
Aberrant Glycosylation of Anchor-Optimized MUC1 Peptides Can Enhance Antigen Binding Affinity and Reverse Tolerance to Cytotoxic T Lymphocytes

 ,
, 
Abstract
:1. Introduction
2. Results
2.1. Identification of Heteroclitic and Aberrantly Glycosylated Peptides for a Human Vaccine
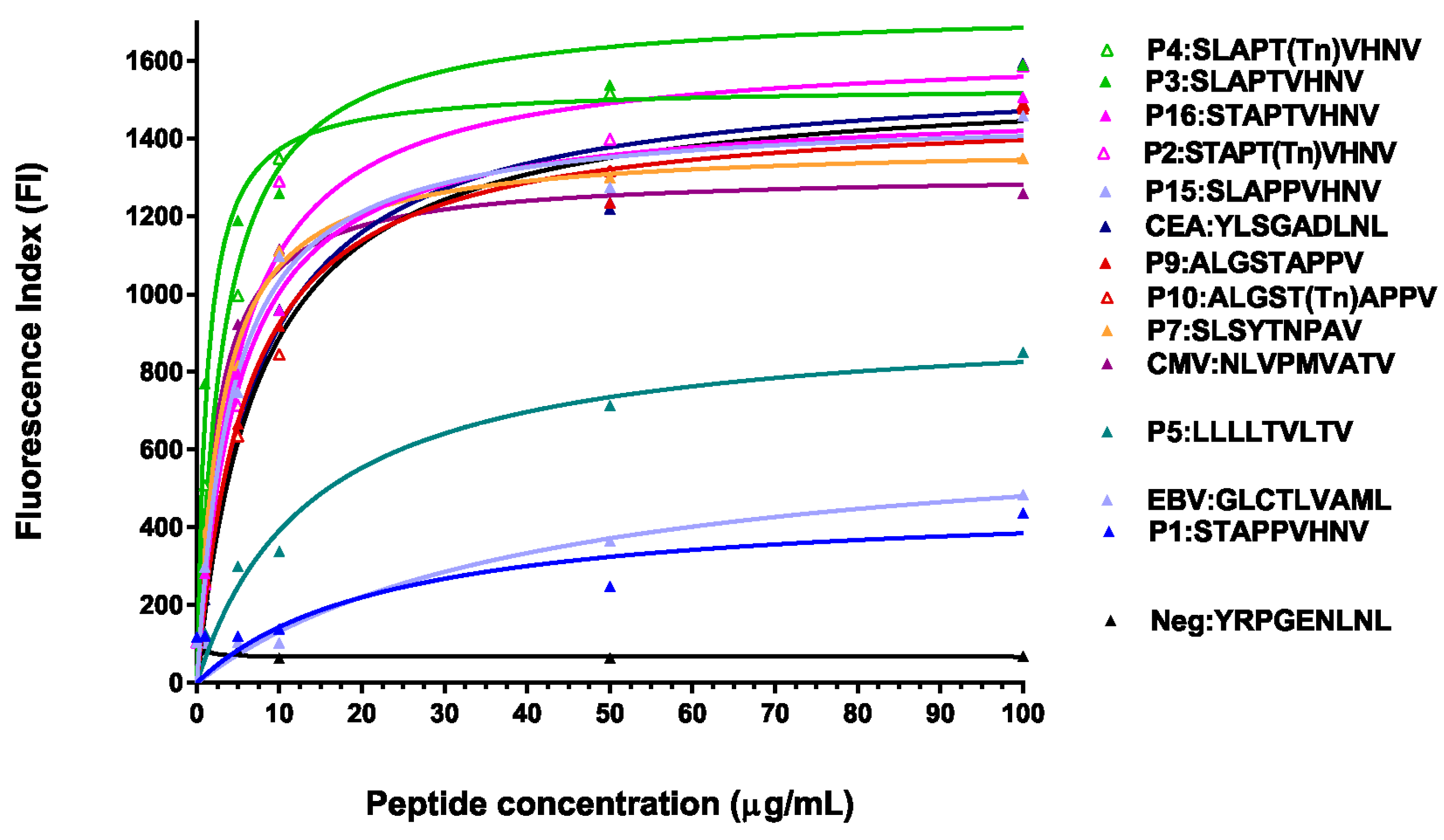
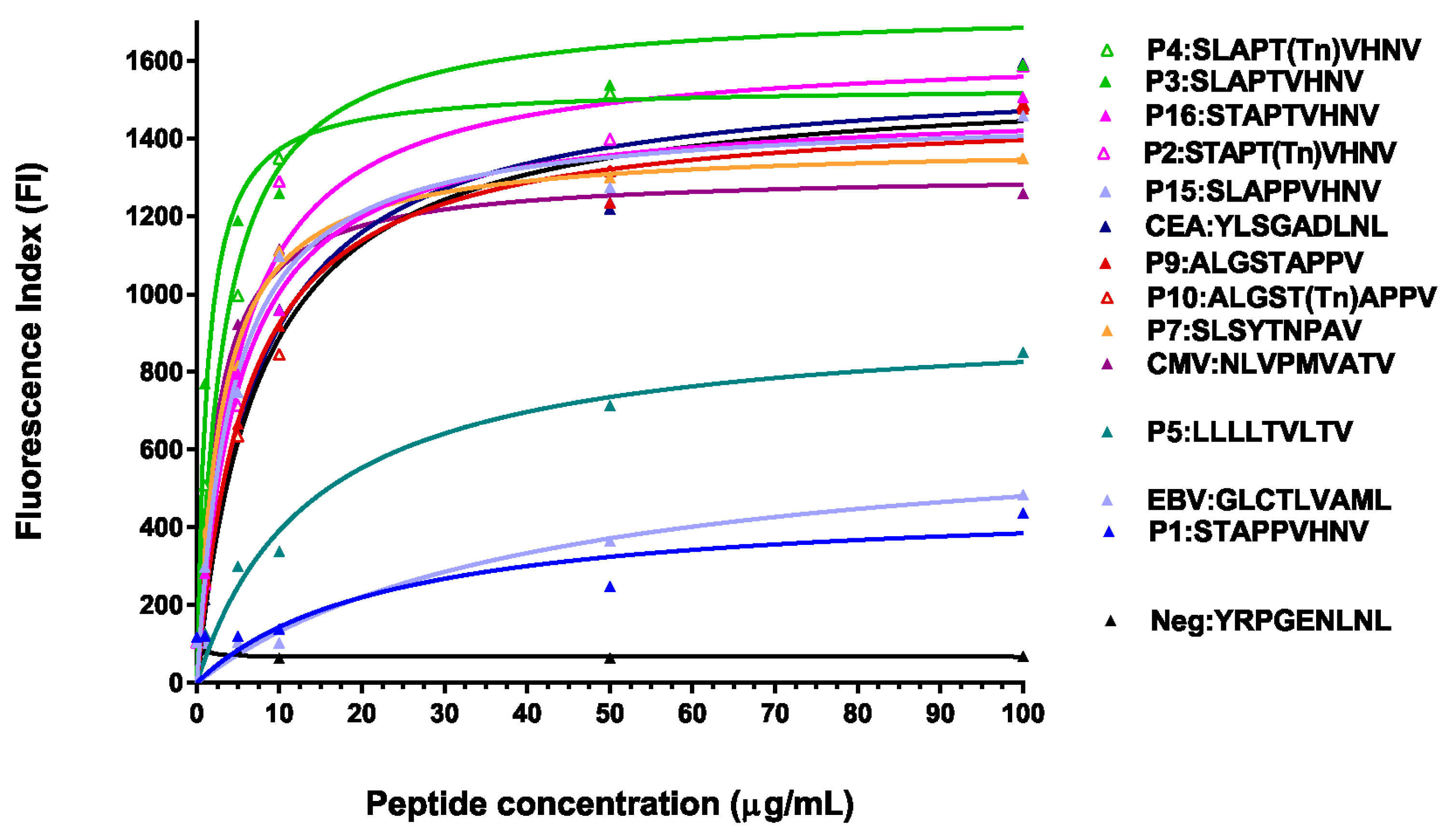
2.2. Glycosylated and/or Anchor-Optimized Peptides Show Increased Stabilization of HLA-A*0201 Molecules on T2 Cells
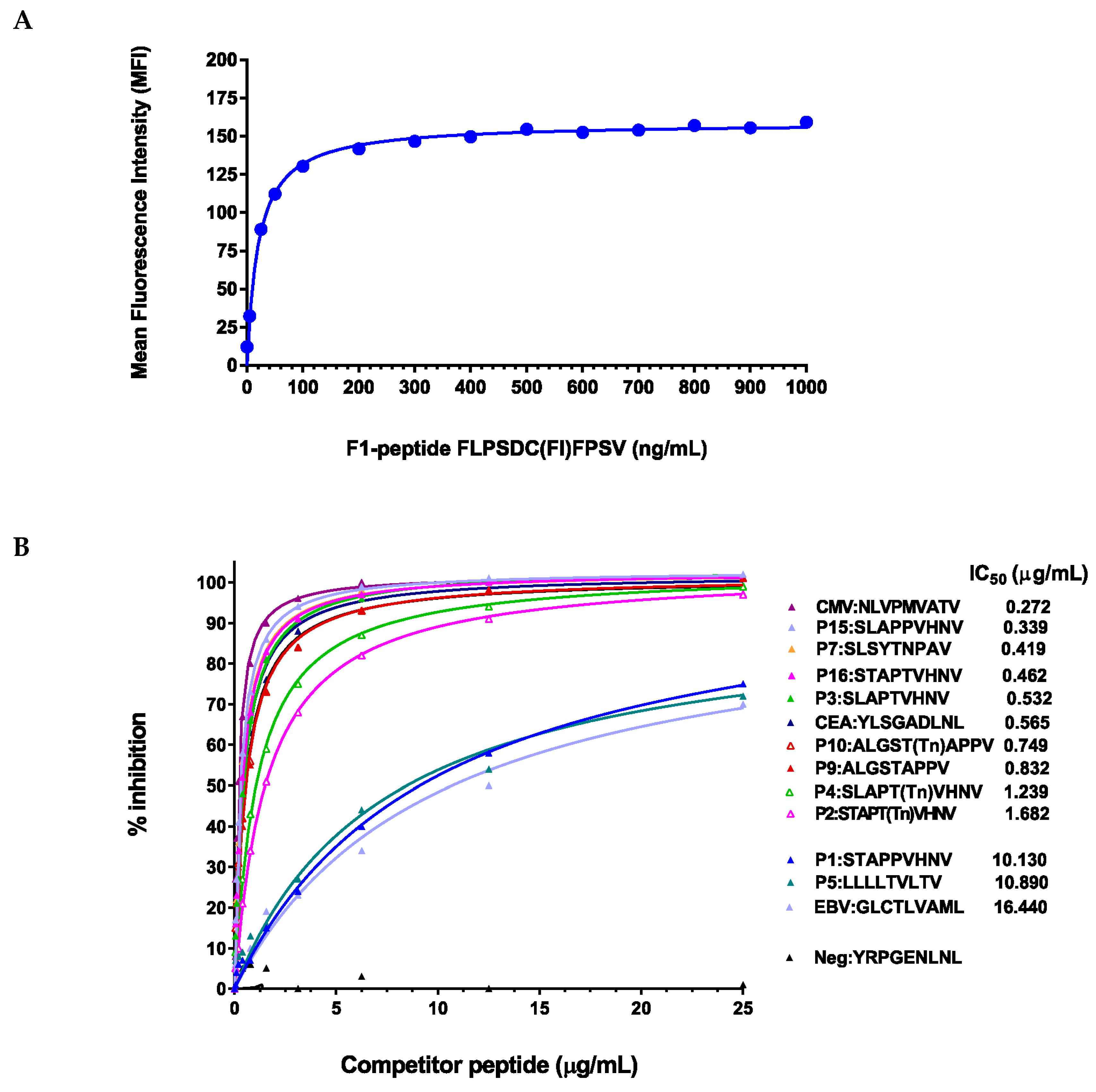
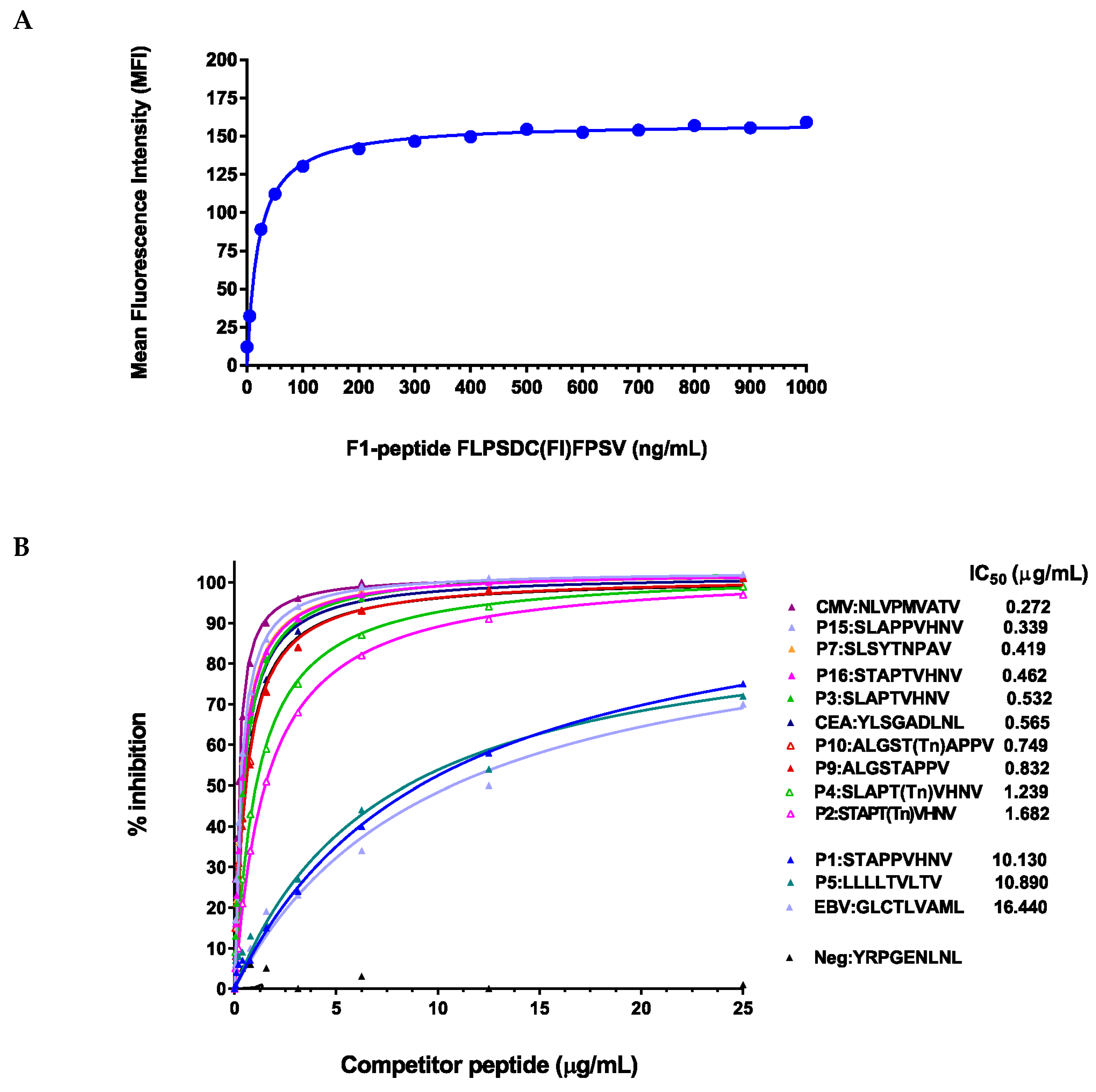
2.3. Peptide Modifications Increased Binding Affinities for HLA-A*0201 Molecules
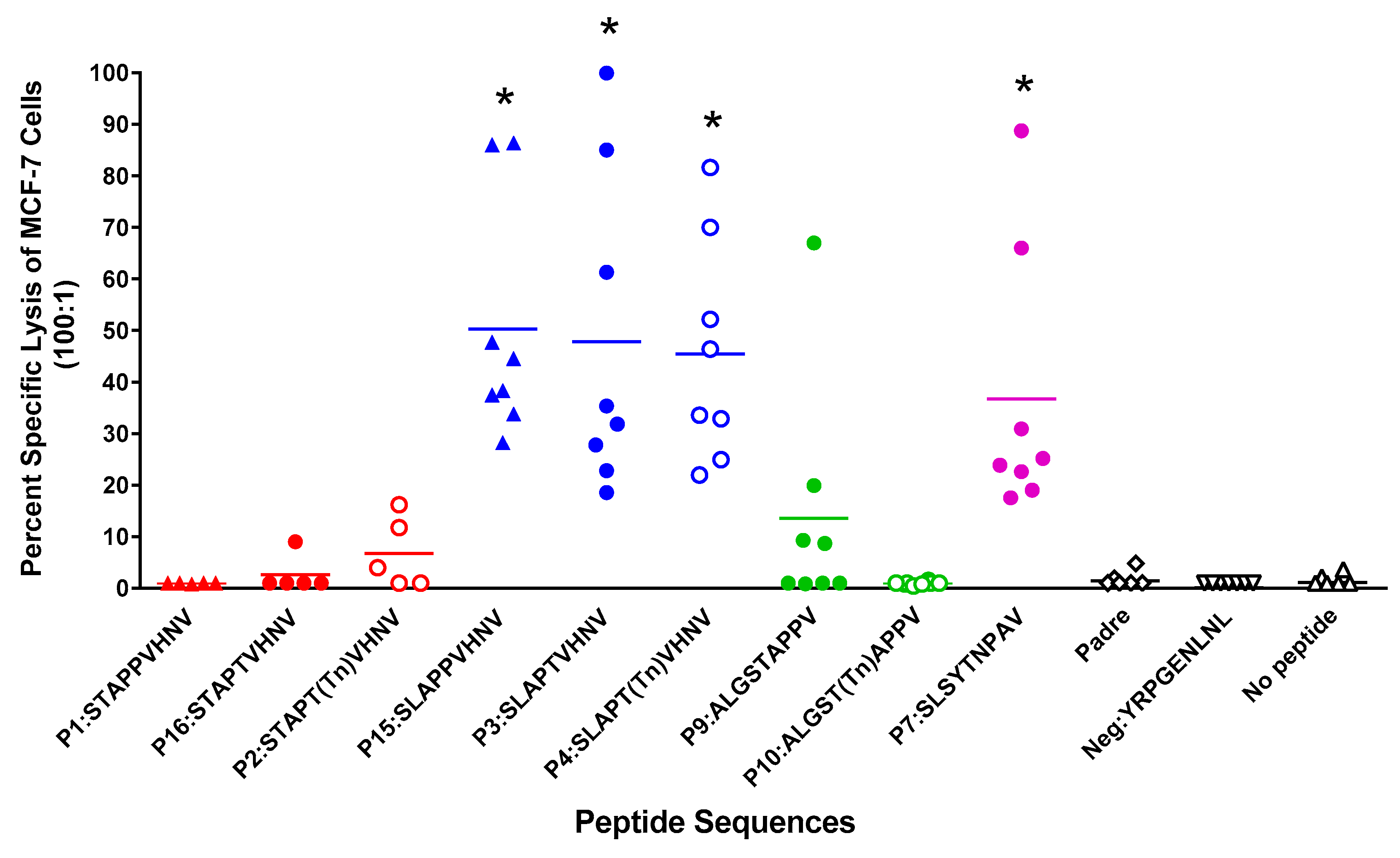
2.4. In Vitro Stimulation of T Cells from Normal HLA-A*0201 Women Elicited Strong MUC1-Specific CTL Responses
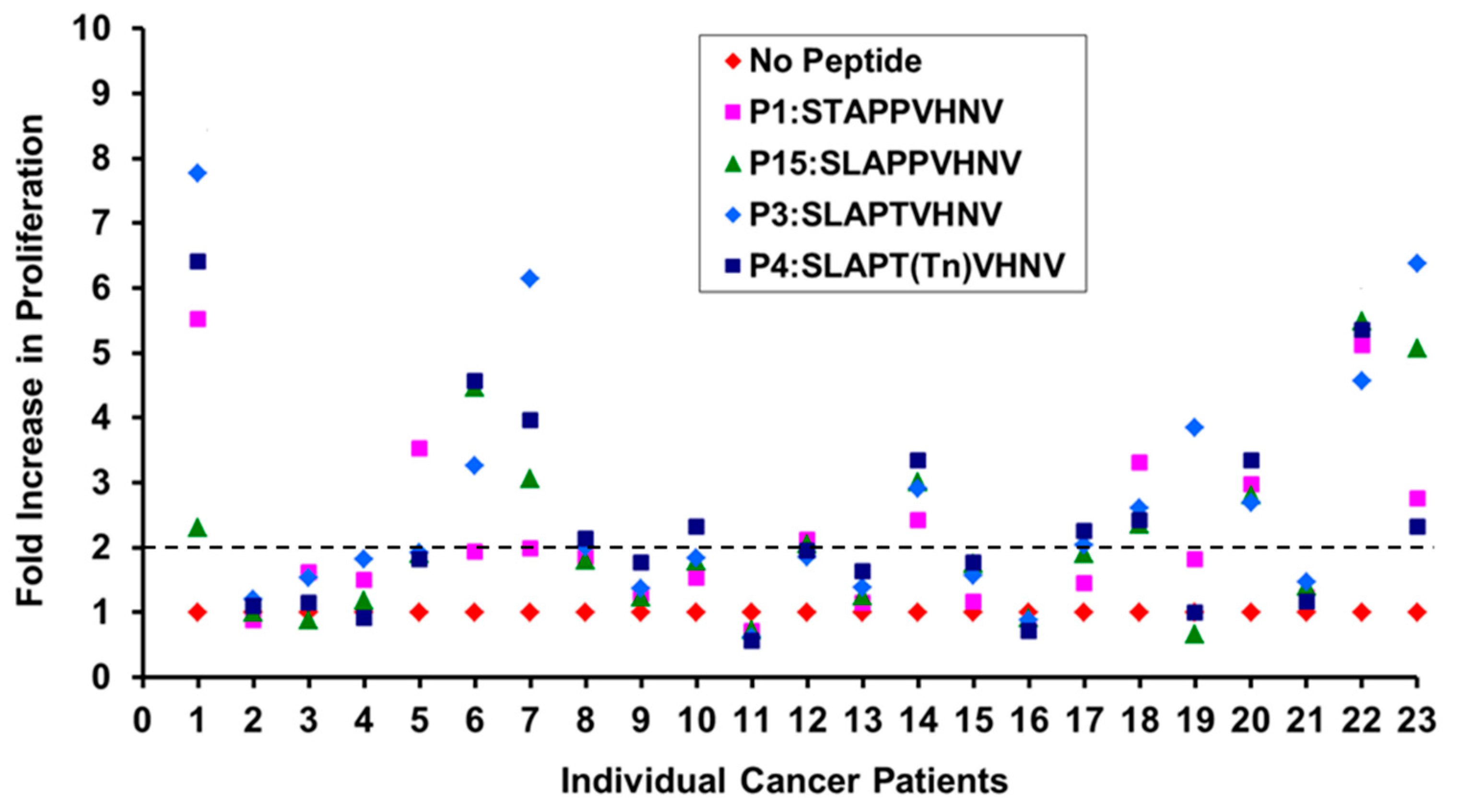
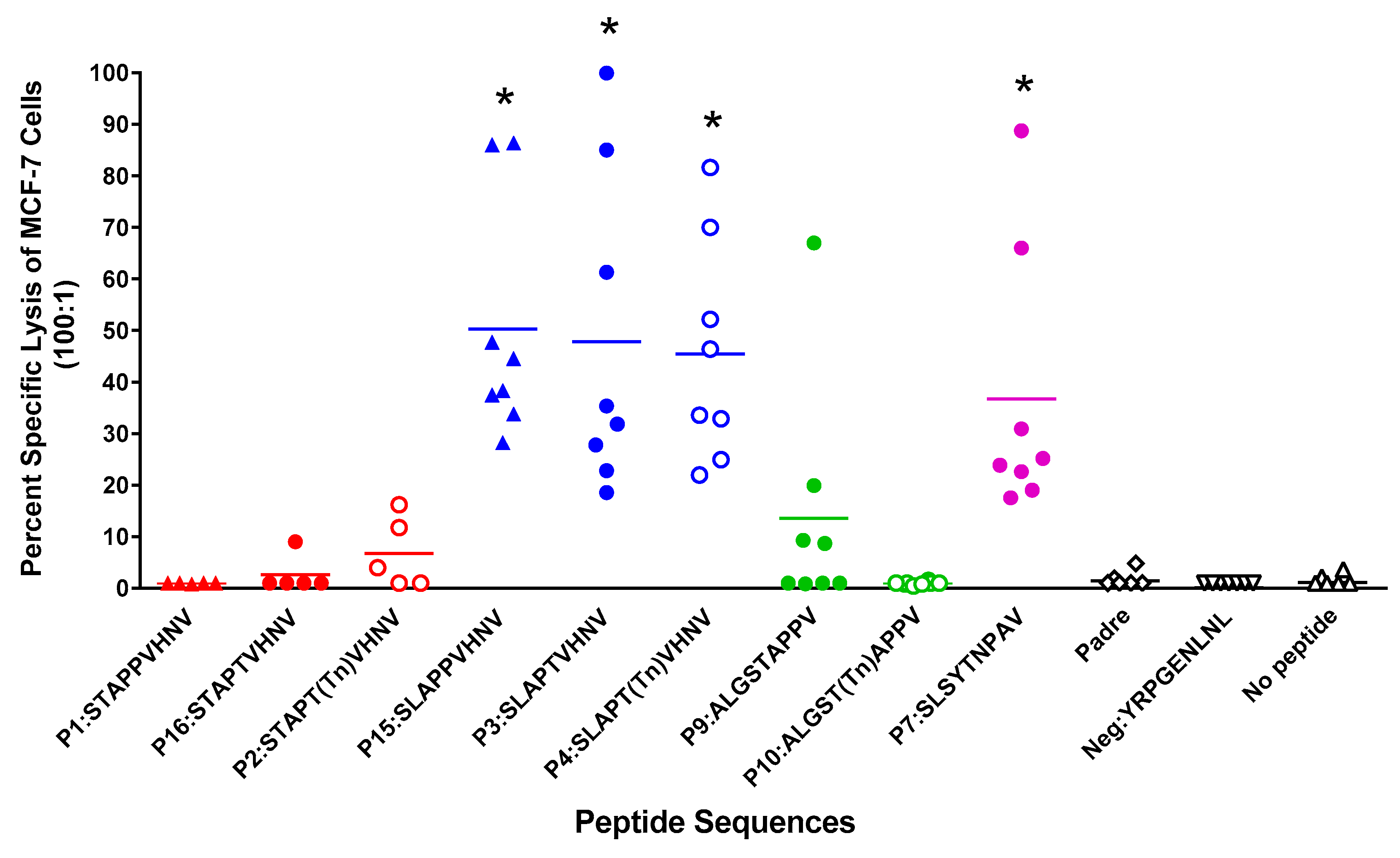
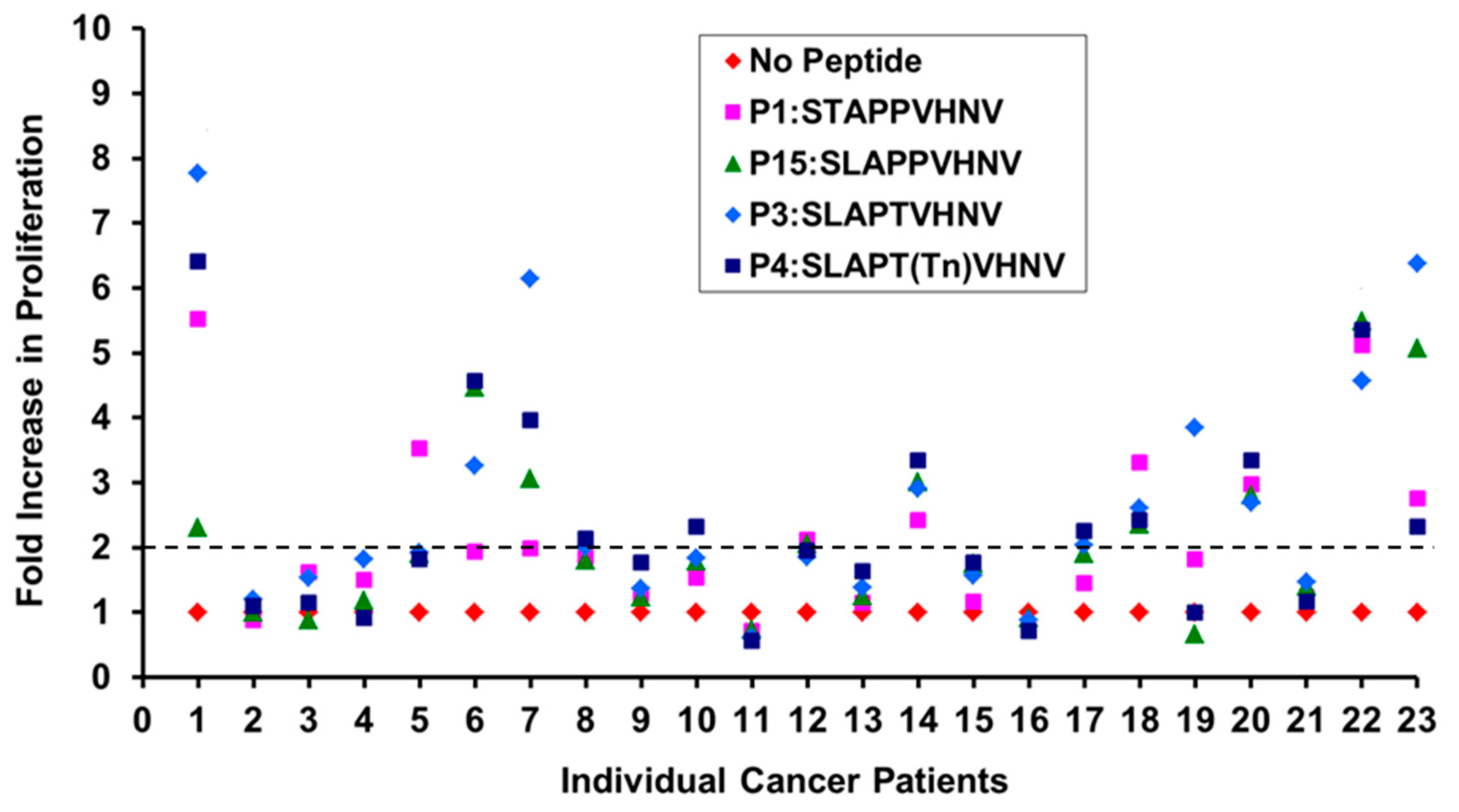
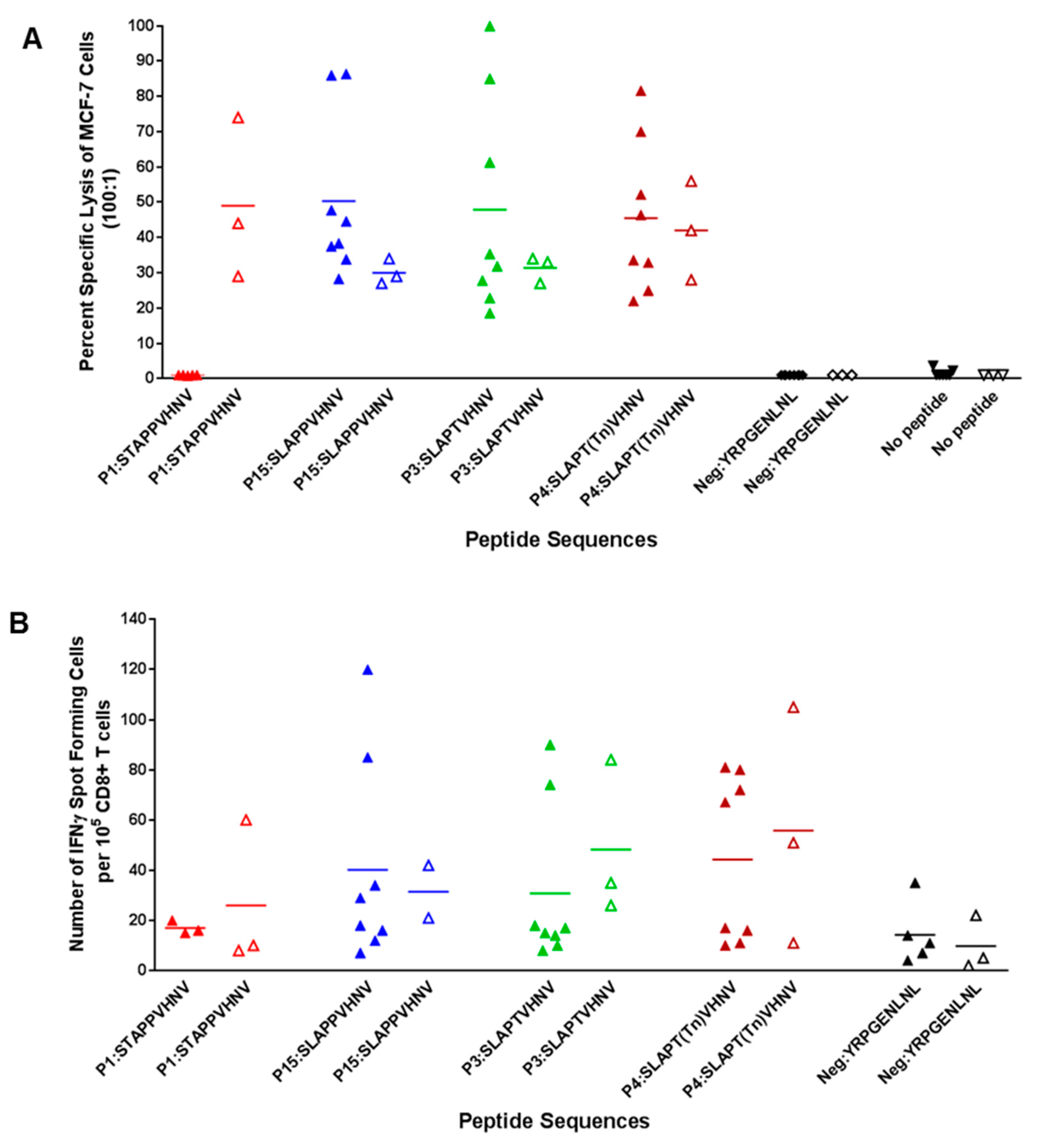
2.5. Breast Cancer Patients Recognize and Proliferate to the MUC1 Peptides in Vitro
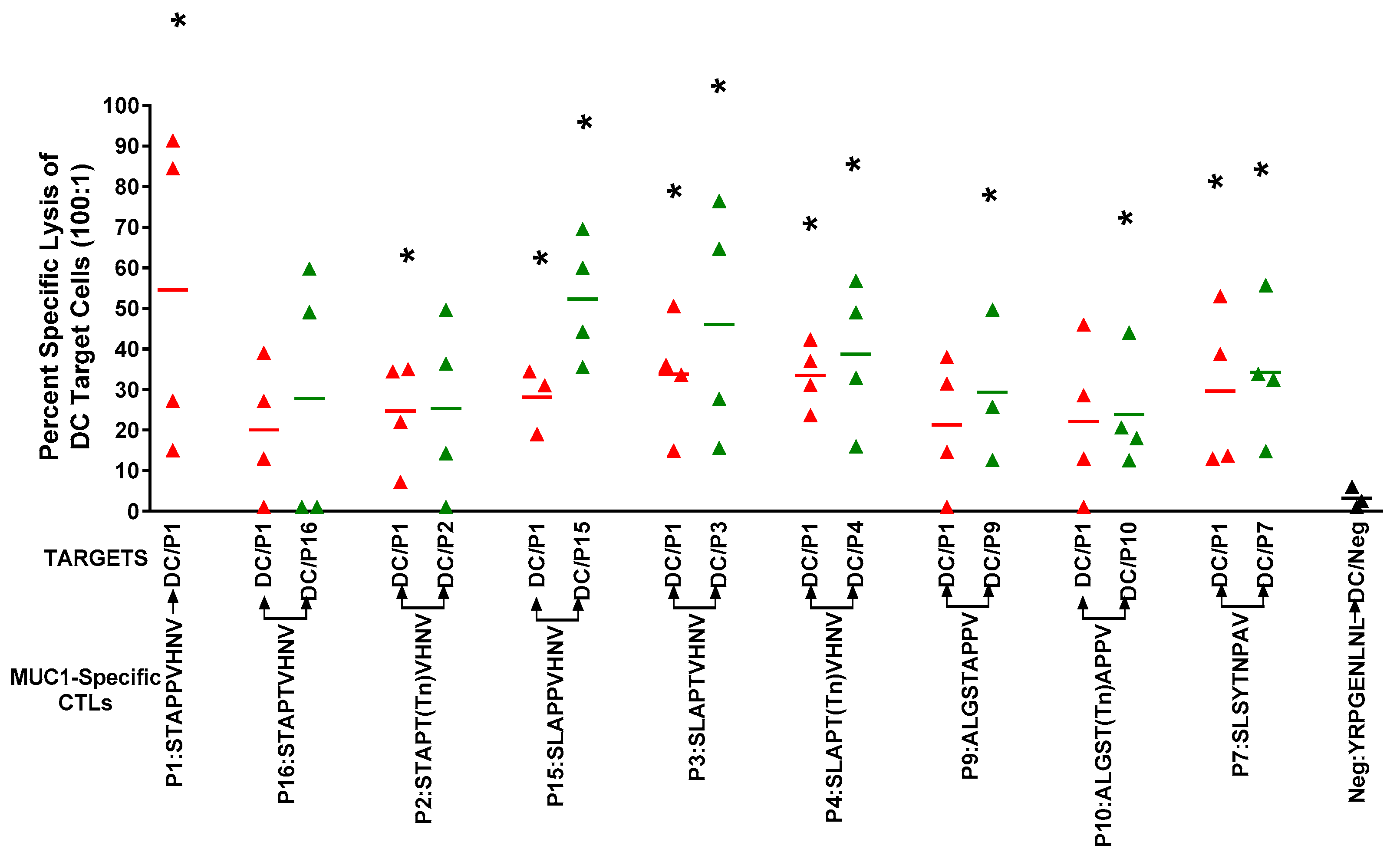
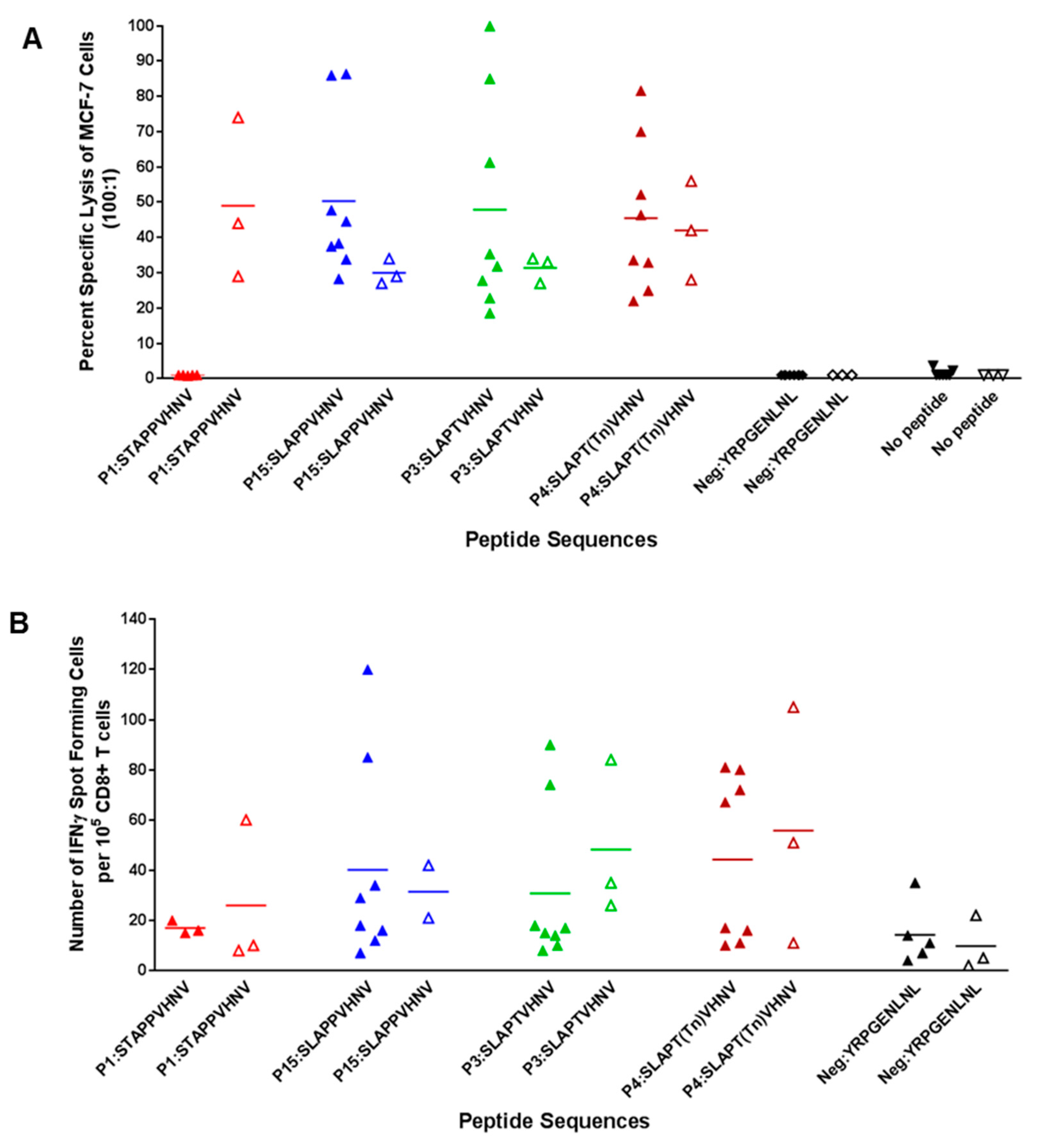
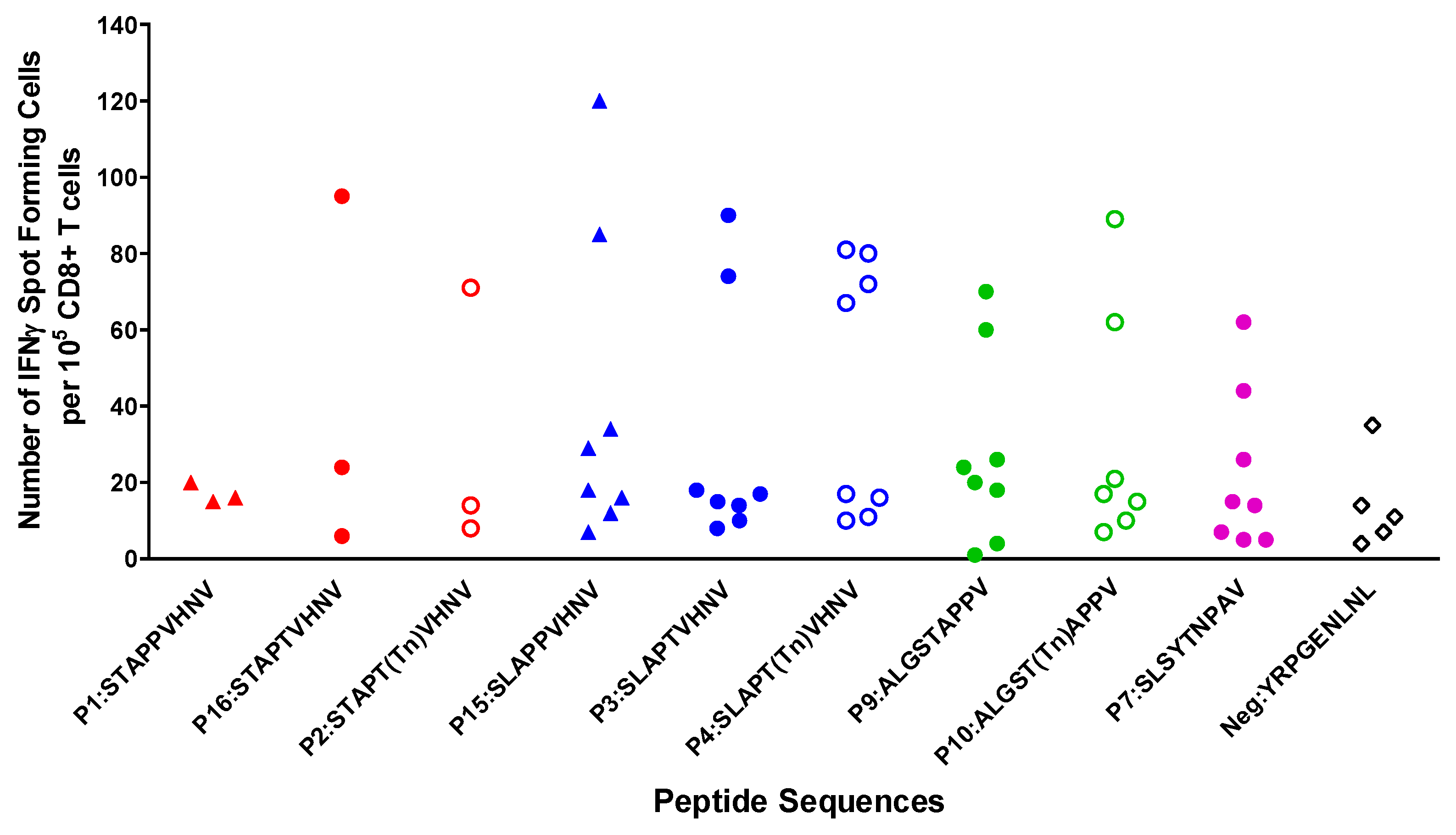
2.6. In Vitro Stimulation of T Cells from Breast Cancer Patients Elicited a Strong CTL Response
3. Discussion
4. Materials and Methods
4.1. Peptide Synthesis
4.2. MHC Stabilization Assay
4.3. Competitive Peptide Inhibition Assay
4.4. Isolation of PBMCs and HLA Testing
4.5. In Vitro Induction of Peptide-Specific CTLs
4.6. Measurement of Cytolytic Activity of CTLs by 51Cr-Release Assay
4.7. Detection of IFNγ Secreting MUC1-Specific Cytotoxic CD8+ T Cells by ELISpot
4.8. Ethics Statement
4.9. Statistics
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Moreno, B.H.; Ribas, A. Anti-programmed cell death protein-1/ligand-1 therapy in different cancers. Br. J. Cancer 2015, 112, 1421–1427. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Horn, L.; Antonia, S.; Spigel, D.R.; Gandhi, L.; Sequist, L.V.; Wigginton, J.; McDonald, D.; Kollia, G.; Gupta, A.K.; et al. Clinical activity and safety of anti-PD1 (BMS-936558, MDX-1106) in patients with advanced non-small-cell lung cancer (NSCLC). J. Clin. Oncol. 2012, 30. Abstr. 7509. [Google Scholar]
- Foley, K.; Kim, V.; Jaffee, E.; Zheng, L. Current progress in immunotherapy for pancreatic cancer. Cancer Lett. 2015. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Pusztai, L.; Ladanyi, A.; Szekely, B.; Dank, M. Immunotherapy opportunities in breast cancer. Magy. Onkol. 2016, 60, 34–40. [Google Scholar] [PubMed]
- Strickland, K.C.; Howitt, B.E.; Shukla, S.A.; Rodig, S.; Ritterhouse, L.L.; Liu, J.F.; Garber, J.E.; Chowdhury, D.; Wu, C.J.; D’Andrea, A.D.; et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget 2016, 7, 13587–13598. [Google Scholar] [PubMed]
- Soo, R.A. Shedding light on the molecular determinants of response to anti-PD-1 therapy. Transl. Lung Cancer Res. 2015, 4, 816–819. [Google Scholar] [PubMed]
- Lutz, E.R.; Wu, A.A.; Bigelow, E.; Sharma, R.; Mo, G.; Soares, K.; Solt, S.; Dorman, A.; Wamwea, A.; Yager, A.; et al. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunol. Res. 2014, 2, 616–631. [Google Scholar] [CrossRef] [PubMed]
- Siroy, A.; Abdul-Karim, F.W.; Miedler, J.; Fong, N.; Fu, P.; Gilmore, H.; Baar, J. MUC1 is expressed at high frequency in early-stage basal-like triple-negative breast cancer. Hum. Pathol. 2013, 44, 2159–2166. [Google Scholar] [CrossRef] [PubMed]
- Garbar, C.; Mascaux, C.; Giustiniani, J.; Salesse, S.; Debelle, L.; Antonicelli, F.; Merrouche, Y.; Bensussan, A. Autophagy is decreased in triple-negative breast carcinoma involving likely the MUC1-EGFR-NEU1 signalling pathway. Int. J. Clin. Exp. Pathol. 2015, 8, 4344–4355. [Google Scholar] [PubMed]
- Cheever, M.A.; Allison, J.P.; Ferris, A.S.; Finn, O.J.; Hastings, B.M.; Hecht, T.T.; Mellman, I.; Prindiville, S.A.; Viner, J.L.; Weiner, L.M.; et al. The Prioritization of Cancer Antigens: A National Cancer Institute Pilot Project for the Acceleration of Translational Research. Clin. Cancer Res. 2009, 15, 5323–5337. [Google Scholar] [CrossRef] [PubMed]
- Cazet, A.; Julien, S.; Bobowski, M.; Burchell, J.; Delannoy, P. Tumour-associated carbohydrate antigens in breast cancer. Breast Cancer Res. 2010, 12, 204. [Google Scholar] [CrossRef] [PubMed]
- Pinho, S.S.; Reis, C.A. Glycosylation in cancer: Mechanisms and clinical implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Beatson, R.; Maurstad, G.; Picco, G.; Arulappu, A.; Coleman, J.; Wandell, H.H.; Clausen, H.; Mandel, U.; Taylor-Papadimitriou, J.; Sletmoen, M.; et al. The Breast Cancer-Associated Glycoforms of MUC1, MUC1-Tn and sialyl-Tn, Are Expressed in COSMC Wild-Type Cells and Bind the C-Type Lectin MGL. PLoS ONE 2015, 10, e0125994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butts, C.; Socinski, M.A.; Mitchell, P.L.; Thatcher, N.; Havel, L.; Krzakowski, M.; Nawrocki, S.; Ciuleanu, T.E.; Bosquee, L.; Trigo, J.M.; et al. Tecemotide (L-BLP25) versus placebo after chemoradiotherapy for stage III non-small-cell lung cancer (START): A randomised, double-blind, phase 3 trial. Lancet Oncol. 2014, 15, 59–68. [Google Scholar] [CrossRef]
- Nemunaitis, J.; Bedell, C.; Klucher, K.; Vo, A.; Whiting, S. Phase 1 dose escalation of ONT-10, a therapeutic MUC1 vaccine, in patients with advanced cancer. J. Immunother. Cancer 2013, 240. [Google Scholar] [CrossRef]
- Carmon, L.; Avivi, I.; Kovjazin, R.; Zuckerman, T.; Dray, L.; Gatt, M.E.; Or, R.; Shapira, M.Y. Phase I/II study exploring ImMucin, a pan-major histocompatibility complex, anti-MUC1 signal peptide vaccine, in multiple myeloma patients. Br. J. Haematol. 2015, 169, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Kondo, H.; Hazama, S.; Kawaoka, T.; Yoshino, S.; Yoshida, S.; Tokuno, K.; Takashima, M.; Ueno, T.; Hinoda, Y.; Oka, M. Adoptive immunotherapy for pancreatic cancer using MUC1 peptide-pulsed dendritic cells and activated T lymphocytes. Anticancer Res. 2008, 28, 379–387. [Google Scholar] [PubMed]
- Quoix, E.; Ramlau, R.; Westeel, V.; Papai, Z.; Madroszyk, A.; Riviere, A.; Koralewski, P.; Breton, J.L.; Stoelben, E.; Braun, D.; et al. Therapeutic vaccination with TG4010 and first-line chemotherapy in advanced non-small-cell lung cancer: A controlled phase 2B trial. Lancet Oncol. 2011, 12, 1125–1133. [Google Scholar] [CrossRef]
- Vassilaros, S.; Tsibanis, A.; Tsikkinis, A.; Pietersz, G.A.; McKenzie, I.F.; Apostolopoulos, V. Up to 15-year clinical follow-up of a pilot Phase III immunotherapy study in stage II breast cancer patients using oxidized mannan-MUC1. Immunotherapy 2013, 5, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, S.; Mead, A.; Malinovskis, A.; Hardwick, N.R.; Guinn, B.A. Analogue peptides for the immunotherapy of human acute myeloid leukemia. Cancer Immunol. Immunother. 2015, 64, 1357–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salazar, E.; Zaremba, S.; Arlen, P.M.; Tsang, K.Y.; Schlom, J. Agonist peptide from a cytotoxic t-lymphocyte epitope of human carcinoembryonic antigen stimulates production of tc1-type cytokines and increases tyrosine phosphorylation more efficiently than cognate peptide. Int. J. Cancer 2000, 85, 829–838. [Google Scholar]
- Tangri, S.; Ishioka, G.Y.; Huang, X.; Sidney, J.; Southwood, S.; Fikes, J.; Sette, A. Structural features of peptide analogs of human histocompatibility leukocyte antigen class I epitopes that are more potent and immunogenic than wild-type peptide. J. Exp. Med. 2001, 194, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Tsang, K.Y.; Palena, C.; Gulley, J.; Arlen, P.; Schlom, J. A human cytotoxic T-lymphocyte epitope and its agonist epitope from the nonvariable number of tandem repeat sequence of MUC-1. Clin. Cancer Res. 2004, 10, 2139–2149. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.; Samur, M.; Munshi, A.; Hideshima, T.; Keskin, D.; Kimmelman, A.; Lee, A.H.; Dranoff, G.; Anderson, K.C.; Munshi, N.C. Heteroclitic XBP1 peptides evoke tumor-specific memory cytotoxic T lymphocytes against breast cancer, colon cancer, and pancreatic cancer cells. Oncoimmunology 2014, 3, e970914. [Google Scholar] [CrossRef] [PubMed]
- Adegoke, A.O.; Grant, M.D. Enhancing Human Immunodeficiency Virus-Specific CD8(+) T Cell Responses with Heteroclitic Peptides. Front. Immunol. 2015, 6, 377. [Google Scholar] [CrossRef] [PubMed]
- Apostolopoulos, V.; Yu, M.; Corper, A.L.; Teyton, L.; Pietersz, G.A.; McKenzie, I.F.; Wilson, I.A.; Plebanski, M. Crystal structure of a non-canonical low-affinity peptide complexed with MHC class I: A new approach for vaccine design. J. Mol. Biol. 2002, 318, 1293–1305. [Google Scholar] [CrossRef]
- Chen, J.L.; Stewart-Jones, G.; Bossi, G.; Lissin, N.M.; Wooldridge, L.; Choi, E.M.; Held, G.; Dunbar, P.R.; Esnouf, R.M.; Sami, M.; et al. Structural and kinetic basis for heightened immunogenicity of T cell vaccines. J. Exp. Med. 2005, 201, 1243–1255. [Google Scholar] [CrossRef] [PubMed]
- Engels, B.; Engelhard, V.H.; Sidney, J.; Sette, A.; Binder, D.C.; Liu, R.B.; Kranz, D.M.; Meredith, S.C.; Rowley, D.A.; Schreiber, H. Relapse or eradication of cancer is predicted by peptide-major histocompatibility complex affinity. Cancer Cell 2013, 23, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Gendler, S.J.; Franco, A. Designer glycopeptides for cytotoxic T cell-based elimination of carcinomas. J. Exp. Med. 2004, 199, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Lakshminarayanan, V.; Thompson, P.; Wolfert, M.A.; Buskas, T.; Bradley, J.M.; Pathangey, L.B.; Madsen, C.S.; Cohen, P.A.; Gendler, S.J.; Boons, G.J. Immune recognition of tumor-associated mucin MUC1 is achieved by a fully synthetic aberrantly glycosylated MUC1 tripartite vaccine. Proc. Natl. Acad. Sci. USA 2012, 109, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Aal, A.M.; Lakshminarayanan, V.; Thompson, P.; Supekar, N.; Bradley, J.M.; Wolfert, M.A.; Cohen, P.A.; Gendler, S.J.; Boons, G.J. Immune and Anticancer Responses Elicited by Fully Synthetic Aberrantly Glycosylated MUC1 Tripartite Vaccines Modified by a TLR2 or TLR9 Agonist. Chembiochem 2014, 15, 1508–1513. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.; Lakshminarayanan, V.; Supekar, N.T.; Bradley, J.M.; Cohen, P.A.; Wolfert, M.A.; Gendler, S.J.; Boons, G.J. Linear synthesis and immunological properties of a fully synthetic vaccine candidate containing a sialylated MUC1 glycopeptide. Chem. Commun. 2015, 51, 10214–10217. [Google Scholar] [CrossRef] [PubMed]
- Lakshminarayanan, V.; Supekar, N.T.; Wei, J.; McCurry, D.B.; Dueck, A.C.; Kosiorek, H.E.; Trivedi, P.P.; Bradley, J.M.; Madsen, C.S.; Pathangey, L.B.; et al. MUC1 Vaccines, Comprised of Glycosylated or Non-Glycosylated Peptides or Tumor-Derived MUC1, Can Circumvent Immunoediting to Control Tumor Growth in MUC1 Transgenic Mice. PLoS ONE 2016, 11, e0145920. [Google Scholar] [CrossRef] [PubMed]
- Apostolopoulos, V.; Chelvanayagam, G.; Xing, P.X.; McKenzie, I.F. Anti-MUC1 antibodies react directly with MUC1 peptides presented by class I H2 and HLA molecules. J. Immunol. 1998, 161, 767–775. [Google Scholar] [PubMed]
- Madden, D.R.; Garboczi, D.N.; Wiley, D.C. The antigenic identity of peptide-MHC complexes: A comparison of the conformations of five viral peptides presented by HLA-A2. Cell 1993, 75, 693–708. [Google Scholar] [CrossRef]
- Apostolopoulos, V.; Haurum, J.S.; McKenzie, I.F.C. MUC1 peptide epitopes associated with five different H-2 class I molecules. Eur. J. Immunol. 1997, 27, 2579–2587. [Google Scholar] [CrossRef] [PubMed]
- Falk, K.; Rotzschke, O.; Stevanovic, S.; Jung, G.; Rammensee, H.G. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature 1991, 351, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Brossart, P.; Heinrich, K.S.; Stuhler, G.; Behnke, L.; Reichardt, V.L.; Stevanovic, S.; Muhm, A.; Rammensee, H.G.; Kanz, L.; Brugger, W. Identification of HLA-A2-restricted T-cell epitopes derived from the MUC1 tumor antigen for broadly applicable vaccine therapies. Blood 1999, 93, 4309–4317. [Google Scholar] [PubMed]
- Kessler, J.H.; Mommaas, B.; Mutis, T.; Huijbers, I.; Vissers, D.; Benckhuijsen, W.E.; Schreuder, G.M.; Offringa, R.; Goulmy, E.; Melief, C.J.; et al. Competition-based cellular peptide binding assays for 13 prevalent HLA class I alleles using fluorescein-labeled synthetic peptides. Hum. Immunol. 2003, 64, 245–255. [Google Scholar] [CrossRef]
- Zaremba, S.; Barzaga, E.; Zhu, M.; Soares, N.; Tsang, K.Y.; Schlom, J. Identification of an enhancer agonist cytotoxic T lymphocyte peptide from human carcinoembryonic antigen. Cancer Res. 1997, 57, 4570–4577. [Google Scholar] [PubMed]
- Brossart, P.; Wirths, S.; Stuhler, G.; Reichardt, V.L.; Kanz, L.; Brugger, W. Induction of cytotoxic T-lymphocyte responses in vivo after vaccinations with peptide-pulsed dendritic cells. Blood 2000, 96, 3102–3108. [Google Scholar] [PubMed]
- Slifka, M.K.; Rodriguez, F.; Whitton, J.L. Rapid on/off cycling of cytokine production by virus-specific CD8+ T cells. Nature 1999, 401, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Krauss, J.C.; Plautz, G.E.; Mukai, S.; Shu, S.; Cohen, P.A. T cell-mediated tumor rejection displays diverse dependence upon perforin and IFN-gamma mechanisms that cannot be predicted from in vitro T cell characteristics. J. Immunol. 2000, 165, 7116–7124. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Elford, A.R.; Murakami, K.; Garza, K.M.; Schoenberger, S.P.; Odermatt, B.; Speiser, D.E.; Ohashi, P.S. Tumor growth enhances cross-presentation leading to limited T cell activation without tolerance. J. Exp. Med. 2002, 195, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Apostolopoulos, V.; Yuriev, E.; Ramsland, P.A.; Halton, J.; Osinski, C.; Li, W.; Plebanski, M.; Paulsen, H.; McKenzie, I.F. A glycopeptide in complex with MHC class I uses the GalNAc residue as an anchor. Proc. Natl. Acad. Sci. USA 2003, 100, 15029–15034. [Google Scholar] [CrossRef] [PubMed]
- Van der Burg, S.H.; Arens, R.; Ossendorp, F.; van Hall, T.; Melief, C.J. Vaccines for established cancer: Overcoming the challenges posed by immune evasion. Nat. Rev. Cancer 2016, 16, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Weiss, V.L.; Lee, T.H.; Song, H.; Kouo, T.S.; Black, C.M.; Sgouros, G.; Jaffee, E.M.; Armstrong, T.D. Trafficking of high avidity HER-2/neu-specific T cells into HER-2/neu-expressing tumors after depletion of effector/memory-like regulatory T cells. PLoS ONE 2012, 7, e31962. [Google Scholar] [CrossRef] [PubMed]
- Twyman-Saint Victor, C.; Rech, A.J.; Maity, A.; Rengan, R.; Pauken, K.E.; Stelekati, E.; Benci, J.L.; Xu, B.; Dada, H.; Odorizzi, P.M.; et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015, 520, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Sette, A.; Vitiello, A.; Reherman, B.; Fowler, P.; Nayersina, R.; Kast, W.M.; Melief, C.J.; Oseroff, C.; Yuan, L.; Ruppert, J.; et al. The relationship between class I binding affinity and immunogenicity of potential cytotoxic T cell epitopes. J. Immunol. 1994, 153, 5586–5592. [Google Scholar] [PubMed]
- Heukamp, L.C.; van der Burg, S.H.; Drijfhout, J.W.; Melief, C.J.; Taylor-Papadimitriou, J.; Offringa, R. Identification of three non-VNTR MUC1-derived HLA-A*0201-restricted T-cell epitopes that induce protective anti-tumor immunity in HLA-A2/K(b)-transgenic mice. Int. J. Cancer 2001, 91, 385–392. [Google Scholar] [CrossRef]
- Guan, P.; Doytchinova, I.A.; Walshe, V.A.; Borrow, P.; Flower, D.R. Analysis of peptide-protein binding using amino acid descriptors: Prediction and experimental verification for human histocompatibility complex HLA-A0201. J. Med. Chem. 2005, 48, 7418–7425. [Google Scholar] [CrossRef] [PubMed]
- Dietz, A.B.; Bulur, P.A.; Emery, R.L.; Winters, J.L.; Epps, D.E.; Zubair, A.C.; Vuk-Pavlovic, S. A novel source of viable peripheral blood mononuclear cells from leukoreduction system chambers. Transfusion 2006, 46, 2083–2089. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Human Peptide | Sequence | Gene | Modification |
|---|---|---|---|
| P:1 | STAPPVHNV | MUC1 | Degenerate TR |
| P:16 | STAPTVHNV | MUC1 | Modified |
| P: 2 | STAPT(Tn)VHNV | MUC1 | Glycosylated |
| P:15 | SLAPPVHNV | MUC1 | Anchor-optimized |
| P: 3 | SLAPTVHNV | MUC1 | Anchor-optimized |
| P: 4 | SLAPT(Tn)VHNV | MUC1 | Glycosylated |
| P: 9 | ALGSTAPPV | MUC1 | Degenerate TR |
| P:10 | ALGST(Tn)APPV | MUC1 | Glycosylated |
| P: 7 | SLSYTNPAV | MUC1 | Cytoplasmic tail |
| P: 5 | LLLLTVLTV | MUC1 | Signal peptide |
| P:11 | YRPGENLNL | None (Negative control) | |
| P:12 | YLSGADLNL | CEA | None (Positive control) |
| P:13 | GLCTLVAML | EBV | None (Positive control) |
| P:14 | NLVPMVATV | CMV | None (Positive control) |
| Peptide Sequence | % of Donors with at Least 30% Cell Kill | 95% CI |
|---|---|---|
| SLAPPVHNV | 87.50% | 47.3%–99.7% |
| SLAPT(Tn)VHNV | 75% | 34.9%–96.8% |
| SLAPTVHNV | 62.50% | 24.5%–91.48% |
| SLSYTNPAV | 37.50% | 8.5%–75.5% |
| ALGSTAPPV | 12.50% | 0.32%–52.6% |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pathangey, L.B.; Lakshminarayanan, V.; Suman, V.J.; Pockaj, B.A.; Mukherjee, P.; Gendler, S.J. Aberrant Glycosylation of Anchor-Optimized MUC1 Peptides Can Enhance Antigen Binding Affinity and Reverse Tolerance to Cytotoxic T Lymphocytes. Biomolecules 2016, 6, 31. https://doi.org/10.3390/biom6030031
Pathangey LB, Lakshminarayanan V, Suman VJ, Pockaj BA, Mukherjee P, Gendler SJ. Aberrant Glycosylation of Anchor-Optimized MUC1 Peptides Can Enhance Antigen Binding Affinity and Reverse Tolerance to Cytotoxic T Lymphocytes. Biomolecules. 2016; 6(3):31. https://doi.org/10.3390/biom6030031
Chicago/Turabian StylePathangey, Latha B., Vani Lakshminarayanan, Vera J. Suman, Barbara A. Pockaj, Pinku Mukherjee, and Sandra J. Gendler. 2016. "Aberrant Glycosylation of Anchor-Optimized MUC1 Peptides Can Enhance Antigen Binding Affinity and Reverse Tolerance to Cytotoxic T Lymphocytes" Biomolecules 6, no. 3: 31. https://doi.org/10.3390/biom6030031