonde, S. Visualization of maltose uptake in living yeast cells by fluorescent nanosensors. Proc. Natl. Acad. Sci. USA 2002, 99, 9846–9851. [Google Scholar] [CrossRef]
- Medintz, I.L.; Deschamps, J.R. Maltose-binding protein: A versitile platform for prototyping biosensing. Curr. Opin. Biotechnol. 2006, 17, 17–27. [Google Scholar] [CrossRef]
- Mochizuki, N.; Yamashita, S.; Kurokawa, K.; Ohba, Y.; Nagal, T.; Miyawaki, A.; Matsuda, M. Spatio-temporal images of growth-factor-induced activation of Ras and Rap1. Nature 2001, 411, 1065–1068. [Google Scholar] [CrossRef]
- Tyagi, S.; Kramer, F.R. Molecular beacons: Probes that fluoresce upon hybridization. Nat. Biotechol. 1996, 14, 303–308. [Google Scholar] [CrossRef]
- Vallée-Bélisle, A.; Plaxco, K.W. Structure-switching biosensors: Inspired by nature. Curr. Opin. Struct. Biol. 2010, 20, 518–526. [Google Scholar] [CrossRef]
- Monod, J.; Changeux, J.-P.; Jacob, F. Allosteric proteins and cellular control systems. J. Mol. Biol. 1963, 6, 306–329. [Google Scholar] [CrossRef]
- Engvall, E.; Perlmann, P. Enzyme-linked immunosorbent assay (ELISA). Quantitative assay of immunoglobin G. Immunochemistry 1971, 8, 871–874. [Google Scholar]
- Gosling, J.P. A decade of development in immunoassay methodology. Clin. Chem. 1990, 36, 1408–1427. [Google Scholar]
- Van Weemen, B.K.; Schuurs, A.H.W.M. Immunoassay using antigen-enzyme conjugates. FEBS Lett. 1971, 15, 232–236. [Google Scholar] [CrossRef]
- Saiki, R.K.; Scharf, S.; Faloona, F.; Mullis, K.B.; Horn, G.T.; Erlich, H.A.; Arnheim, N. Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 1985, 230, 1350–1354. [Google Scholar]
- Saiki, R.K.; Bugawan, T.L.; Horn, G.T.; Mullis, K.B.; Erlich, H.A. Analysis of enzymatically amplified beta-globin and HLA-DQα DNA with allele-specific oligonucleotide probes. Nature 1986, 324, 163–166. [Google Scholar] [CrossRef]
- Sano, T.; Smith, C.T.; Cantor, C.R. Immuno-PCR: Very sensitive antigen detection by means of specific antibody-DNA conjugates. Science 1992, 258, 120–122. [Google Scholar]
- Niemeyer, C.M.; Adler, M.; Wacker, R. Detecting antigens by quantitative immuno-PCR. Nat. Protoc. 2007, 2, 1918–1930. [Google Scholar] [CrossRef]
- Niemeyer, C.M.; Adler, M.; Wacker, R. Immuno-PCR: High sensitivity detection of proteins by nucleic acid amplification. Trends Biotechnol. 2005, 23, 208–216. [Google Scholar] [CrossRef]
- Heid, C.A.; Stevens, J.; Livak, K.J.; Williams, P.M. Real time quantitative PCR. Genome Res. 1996, 6, 986–994. [Google Scholar]
- Scrimin, P.; Prins, L.J. Sensing through signal amplification. Chem. Soc. Rev. 2011, 40, 4488–4505. [Google Scholar] [CrossRef]
- Zhu, L.; Anslyn, E.V. Signal ampliifcation by allosteric catalysis. Angew. Chem. Int. Ed. 2006, 45, 1190–1196. [Google Scholar] [CrossRef]
- Saghatelian, A.; Guckian, K.M.; Thayer, D.A.; Ghadiri, M.R. DNA detection and signal amplification via an engineered allosteric enzyme. J. Am. Chem. Soc. 2003, 125, 344–345. [Google Scholar] [CrossRef]
- Mitrea, D.M.; Parsons, L.S.; Loh, S.N. Engineering an artificial zymogen by alternate frame protein folding. Proc. Natl. Acad. Sci. USA 2010, 107, 2824–2829. [Google Scholar] [CrossRef]
- Hartig, J.S.; Grüne, I.; Najafi-Shoushtari, H.; Famulok, M. Sequence-specific detection of micrornas by signal-amplifying ribozymes. J. Am. Chem. Soc. 2004, 126, 722–723. [Google Scholar]
- Graf, N.; Krämer, R. Enzymatic amplification in a bioinspired, autonomous signal cascade. Chem. Commun. 2006, 4375–4376. [Google Scholar] [CrossRef]
- Graf, N.; Göritz, M.; Krämer, R. A metal-ion-releasing probe for DNA detection by catalytic signal amplification. Angew. Chem. Int. Ed. 2006, 45, 4013–4015. [Google Scholar] [CrossRef]
- Miranda, O.R.; Chen, H.-T.; You, C.-C.; Mortenson, D.E.; Yang, X.-C.; Bunz, U.H.F.; Rotello, V.M. Enzyme-amplified array sensing of proteins in solution and in biofluids. J. Am. Chem. Soc. 2010, 132, 5285–5289. [Google Scholar]
- Bonomi, R.; Cazzolaro, A.; Sansone, A.; Scrimin, P.; Prins, L.J. Detection of enzyme activity through catalytic signal amplification with functionalized gold nanoparticles. Angew. Chem. Int. Ed. 2011, 50, 2307–2312. [Google Scholar] [CrossRef]
- Wright, C.M.; Heins, R.A.; Ostermeier, M. As easy as flipping a switch? Curr. Opin. Chem. Biol. 2007, 11, 342–346. [Google Scholar] [CrossRef]
- Ambroggio, X.I.; Kuhlman, B. Design of protein conformational switches. Curr. Opin. Struct. Biol. 2006, 16, 525–530. [Google Scholar] [CrossRef]
- Ha, J.-H.; Loh, S.N. Protein conformational switches: From nature to design. Chem. Eur. J. 2012, 18, 7984–7999. [Google Scholar] [CrossRef]
- Ostermeier, M. Designing switchable enzymes. Curr. Opin. Struct. Biol. 2009, 19, 442–448. [Google Scholar] [CrossRef]
- Koide, S. Generation of new protein functions by nonhomologous combinations and rearrangements of domains and modules. Curr. Opin. Biotechnol. 2009, 20, 398–404. [Google Scholar] [CrossRef]
- Stratton, M.M.; Loh, S.N. Converting a protein into a switch for biosensing and functional regulation. Protein Sci. 2011, 20, 19–29. [Google Scholar] [CrossRef]
- Ostermeier, M. Engineering allosteric protein switches by domain insertion. Protein Eng. Des. Sel. 2005, 18, 359–364. [Google Scholar] [CrossRef]
- Kim, S.B.; Otani, Y.; Umezawa, Y.; Tao, H. Bioluminescence indicator for determining protein-protein interactions using intramolecular complementation of split click beetle luciferase. Analyt. Chem. 2007, 79, 4820–4826. [Google Scholar] [CrossRef]
- Kim, S.B.; Umezawa, Y.; Kanno, K.A.; Tao, H. An integrated-molecule-format multicolor probe for monitoring multiple activities of a bioactive small molecule. ACS Chem. Biol. 2008, 3, 359–372. [Google Scholar] [CrossRef]
- Guntas, G.; Mitchell, S.F.; Ostermeier, M. A molecular switch created by in vitro recombination of nonhomologous genes. Chem. Biol. 2004, 11, 1483–1487. [Google Scholar] [CrossRef]
- Guntas, G.; Mansell, T.J.; Kim, J.R.; Ostermeier, M. Directed evolution of protein switches and their application to the creation of ligand-binding proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 11224–11229. [Google Scholar] [CrossRef]
- Edwards, W.R.; Busse, K.; Allemann, R.K.; Jones, D.D. Linking the functions of unrelated proteins using a novel directed evolution domain insertion method. Nucleic Acids Res. 2008. [Google Scholar] [CrossRef]
- Edwards, W.R.; Williams, A.J.; Morris, J.L.; Baldwin, A.J.; Allemann, R.K.; Jones, D.D. Regulation of beta-lactamase activity by remote binding of heme: Functional coupling of unrelated proteins through domain insertion. Biochemistry 2010, 49, 6541–6549. [Google Scholar]
- Fan, F.; Binkowski, B.F.; Butler, B.L.; Stecha, P.F.; Lewis, M.K.; Wood, K.V. Novel genetically encoded biosensors using firefly luciferase. ACS Chem. Biol. 2008, 3, 346–351. [Google Scholar] [CrossRef]
- Buskirk, A.R.; Ong, Y.-C.; Gartner, Z.J.; Liu, D.R. Directed evolution of ligand dependence: Small-molecule-activated protein splicing. Proc. Natl. Acad. Sci. USA 2004, 101, 10505–10510. [Google Scholar]
- Skretas, G.; Wood, D.W. Regulation of protein activity with small-molecule-controlled inteins. Protein Sci. 2005, 14, 523–532. [Google Scholar] [CrossRef]
- Deckert, K.; Budiardjo, S.J.; Brunner, L.C.; Lovell, S.; Karanicolas, J. Designing alllosteric control into enzymes by chemical rescue of structure. J. Am. Chem. Soc. 2012, 134, 10055–10060. [Google Scholar]
- Xia, Y.; DiPrimo, N.; Keppel, T.R.; Vo, B.; Fraser, K.; Battaile, K.P.; Egan, C.; Bystroff, C.; Lovell, S.; Weis, D.D.; et al. The designability of protein switches by chemical rescue of structure: Mechanism of inactivation and reactivation. J. Am. Chem. Soc. 2013, 135, 18840–18849. [Google Scholar] [CrossRef]
- Lin, Q.; Barbas, C.F., III; Schultz, P.G. Small-molecule switches for zinc finger transcription factors. J. Am. Chem. Soc. 2002, 125, 612–613. [Google Scholar]
- Korendovych, I.V.; Kulp, D.W.; Wu, Y.; Cheng, H.; Roder, H.; DeGrado, W.F. Design of a switchable eliminase. Proc. Natl. Acad. Sci. USA 2011, 108, 6823–6827. [Google Scholar]
- Moroz, O.V.; Moroz, Y.S.; Wu, Y.; Olsen, A.B.; Cheng, H.; Mack, K.L.; McLaughlin, J.M.; Raymond, E.A.; Zhezherya, K.; Roder, H.; et al. A single mutation in a regulatory protein produces evolvable allosterically regulated catalyst of nonnatural reaction. Angew. Chem. Int. Ed. 2013, 52, 6246–6249. [Google Scholar] [CrossRef]
- Mack, K.L.; Moroz, O.V.; Moroz, Y.S.; Olsen, A.B.; McLaughlin, J.M.; Korendovych, I.V. Reprogramming EF-hands for design of catalytically amplified lanthanide sensors. J. Biol. Inorg. Chem. 2013, 18, 411–418. [Google Scholar] [CrossRef]
- Dwyer, M.A.; Looger, L.L.; Hellinga, H.W. Computational design of a Zn2+ receptor that controls bacterial gene expression. Proc. Natl. Acad. Sci. USA 2003, 100, 11255–11260. [Google Scholar] [CrossRef]
- Looger, L.L.; Dwyer, M.A.; Smith, J.J.; Hellinga, H.W. Computational design of receptor and sensor proteins with novel functions. Nature 2003, 423, 185–190. [Google Scholar] [CrossRef]
- Schreier, B.; Stumpp, C.; Wiesner, S.; Höcker, B. Computational design of ligand binding is not a solved problem. Proc. Natl. Acad. Sci. USA 2009, 106, 18491–18496. [Google Scholar] [CrossRef]
- Liang, H.; Deng, X.; Bosscher, M.; Ji, Q.; Jensen, M.P.; He, C. Enginering bacterial two-component system PmrA/PmrB to sense lanthanide ions. J. Am. Chem. Soc. 2013, 135, 2037–2039. [Google Scholar] [CrossRef]
- Mensa, B.; Kim, Y.H.; Choi, S.; Scott, R.; Caputo, G.A.; DeGrado, W.F. Antibacterial mechanism of action of arylamide foldamers. Antimicrob. Agents Chemother. 2011, 55, 5043–5053. [Google Scholar] [CrossRef]
- Hancock, R.E.W.; Wong, P.W. Compounds which increase the permeability of the pseudomonas aeruginosa outer membrane. Antimicrob. Agents Chemother. 1984, 26, 48–52. [Google Scholar] [CrossRef]
- Gibney, K.A.; Sovadinova, I.; Lopez, A.I.; Urban, M.; Ridgway, Z.; Caputo, G.A.; Kuroda, K. Poly(ethylene imine)s as antimicrobial agents with selective activity. Macromol. Biosci. 2012, 12, 1279–1289. [Google Scholar] [CrossRef]
- Wiester, M.J.; Ulmann, P.A.; Mirkin, C.A. Enzyme mimics based upon supramolecular coordination chemistry. Angew. Chem. Int. Ed. 2011, 50, 114–137. [Google Scholar] [CrossRef]
- Kremer, C.; Lützen, A. Artificial allosteric receptors. Chem. Eur. J. 2013, 19, 6162–6196. [Google Scholar] [CrossRef]
- Kovbasyuk, L.; Krämer, R. Allosteric supramolecular receptors and catalysts. Chem. Rev. 2004, 104, 3161–3187. [Google Scholar] [CrossRef]
- Raynal, M.; Ballester, P.; Vidal-Ferran, A.; van Leeuwen, P.W.N.M. Supramolecular catalysis. Part 2: Artificial enzyme mimics. Chem. Soc. Rev. 2014, 43, 1734–1787. [Google Scholar] [CrossRef]
- Gianneschi, N.C.; Masar, M.S., III; Mirkin, C.A. Development of a coordination chemistry-based approach for functional supramolecular structures. Acc. Chem. Res. 2005, 38, 825–837. [Google Scholar] [CrossRef]
- Holliday, B.J.; Mirkin, C.A. Strategies for the construction of supramolecular compounds through coordination chemistry. Angew. Chem. Int. Ed. 2001, 40, 2022–2043. [Google Scholar] [CrossRef]
- Gianneschi, N.C.; Nguyen, S.T.; Mirkin, C.A. Signal amplification and detection via a supramolecular allosteric catalyst. J. Am. Chem. Soc. 2005, 127, 1644–1645. [Google Scholar] [CrossRef]
- Yoon, H.J.; Mirkin, C.A. PCR-like cascade reactions in the context of an allosteric enzyme mimic. J. Am. Chem. Soc. 2008, 130, 11590–11591. [Google Scholar] [CrossRef]
- Masar, M.S., III; Gianneschi, N.C.; Oliveri, C.G.; Stern, C.L.; Nguyen, S.T.; Mirkin, C.A. Allosterically regulated supramolecular catalysis of acyl transfer reactions for signal amplification and detection of small molecules. J. Am. Chem. Soc. 2007, 129, 10149–10158. [Google Scholar]
- Yoon, H.J.; Heo, J.; Mirkin, C.A. Allosteric regulation of phosphate diester transesterification based upon a dinuclear zinc catalyst assembled via the weak-link approach. J. Am. Chem. Soc. 2007, 129, 14182–14183. [Google Scholar] [CrossRef]
- Wu, Q.; Anslyn, E.V. Catalytic signal amplification using a heck reaction. An axample in the fluorescence sensing of Cu(II). J. Am. Chem. Soc. 2004, 126, 14682–14683. [Google Scholar] [CrossRef]
- Wu, Q.; Anslyn, E.V. Heavy metal analysis using a heck-catalyzed cyclization to create coumarin. J. Mat. Chem. 2005, 15, 2815–2819. [Google Scholar] [CrossRef]
- Zhu, L.; Lynch, V.M.; Anslyn, E.V. Fret induced by an “allosteric” cycloaddition reaction regulated with exogenous inhibitor and effectors. Tetrahedron 2004, 60, 7267–7275. [Google Scholar] [CrossRef]
- Sella, E.; Lubelski, A.; Klafter, J.; Shabat, D. Two-component dendritic chain reactions: Experiment and theory. J. Am. Chem. Soc. 2010, 132, 3945–3952. [Google Scholar] [CrossRef]
- Perry-Feigenbaum, R.; Sella, E.; Shabat, D. Autoinductive exponential signal amplification: A diagnostic probe for direct detection of fluoride. Chem. Eur. J. 2011, 17, 12123–12128. [Google Scholar] [CrossRef]
- Baker, M.S.; Phillips, S.T. A two-component small molecule system for activity-based detection and signal amplification: Application to the visual detection of threshold levels of Pd(II). J. Am. Chem. Soc. 2011, 133, 5170–5173. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Makhlynets, O.V.; Korendovych, I.V. Design of Catalytically Amplified Sensors for Small Molecules. Biomolecules 2014, 4, 402-418. https://doi.org/10.3390/biom4020402
Makhlynets OV, Korendovych IV. Design of Catalytically Amplified Sensors for Small Molecules. Biomolecules. 2014; 4(2):402-418. https://doi.org/10.3390/biom4020402
Chicago/Turabian StyleMakhlynets, Olga V., and Ivan V. Korendovych. 2014. "Design of Catalytically Amplified Sensors for Small Molecules" Biomolecules 4, no. 2: 402-418. https://doi.org/10.3390/biom4020402
APA StyleMakhlynets, O. V., & Korendovych, I. V. (2014). Design of Catalytically Amplified Sensors for Small Molecules. Biomolecules, 4(2), 402-418. https://doi.org/10.3390/biom4020402