
Role of Protein Quality Control Failure in Alcoholic Hepatitis Pathogenesis

Abstract
:1. Introduction
2. Role of the Ufmylation and FAT10ylation Pathways to Protein Degradation by the 26S Proteasome
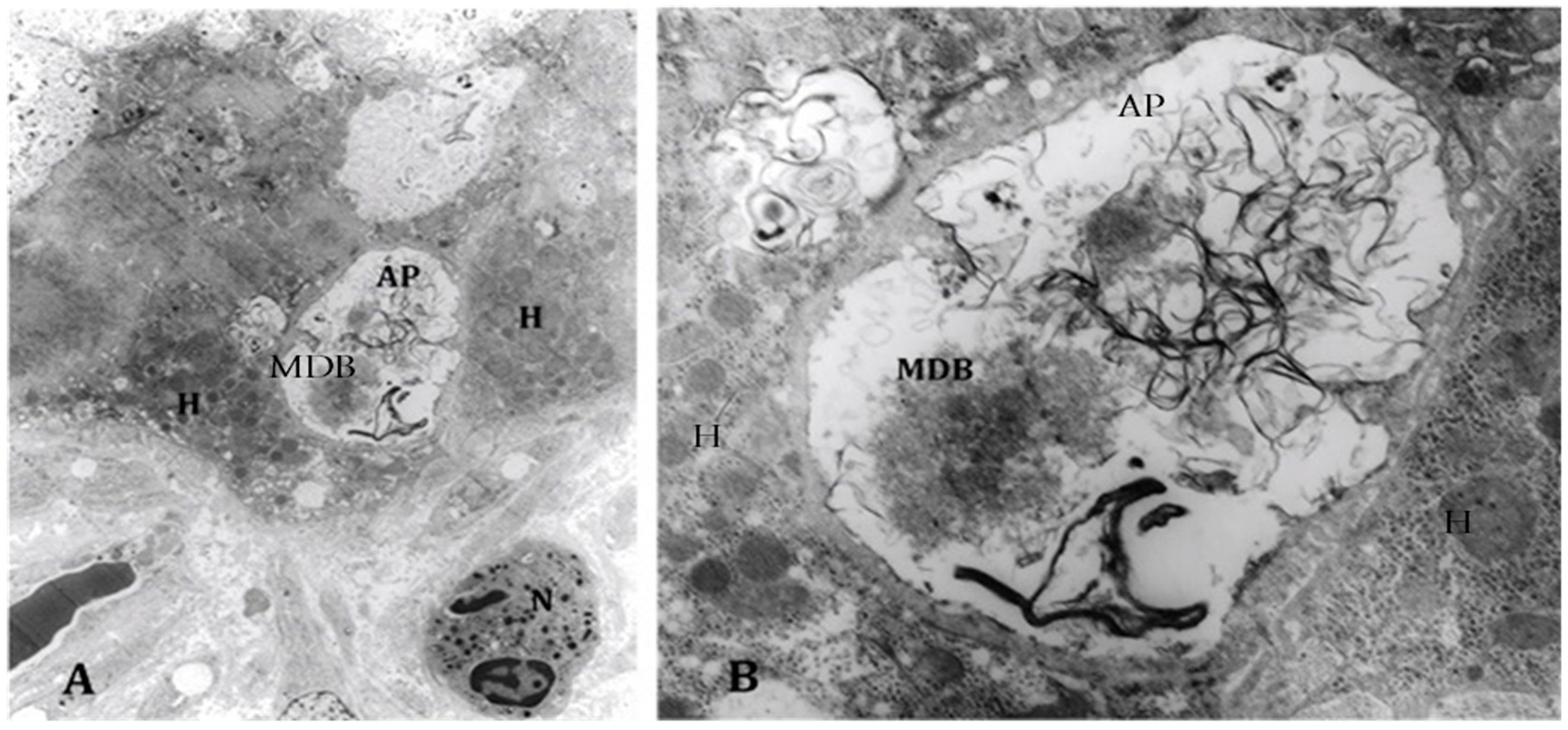
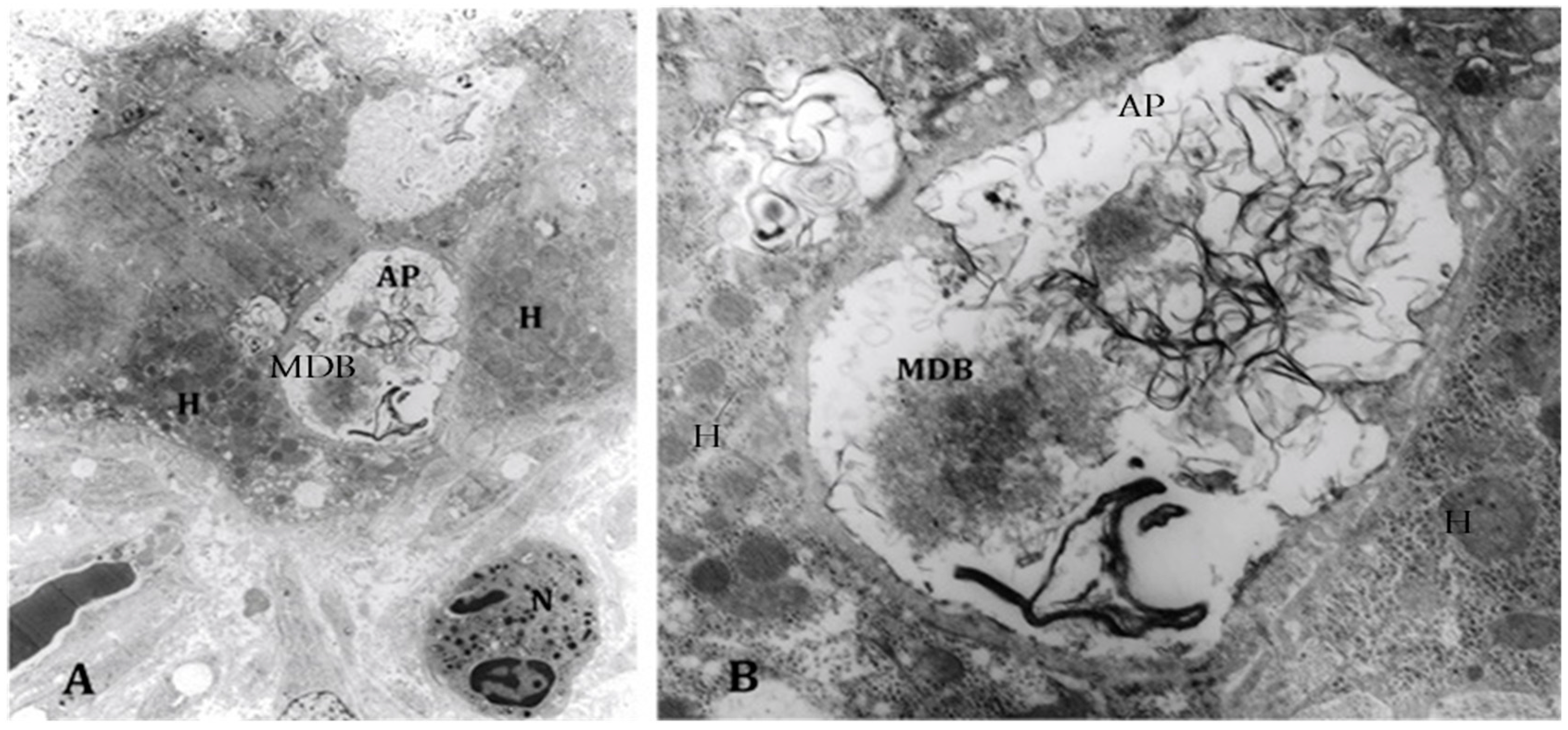
3. Changes in the Activity of Metacaspase 1 and Chaperones Involved in Protein Quality Control in Alcoholic Hepatitis
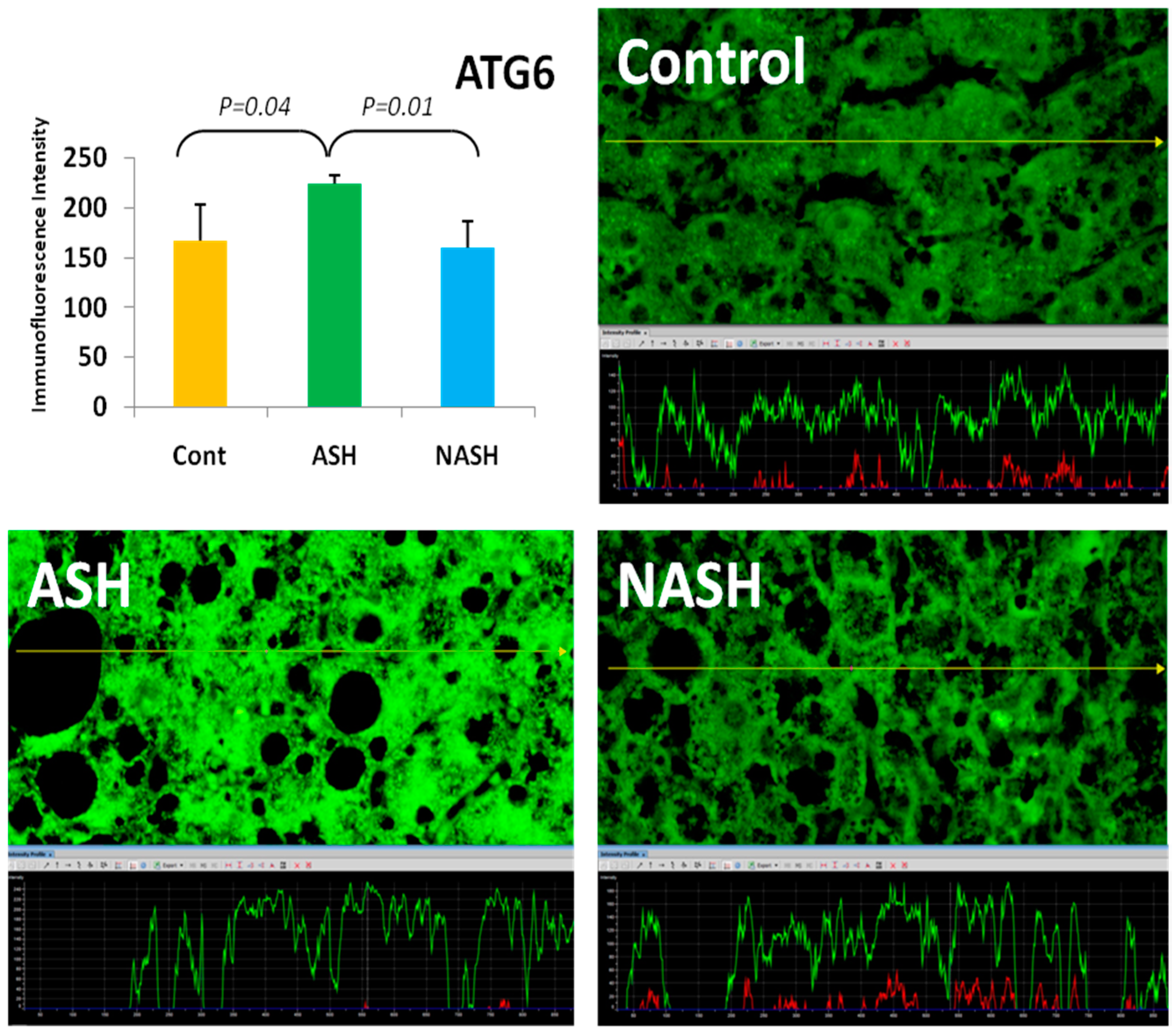
4. Components of Autophagocytosis are Upregulated in Alcoholic Hepatitis
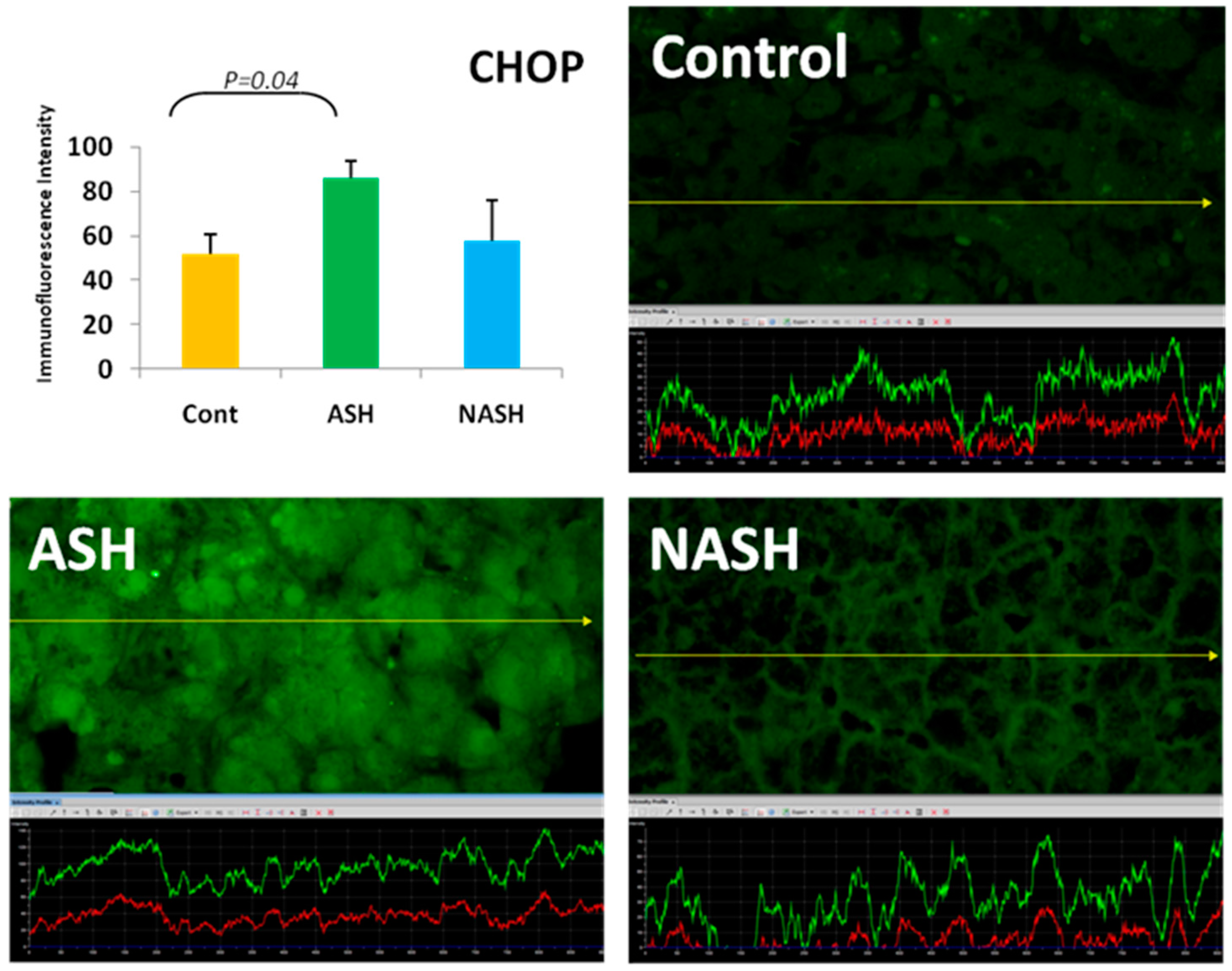
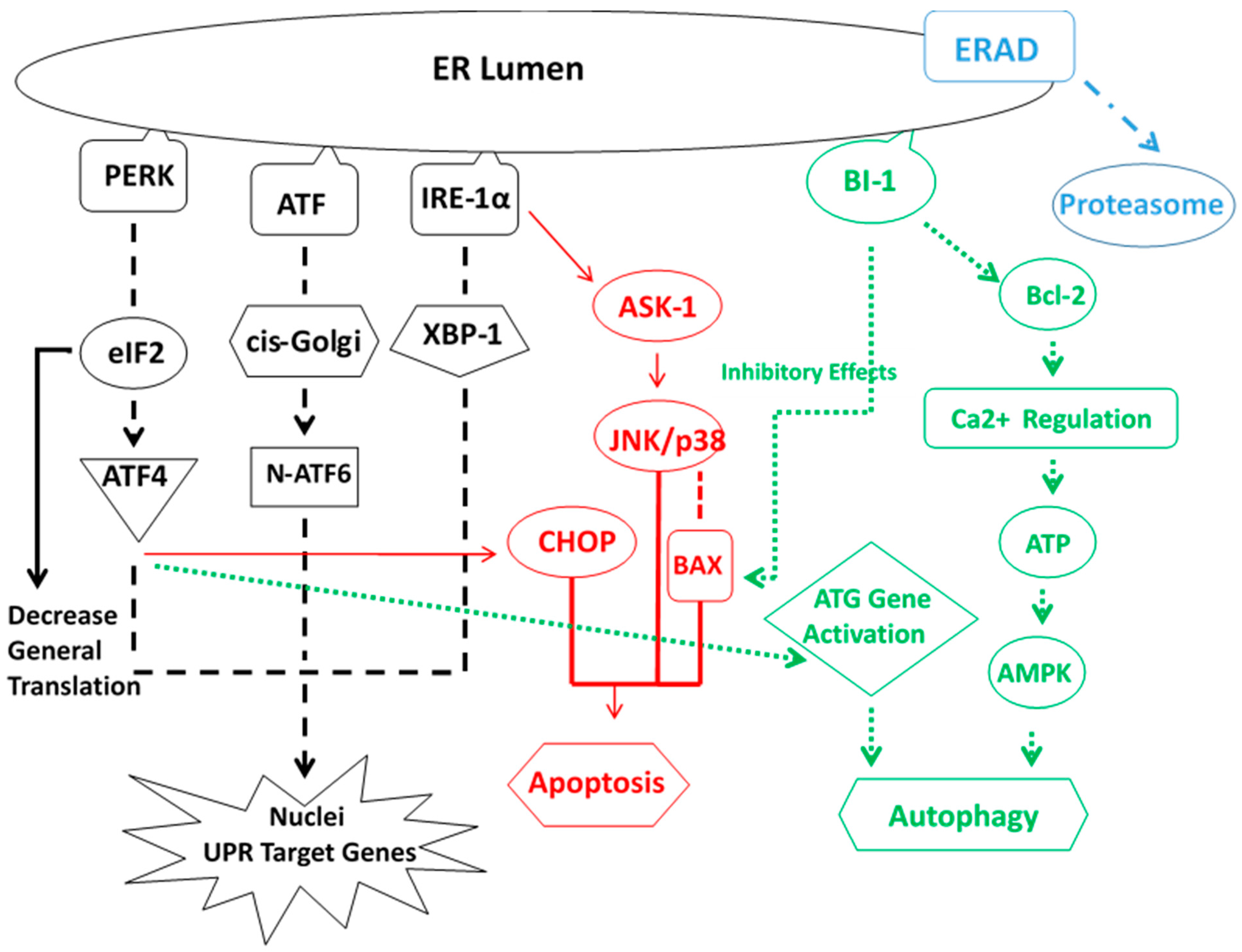
5. ER Stress Response
6. Conclusions
Acknowledgments
Author contributions
Conflicts of Interest
References
- Liu, H.; Tillman, B.; French, S.W. UFmylation and FATylation pathways are downregulated in human alcoholic and steatohepatitis and mice fed DDC, where Mallory-Denk bodies (MDBs) form. Exp. Mol. Pathol. 2014, 97, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Oliva, J.; Bardag-Gorce, F.; Lin, A.; French, B.A.; French, S.W. The role of cytokines in UBD promoter regulation and Mallory-Denk body-like aggresomes. Exp. Mol. Pathol. 2010, 89, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bardag-Gorce, F.; Oliva, J.; Li, J.; French, B.A.; French, S.W. SAMe prevents the induction of the immunoproteasome and preserves the 26S proteasome in the DDC induced MDB mouse model. Exp. Mol. Pathol. 2010, 88, 353–362. [Google Scholar] [CrossRef] [PubMed]
- French, S.W.; French, B.A.; Oliva, J.; Li, J.; Bardag- Gorce, F.; Tillman, B.; Canaan, A. FAT10 Knockout Mice Livers Fail to develop Mallory-Denk Bodies in the DDC Mouse Model. Exp. Mol. Pathol. 2012, 93, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Gong, M.; French, B.A.; Li, J.; Tillman, B.; French, S.W. Mallory-Denk Body (MDB) formation modulation Ufmylation expression epigenetically in alcoholic hepatitis (AH) and non-alcoholic hepatitis (NASH). Exp. Mol. Pathol. 2014, 97, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Oliva, J.; Bardag-Gorce, F.; Li, J.; French, B.A.; Nguyen, S.C.; Lu, S.C.; French, S.W. Betaine prevents Mallory-Denk body formation in drug-primed mice by epigenetic mechanisms. Exp. Mol. Pathol. 2009, 86, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Bardag-Gorce, F.; Oliva, J.; Villegas, J.; Fraley, S.; Amidi, F.; Li, J.; Dedes, J.; French, S.W. Epigenetic mechanisms regulate Mallory-Denk body formation in the livers of drug-primed mice. Exp. Mol. Pathol. 2008, 84, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, A.S.; Dorce, J.; Peng, Y.; French, B.A.; Tillman, B.; Li, J.; French, S.W. Levels of metacaspase 1 and chaperones related to protein quality control in alcoholic and nonalcoholic steatohepatitis. Exp. Mol. Pathol. 2015, 98, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.M.; Hao, X.; Liu, B.; Nystrom, T. Life-span extension by a metacaspase in the yeast Saccharomyces cerevisiae. Science 2014, 344, 1389–1392. [Google Scholar] [CrossRef]
- Bjerkey, G.; Lemark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stemark, H.; Johanson, T. p62/SQSTM1 forms protein aggregates degraded by autophages and has a protective effect on huntingtin-induced cell death. J. Cell. Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Sherman, M.Y.; Goldberg, A.L. Involvement of the molecular chaperone Ydj1 in the ubiquitin-dependent degradation of short-lived and abnormal proteins in Saccharomyces cerevisiae. Mol. Cell. Biol. 1996, 16, 4773–4781. [Google Scholar] [CrossRef] [PubMed]
- Bug, M. Expanding into new markets-VCP/p97 in endocytosis and autophagy. J. Struct. Biol. 2012, 179, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Kaganovich, D.; Kopito, R.; Frydman, J. Misfolded protein partition between two distinct quality control compartments. Nature 2008, 454, 1088–1095. [Google Scholar] [CrossRef]
- Klonsky, D.J.; Cregg, J.M.; Dunn, W.A., Jr.; Emr, S.D.; Sakai, Y.; Scandoval, I.V.; Sibirnya, A.; Subramani, S.; Thumam, M.; Veenhuis, M. A unified nomenclature for yeast autophagy-related genes. Dev. Cell. 2003, 5, 539–545. [Google Scholar] [CrossRef]
- Nakatogawa, H.; Suzuki, K.; Kamada, Y.; Ohsumi, Y. Dynamics and diversity in autophagy mechanisms: Lesions from yeast. Rev. Molec. Cell. Biol. 2011, 9, 1016–1123. [Google Scholar] [CrossRef] [PubMed]
- Miyaylova, M.; Shaw, R.J. The AMPK signaling pathway coordinates cell growth, autophagy and metabolism. Natl. Cell. Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell. Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Takamura, A.; Kishi, C.; Lemura, S.; Natsume, T.; Guan, J.L.; Mizushima, N. FIP200, a ULK-interacting protein, is required for autophagasome formation in mammalian cells. J. Cell. Biol. 2008, 181, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ouyang, L.; Liu, B.; Cheng, Y. Unveiling the roles of ATG4 proteases from autophagy modulation in targeted cancer therapy. Cancer Lett. 2016, 373, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Routou, R.E.; Mansouri, A.; Lihrec, D.; Durand, F.; Valla, D.; Moreau, R. Autophagy in liver diseases. J. Hepatol. 2010, 53, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
- Masouminia, M.; Samadzadeh, S.; Mendoza, A.S.; French, B.A.; Tillman, B.; French, S.W. Up reduction of autophagia components in alcoholic hepatitis and non-alcoholic hepatitis. Exp. Mol. Pathol. 2016, 101, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Cheng Ji, L.D.; Kaplowitz, N. The contribution of endoplasmic reticulum stress to liver diseases. Hepatology 2011, 53, 1752–1763. [Google Scholar]
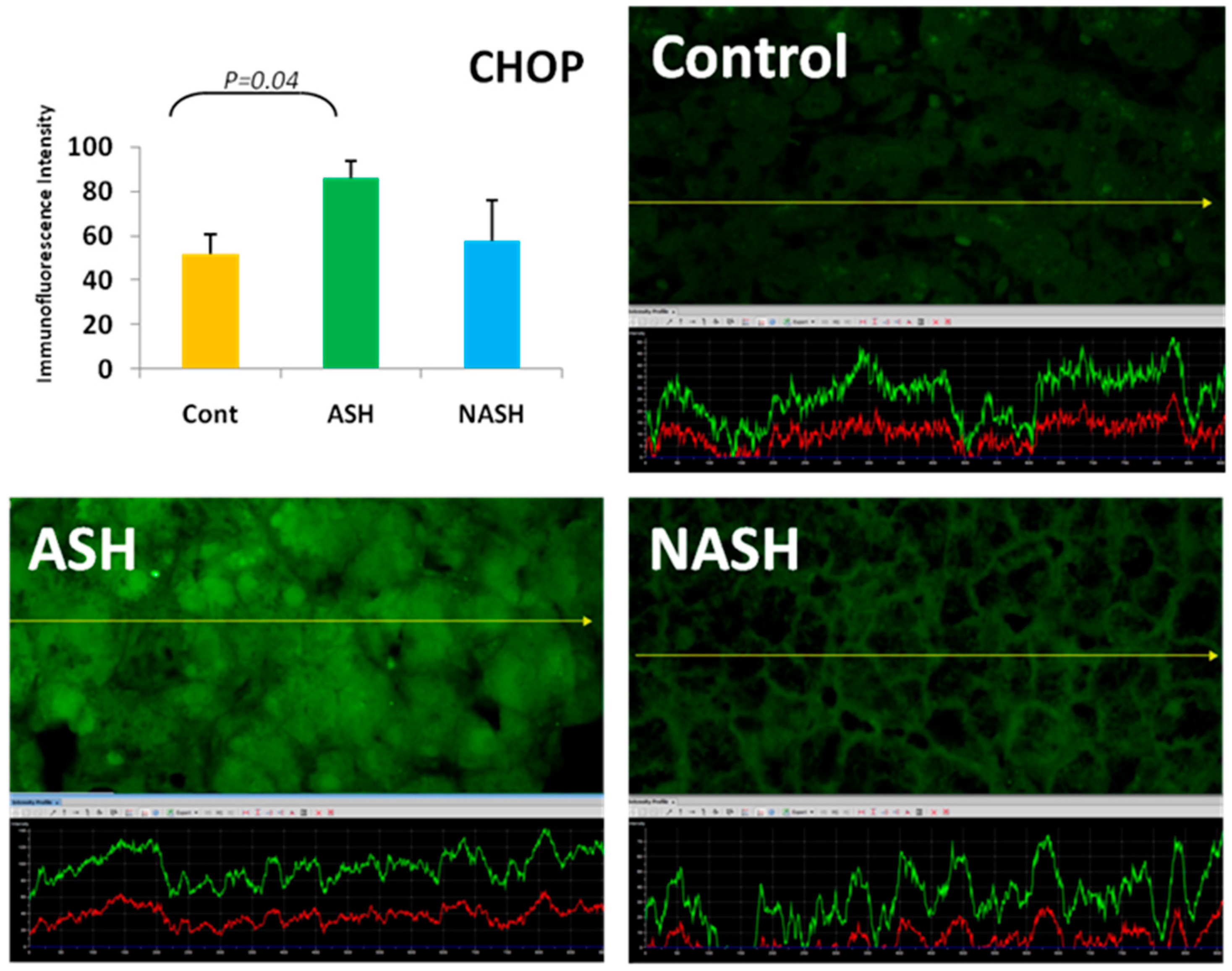
- Masouminia, M.; Samadzadeh, M.D.; Ebaee, A.; French, B.A.; Tillman, B.S.; French, S.W. Alcoholic steatohepatitis (ASH) causes more UPR-ER stress than non-alcoholic steatohepatitis. Exp. Mol. Pathol. 2016, 101, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Stevens, F.J.; Argon, Y. Protein folding in the ER. Semin. Cell. Dev. 1999, 10, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Snapp, E. Endoplasmic reticulum biogenesis. In The Biogenesis of Cellular Organelles, 1st ed.; Mullins, C., Ed.; Springer: New York, NY, USA, 2005; pp. 64–95. [Google Scholar]
- Hardinig, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 197, 271–274. [Google Scholar]
- Harding, H.P.; Navoa, I.; Zhang, Y.; Wek, Q.; Schapira, M.; Ron, D. Regulated translated initiation controls stress-induced gene expression in mammalian cells. Mol. Cell. 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Puthalakath, H.; O’Reilly, L.A.; Cunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michatak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 2008, 129, 1337–1349. [Google Scholar] [CrossRef] [PubMed]
- Cullman, S.B.; Diehl, J.A. Perk-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biochem. 2007, 279, 20108–20117. [Google Scholar]
- Nishitoh, H. Chop is a multifunctional transcription factor in the ER stress response. J. Biochem. 2012, 151, 217–219. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; French, B.A.; Tillman, B.; Morgan, T.R.; French, S.W. The inflammasome in alcoholic hepatitis: Its relationship with Mallory-Denk body formation. Exp. Mol. Pathol. 2014, 97, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Gong, M.; French, B.A.; Liao, G.; Li, J.; Tillman, B.; French, S.W. Aberrant modulation of the BRCA1 and G1/S cell cycle pathways in alcoholic hepatitis patients with Mallory Denk Bodies revealed by RNA sequencing. Oncotarget 2015, 6, 42491–42503. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Function | Reference |
|---|---|---|
| Uba6 | FAT10ylation pathway | [1] |
| USE1 | FAT10ylation pathway | [1] |
| Ufa1 | Ufmylation pathway | [1] |
| Uba5 | Ufmylation pathway | [1] |
| FAT10 | FAT10ylation pathway | [2,3] |
| Ubiquitin | 26S proteasome pathway | [4] |
| LMP7 | Immunoproteasome pathway | [5] |
| DNMT1 | DNA methylation | [7] |
| DNMT3B | DNA methylation | [7] |
| Mca1 | Chaperones pathway | [9] |
| Hsp104 | Autophagia pathway | [9] |
| p62 | Autophagia pathway | [10] |
| Hsp70/Hsp4 | Autophagia pathway | [10] |
| Ydjl/SSa1 | ERAD pathway | [11] |
| VCP/97 | 26S proteasome pathway | [12] |
| JUNQ | Protein sequestration | [13] |
| IPOD | Protein sequestration | [13] |
| ATG 1-10 | Autophagy pathway | [15] |
| TORC1 | Inhibits autophagy | [16] |
| ULK1 | Inhibits autophagy | [16] |
| AMPK | Activates autophagia | [16] |
| mTOR | Inhibits autophagia | [20] |
| PERK | Inhibits ER stress | [26] |
| CHOP IL-1B | Apoptosis, inflammasome, cell cycle arrest | [31] |
| ATM | Inhibit liver regeneration | [32] |
| p21 | Inhibit liver regeneration | [32] |
| p27 | Inhibit liver regeneration | [32] |
| p15 | Inhibit liver regeneration | [32] |
| TGFβ | Inhibit liver regeneration | [32] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
French, S.W.; Masouminia, M.; Samadzadeh, S.; Tillman, B.C.; Mendoza, A.; French, B.A. Role of Protein Quality Control Failure in Alcoholic Hepatitis Pathogenesis. Biomolecules 2017, 7, 11. https://doi.org/10.3390/biom7010011
French SW, Masouminia M, Samadzadeh S, Tillman BC, Mendoza A, French BA. Role of Protein Quality Control Failure in Alcoholic Hepatitis Pathogenesis. Biomolecules. 2017; 7(1):11. https://doi.org/10.3390/biom7010011
Chicago/Turabian StyleFrench, Samuel W., Maryam Masouminia, Sara Samadzadeh, Brittany C. Tillman, Alejandro Mendoza, and Barbara A. French. 2017. "Role of Protein Quality Control Failure in Alcoholic Hepatitis Pathogenesis" Biomolecules 7, no. 1: 11. https://doi.org/10.3390/biom7010011