
Hemoglobin-Based Blood Substitutes and the Treatment of Sickle Cell Disease: More Harm than Help?

Abstract
:1. Introduction
2. HBOC Types and Indications
2.1. Crossed Linked Tetrameric Hemoglobins
2.2. Conjugated “Decorated” Tetrameric Hemoglobins
2.3. Polymerized Human and Bovine Hemoglobins
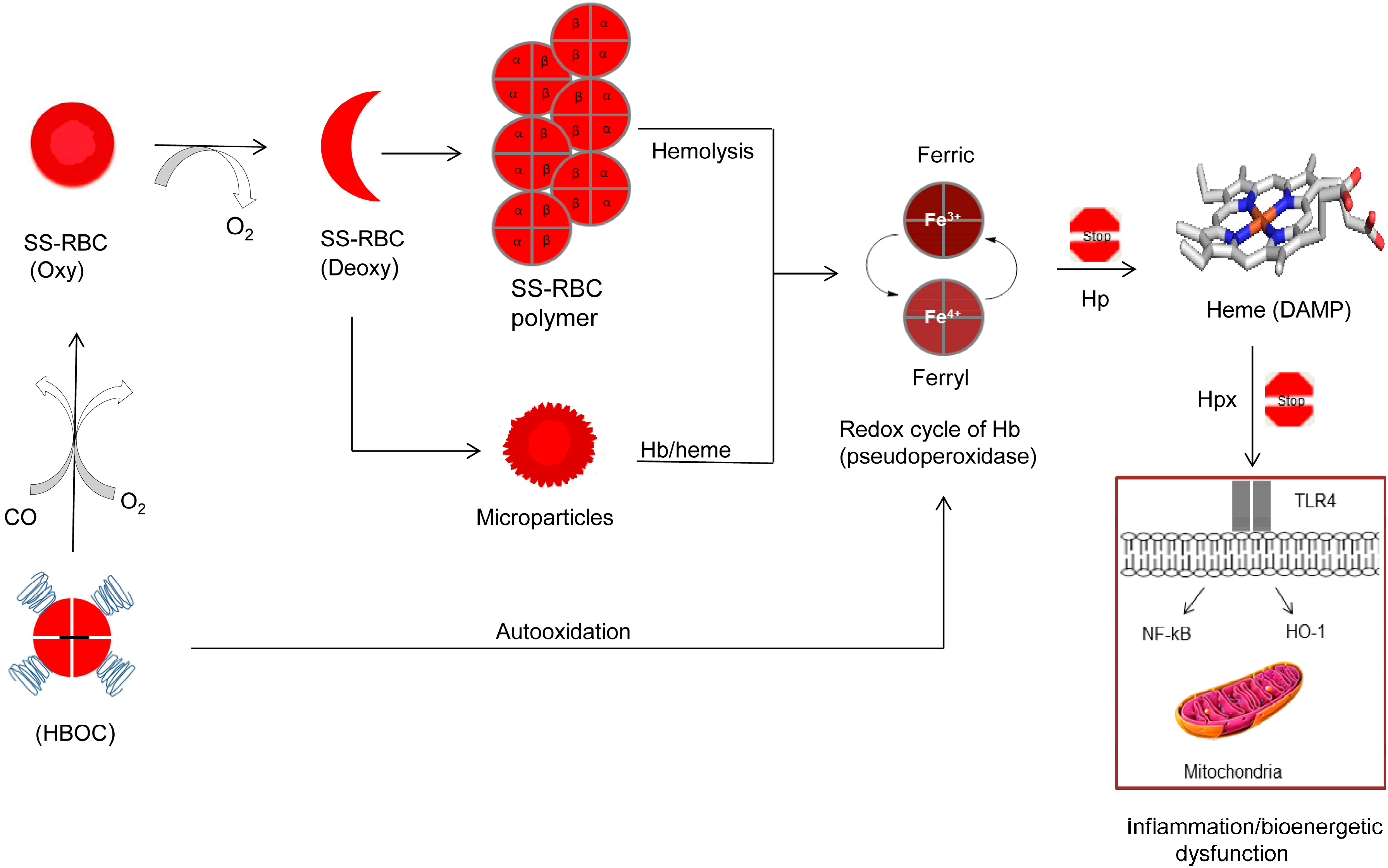
3. Toxicity of Hemoglobin-Based Oxygen Carriers
4. Sickle Cell Disease
5. Do HBOCs Have a Therapeutic Role in Hemoglobinopathies?
6. Second-Generation HBOCs and the Treatment of Sickle Cell Anemia
6.1. Hemospan (MP4)
6.2. Sanguinate
6.3. Hemopure
6.4. HRC 101
7. Do We Need to Be Concerned with Iron Oxidation in Both Sickle Cell and the HBOC Proteins?
7.1. Oxidation Reactions of HBOCs
7.2. Oxidation Reactions of Sickle Cell Hemoglobin
8. Summary and Conclusions
Acknowledgments
Conflicts of Interest
References
- Kim, H.W.; Greenburg, A.G. Artificial oxygen carriers as red blood cell substitutes: A selected review and current status. Artif. Organs 2004, 28, 813–828. [Google Scholar] [CrossRef] [PubMed]
- Winslow, R.M. Oxygen: The poison is in the dose. Transfusion 2013, 53, 424–437. [Google Scholar] [CrossRef] [PubMed]
- Winslow, R.M. Cell-free oxygen carriers: Scientific foundations, clinical development, and new directions. Biochim. Biophys. Acta 2008, 1784, 1382–1386. [Google Scholar] [CrossRef] [PubMed]
- Alayash, A.I. Hemoglobin-based blood substitutes: Oxygen carriers, pressor agents, or oxidants? Nat. Biotechnol. 1999, 17, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Alayash, A.I.; Cashon, R.E. Hemoglobin and free radicals: Implications for the development of a safe blood substitute. Mol. Med. Today 1995, 1, 122–127. [Google Scholar] [CrossRef]
- Weiskopf, R.B. Hemoglobin-based oxygen carriers: Disclosed history and the way ahead: The relativity of safety. Anesth. Analg. 2014, 119, 758–760. [Google Scholar] [CrossRef] [PubMed]
- Alayash, A.I. Setbacks in blood substitutes research and development: A biochemical perspective. Clin. Lab. Med. 2010, 30, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Freilich, D.; Pearce, L.B.; Pitman, A.; Greenburg, G.; Berzins, M.; Bebris, L.; Ahlers, S.; McCarron, R.J. HBOC-201 vasoactivity in a phase III clinical trial in orthopedic surgery subjects—Extrapolation of potential risk for acute trauma trials. J. Trauma 2009, 66, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Alayash, A.I. Blood substitutes: Why haven’t we been more successful? Trends Biotechnol. 2014, 32, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Reeder, B.J. The redox activity of hemoglobins: From physiologic functions to pathologic mechanisms. Antioxid. Redox Signal. 2010, 13, 1087–1123. [Google Scholar] [CrossRef] [PubMed]
- Buehler, P.W.; Alayash, A.I. Toxicities of hemoglobin solutions: In search of in vitro and in vivo model systems. Transfusion 2004, 44, 1516–1530. [Google Scholar] [CrossRef] [PubMed]
- Alayash, A.I. Oxygen therapeutics: Can we tame haemoglobin? Nat. Rev. Drug Discov. 2004, 3, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.; Azari, M.; Brown, R.; Burhop, K.; Bush, S.; Catarello, J.; Chuang, H.; Downing, C.; Estep, T.; Loewen, A.; et al. Preparation and characterization of diaspirin cross-linked hemoglobin solutions for preclinical studies. Biomater. Artif. Cells Immobil. Biotechnol. 1992, 20, 423–427. [Google Scholar] [CrossRef]
- Sloan, E.P.; Koenigsberg, M.; Weir, W.B.; Clark, J.M.; O’Connor, R.; Olinger, M.; Cydulka, R. Emergency resuscitation of patients enrolled in the US diaspirin cross-linked hemoglobin (DCLHb) Clinical Efficacy Trial. Prehosp. Disaster Med. 2015, 1, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, S.J.; Looker, D.L.; Roehrich, J.M.; Cozart, P.E.; Durfee, S.L.; Tedesco, J.L.; Stetler, G.L. Expression of fully functional tetrameric human hemoglobin in coli. Proc. Natl. Acad. Sci. USA 1990, 87, 8521–8525. [Google Scholar] [CrossRef] [PubMed]
- Sloan, E.P.; Koenigsberg, M.; Clark, J.M.; Weir, W.B.; Philbin, N. Shock index and prediction of traumatic hemorrhagic shock 28-day mortality: Data from the DCLHb resuscitation clinical trials. West J. Emerg. Med. 2014, 7, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Washita, Y. Relationship between chemical properties and biological properties of pyridoxalated hemoglobin polyoxyethylene. Biomat. Artif. Cell Immbol. 1992, 20, 299–307. [Google Scholar]
- Kinasewitz, G.T.; Privalle, C.T.; Imm, A.; Steingrub, J.S.; Malcynski, J.T.; Balk, R.A.; DeAngelo, J. Multicenter, randomized, placebo-controlled study of the nitric oxide scavenger pyridoxalated hemoglobin polyoxyethylene in distributive shock. Crit. Care Med. 2008, 36, 1999–2007. [Google Scholar] [CrossRef] [PubMed]
- Winslow, R.M. MP4, a new nonvasoactive polyethylene glycol-hemoglobin conjugate. Artif. Organs 2004, 9, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, C.I.; Górecki, A.Z.; Dirksen, R.; Kofranek, I.; Majewski, J.A.; Mazurkiewicz, T.; Jahoda, D.; Fagrell, B.; Keipert, P.E.; Hardiman, Y.J.; et al. Study 6084 Clinical Investigators. Evaluation of MP4OX for prevention of perioperative hypotension in patients undergoing primary hip arthroplasty with spinal anesthesia: A randomized, double-blind, multicenter study. Anesthesiology 2011, 114, 1048–1063. [Google Scholar] [CrossRef] [PubMed]
- Gould, S.A.; Moore, E.E.; Hoyt, D.B.; Burch, J.M.; Haenel, J.B.; Garcia, J.; DeWoskin, R.; Moss, G.S. The first randomized trial of human polymerized hemoglobin as a blood substitute in acute trauma and emergent surgery. J. Am. Coll. Surg. 1998, 187, 113–122. [Google Scholar] [CrossRef]
- Gould, S.A.; Moore, E.E.; Hoyt, D.B.; Ness, P.M.; Norris, E.J.; Carson, J.L.; Hides, G.A.; Freeman, I.H.; DeWoskin, R.; Moss, G.S. The life sustaining capacity of human polymerized hemoglobin when red cells might be unavailable. J. Am. Coll. Surg. 2002, 195, 445–452. [Google Scholar] [CrossRef]
- Levy, J.H.; Goodnough, L.T.; Grelich, P.E.; Parr, G.V.; Stewart, R.W.; Gratz, I.; Wahr, J.; Williams, J.; Comunale, M.E.; Doblar, D.; et al. Polymerized bovine hemoglobin solution as a replacement for allogeneic red blood cell transfusion after cardiac surgery: Results of a randomized, double-blind trial. J. Thorac. Cardiovasc. Surg. 2002, 124, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Sprung, J.; Kindscher, J.D.; Wahr, J.A.; Levy, J.H.; Monk, T.G.; Moritz, M.W.; O’Hara, P.J. The use of bovine hemoglobin glutamer-250 (Hemopure) in surgical patients: Results of a multicenter, randomized, single-blinded trial. Anesth. Analg. 2002, 94, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Boykins, R.A.; Buehler, P.W.; Jia, Y.; Venable, R.; Alayash, A.I. O-raffinose crosslinked hemoglobin lacks site-specific chemistry in the central cavity: Structural and functional consequences of β93Cys modification. Proteins 2005, 59, 840–855. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, F.J.; Ali, A.C.; Campbell, J.A.; Langlois, S.F.; Biro, G.P.; Willan, A.R.; Pierce, C.H.; Greenburg, A.G. A phase I study of oxidized raffinose cross-linked human hemoglobin. Crit. Care Med. 2000, 28, 2283–2292. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.; Gottschalk, L.I.; Grichnik, K. Safety and preliminary efficacy of hemoglobin raffimer for patients undergoing coronary artery bypass surgery. J. Cardiothorac. Vasc. Anesth. 2002, 16, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Silverman, T.A.; Weiskopf, R.B. Hemoglobin-based oxygen carriers: Current status and future directions. Transfusion 2009, 49, 2495–2515. [Google Scholar] [CrossRef] [PubMed]
- Buehler, P.W.; D’Agnillo, F. Toxicological consequences of extracellular hemoglobin: Biochemical and physiological perspectives. Antioxid Redox Signal. 2010, 2, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Buehler, P.W.; Alayash, A.I. All hemoglobin-based oxygen carriers are not created equally. Biochim. Biophys. Acta 2008, 1784, 1378–1381. [Google Scholar] [CrossRef] [PubMed]
- Hess, J.R.; MacDonald, V.W.; Brinkley, W.W. Systemic and pulmonary hypertension after resuscitation with cell-free hemoglobin. J. Appl. Physiol. 1993, 74, 1769–1778. [Google Scholar] [PubMed]
- Burhop, K.; Gordon, D.; Estep, T. Review of hemoglobin-induced myocardial lesions. Artif Cells Blood Substit. Immobil. Biotechnol. 2004, 32, 353–374. [Google Scholar] [CrossRef] [PubMed]
- Rentsendorj, O.; Zhang, X.; Williams, M.C.; Buehler, P.W.; D’Agnillo, F. Transcriptional suppression of renal antioxidant enzyme systems in guinea pigs exposed to polymerized cell-free hemoglobin. Toxics 2016, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Varnado, C.L.; Mollan, T.L.; Birukou, I.; Smith, B.J.; Henderson, D.P.; Olson, J.S. Development of recombinant hemoglobin-based oxygen carriers. Antioxid. Redox Signal. 2013, 18, 2314–2328. [Google Scholar] [CrossRef] [PubMed]
- Engoren, E.M.; Habib, R.H.; Zacharias, A.; Schwann, T.A.; Riordan, C.J.; Durham, S.J. Effect of blood transfusion on long-term survival after cardiac operation. Ann. Thorac. Surg. 2002, 74, 1180–1186. [Google Scholar] [CrossRef]
- Zhang, R.; Hess, D.T.; Qian, Z.; Hausladen, A.; Fonseca, F.; Chaube, R.; Reynolds, J.D.; Stamler, J.S. Hemoglobin βCys93 is essential for cardiovascular function and integrated response to hypoxia. Proc. Natl. Acad. Sci. USA 2015, 112, 6425–6430. [Google Scholar] [CrossRef] [PubMed]
- Kim-Shapiro, D.B.; Gladwin, M.T. Mechanisms of nitrite bioactivation. Nitric Oxide 2014, 38, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Moon-Massat, P.; Scultetus, A.; Arnaud, F.; Brown, A.; Haque, A.; Saha, B.; Kim, B.; Sagini, E.; McGwin, G., Jr.; Auker, C.; et al. The effect HBOC-201 and sodium nitrite resuscitation after uncontrolled haemorrhagic shock in swine. Injury 2012, 43, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.H.; Zhang, X.; Williams, M.C.; Hicks, W.; Buehler, P.W.; D’Agnillo, F. Sodium nitrite potentiates renal oxidative stress and injury in hemoglobin exposed guinea pigs. Toxicology 2015, 333, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Machado, R.F.; Barst, R.J.; Yovetich, N.A.; Hassell, K.L.; Kato, G.J.; Gordeuk, V.R.; Gibbs, J.S.; Little, J.A.; Schraufnagel, D.E.; Krishnamurti, L.; et al. Hospitalization for pain in patients with sickle cell disease treated with sildenafil for elevated TRV and low exercise capacity. Blood 2011, 118, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Alayash, A.I. Haptoglobin: Old protein with new functions. Clin. Chim. Acta 2011, 412, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Schaer, D.J.; Vinchi, F.; Ingoglia, G.; Tolosano, E.; Buehler, P.W. Haptoglobin, hemopexin, and related defense pathways-basic science, clinical perspectives, and drug development. Front. Physiol. 2014, 5, 415. [Google Scholar] [CrossRef] [PubMed]
- Boretti, F.S.; Buehler, P.W.; D’Agnillo, F.; Kluge, K.; Glaus, T.; Butt, O.I.; Jia, Y.; Goede, J.; Pereira, C.P.; Maggiorini, M.; et al. Sequestration of extracellular hemoglobin within a haptoglobin complex decreases its hypertensive and oxidative effects in dogs and guinea pigs. J. Clin. Investig. 2009, 119, 2271–2280. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Wood, F.; Buehler, P.W.; Alayash, A.I. Haptoglobin preferentially binds β but not α subunits cross-linked hemoglobin tetramers with minimal effects on ligand and redox reactions. PLoS ONE 2013, 8, e59841. [Google Scholar] [CrossRef] [PubMed]
- Hebbel, R.P. Reconstructing sickle cell disease: A data-based analysis of the “hyperhemolysis paradigm” for pulmonary hypertension from the perspective of evidence-based medicine. Am. J. Hematol. 2011, 86, 123–154. [Google Scholar] [CrossRef] [PubMed]
- Schaer, D.J.; Buehler, P.W.; Alayash, A.I.; Belcher, J.D.; Vercellotti, G.M. Hemolysis and free hemoglobin revisited: Exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood 2013, 121, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Milbauer, L.; Abdulla, F.; Alayash, A.I.; Smith, A.; Nath, K.A.; Hebbel, R.P.; Vercellotti, G.M. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 2014, 123, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Kassa, T.; Jana, S.; Strader, M.B.; Meng, F.; Jia, Y.; Wilson, M.T.; Alayash, A.I. Sickle cell cemoglobin in the ferryl state promotes βCys-93 oxidation and mitochondrial dysfunction in epithelial lung cells (E10). J. Biol. Chem. 2015, 290, 27939–27958. [Google Scholar] [PubMed]
- Vichinsky, E. Emerging ‘A’ therapies in hemoglobinopathies: Agonists, antagonists, antioxidants, and arginine. Hematol. Am. Soc. Hematol. Educ. Program 2012, 2012, 271–275. [Google Scholar]
- Owusu-Ansah, A.; Ihunnah, C.A.; Walker, A.L.; Ofori-Acquah, S.F. Inflammatory targets of therapy in sickle cell disease. Transl. Res. 2016, 167, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Ferrone, F.A. Sickle cell disease: Its molecular mechanism and the one drug that treats it. Int. J. Biol. Macromol. 2016, 93, 1168–1173. [Google Scholar] [CrossRef] [PubMed]
- Graves, P.E.; Henderson, D.P.; Horstman, M.J.; Solomon, B.J.; Olson, J.S. Enhancing stability and expression of recombinant human hemoglobin in E. coli: Progress in the development of a recombinant HBOC source. Biochim. Biophys. Acta 2008, 1784, 1471–1479. [Google Scholar] [CrossRef] [PubMed]
- Araujo, J.A. HO-1 and CO: Fighters vs sickle cell disease? Blood 2013, 122, 2535–2536. [Google Scholar] [CrossRef] [PubMed]
- Vandegriff, K.D.; Young, M.A.; Lohman, J.; Bellelli, A.; Samaja, M.; Malavalli, A.; Winslow, R.M. CO-MP4, a polyethylene glycol-conjugated haemoglobin derivative and carbon monoxide carrier that reduces myocardial infarct size in rats. Br. J. Pharmacol. 2008, 154, 1649–1661. [Google Scholar] [CrossRef] [PubMed]
- Belcher, J.D.; Young, M.; Chen, C.; Nguyen, J.; Burhop, K.; Tran, P.; Vercellotti, G.M. MP4CO, a pegylated hemoglobin saturated with carbon monoxide, is a modulator of HO-1, inflammation, and vaso-occlusion in transgenic sickle mice. Blood 2013, 122, 2757–2764. [Google Scholar] [CrossRef] [PubMed]
- Telen, M.J. Beyond hydroxyurea: New and old drugs in the pipeline for sickle cell disease. Blood 2016, 127, 810–819. [Google Scholar] [CrossRef] [PubMed]
- Nho, K.; Glower, D.; Bredehoeft, S.; Shankar, H.; Shorr, R.; Abuchowski, A. PEG-bovine hemoglobin: Safety in a canine dehydrated hypovolemic-hemorrhagic shock model. Biomater. Artif. Cells Immobil. Biotechnol. 1992, 20, 511–524. [Google Scholar] [CrossRef]
- Zhang, J.; Cao, S.; Kwansa, H.; Crafa, D.; Kibler, K.K.; Koehler, R.C. Transfusion of hemoglobin-based oxygen carriers in the carboxy state is beneficial during transient focal cerebral ischemia. J. Appl. Physiol. 2012, 113, 1709–1717. [Google Scholar] [CrossRef] [PubMed]
- Abuchowski, A. PEGylated Bovine Carboxyhemoglobin (SANGUINATE™): Results of Clinical Safety Testing and Use in Patients. Adv. Exp. Med. Biol. 2016, 876, 461–467. [Google Scholar] [PubMed]
- Gonzalez, P.; Hackney, A.C.; Jones, S.; Strayhorn, D.; Hoffman, E.B.; Hughes, G.; Jacobs, E.E.; Orringer, E.P. A phase I/II study of polymerized bovine hemoglobin in adult patients with sickle cell disease not in crisis at the time of study. Investig. Med. 1997, 45, 258–264. [Google Scholar]
- Crawford, M.W.; Shichor, T.; Engelhardt, T.; Adamson, G.; Bell, D.; Carmichael, F.J.; Kim, P.C. The novel hemoglobin-based oxygen carrier HRC 101 improves survival in murine sickle cell disease. Anesthesiology 2007, 107, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Mollan, T.L.; Alayash, A.I. Redox reactions of hemoglobin: Mechanisms of toxicity and control. Antioxid. Redox Signal. 2013, 18, 2251–2253. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Buehler, P.W.; Boykins, R.A.; Venable, R.M.; Alayash, A.I. Structural basis of peroxide-mediated changes in human hemoglobin: A novel oxidative pathway. J. Biol. Chem. 2007, 282, 4894–4907. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, C.; Henkens, R.; Alayash, A.I.; Crumbliss, A.L. Allosteric effects on oxidative and nitrosative reactions of cell-free hemoglobins. IUBMB Life 2007, 59, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, C.; Henkens, R.; Alayash, A.I.; Banerjee, S.; Crumbliss, A.L. Molecular controls of the oxygenation and redox reactions of hemoglobin. Antioxid. Redox Signal. 2013, 18, 2298–2313. [Google Scholar] [CrossRef] [PubMed]
- Nagababu, E.; Ramasamy, S.; Rifkind, J.M.; Jia, Y.; Alayash, A.I. Site-specific cross-linking of human and bovine hemoglobins differentially alters oxygen binding and redox side reactions producing rhombic heme and heme degradation. Biochemistry 2002, 41, 7407–7415. [Google Scholar] [CrossRef] [PubMed]
- D’Agnillo, F.; Chang, T.M. Polyhemoglobin-superoxide dismutase-catalase as a blood substitute with antioxidant properties. Nat. Biotechnol. 1998, 16, 667–671. [Google Scholar] [CrossRef] [PubMed]
- Strader, M.B.; Alayash, A.I. Exploring oxidative reactions in hemoglobin variants using mass spectrometry: Lessons for engineering oxidatively stable oxygen therapeutics. Antioxid. Redox Signal. 2016. [Google Scholar] [CrossRef] [PubMed]
- Doherty, D.H.; Doyle, M.P.; Curry, S.R.; Vali, R.J.; Fattor, T.J.; Olson, J.S.; Lemon, D.D. Rate of reaction with nitric oxide determines the hypertensive effect of cell-free hemoglobin. Nat. Biotechnol. 1998, 16, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.S.; Foley, E.W.; Rogge, C.; Tsai, A.L.; Doyle, M.P.; Lemon, D.D. NO scavenging and the hypertensive effect of hemoglobin-based blood substitutes. Free Radic. Biol. Med. 2004, 36, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Buehler, P.W.; D’Agnillo, F.; Hoffman, V.; Alayash, A.I. Effects of endogenous ascorbate on oxidation, oxygenation, and toxicokinetics of cell-free modified hemoglobin after exchange transfusion in rat and guinea pig. J. Pharmacol. Exp. Ther. 2007, 323, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Manalo, D.J.; Buehler, P.W.; Baek, J.H.; Butt, O.; D’Agnillo, F.; Alayash, A.I. Acellular haemoglobin attenuates hypoxia-inducible factor-1α (HIF-1α) and its target genes in haemodiluted rats. Biochem. J. 2008, 414, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Butt, O.I.; Buehler, P.W.; D’Agnillo, F. Differential induction of renal heme oxygenase and ferritin in ascorbate and non-ascorbate producing species transfused with modified cell-free hemoglobin. Antioxid. Redox Signal. 2010, 12, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, M.C.; Chan, J.Y.; Ross, A.W.; Liew, S.M.; Butt, W.W.; Baguley, D.; Salem, H.H.; Russ, M.K.; Deasy, C.; Martin, K.E.; et al. A synthetic haemoglobin-based oxygen carrier and the reversal of cardiac hypoxia secondary to severe anaemia following trauma. Med. J. Aust. 2011, 194, 471–473. [Google Scholar] [PubMed]
- Hebbel, R.P.; Morgan, W.T.; Eaton, J.W.; Hedlund, B.E. Accelerated autoxidation and heme loss due to instability of sickle hemoglobin. Proc. Natl. Acad. Sci. USA 1988, 85, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Hebbel, R.P.; Ney, P.A.; Foker, W. Autoxidation, dehydration, and adhesivity may be related abnormalities of sickle erythrocytes. Am. J. Physiol. 1989, 56, C579–C583. [Google Scholar]
- Marva, E.; Hebbel, R.P. Denaturing interaction between sickle hemoglobin and phosphatidylserine liposomes. Blood 1994, 83, 242–249. [Google Scholar] [PubMed]
- George, A.; Pushkaran, S.; Konstantinidis, D.G.; Koochaki, S.; Malik, P.; Mohandas, N.; Zheng, Y.; Joiner, C.H.; Kalfa, T.A. Erythrocyte NADPH oxidase activity modulated by Rac GTPases, PKC, and plasma cytokines contributes to oxidative stress in sickle cell disease. Blood 2013, 121, 2099–2109. [Google Scholar] [CrossRef] [PubMed]
- Cyrklaff, M.; Sanchez, C.P.; Kilian, N.; Bisseye, C.; Simpore, J.; Frischknecht, F.; Lanzer, M. Hemoglobins S and C interfere with actin remodeling in Plasmodium falciparum-infected erythrocytes. Science 2011, 334, 1283–1286. [Google Scholar] [CrossRef] [PubMed]
- Camus, S.M.; De Moraes, J.A.; Bonnin, P.; Abbyad, P.; Le Jeune, S.; Lionnet, F.; Loufrani, L.; Grimaud, L.; Lambry, J.C.; Charue, D.; et al. Circulating cell membrane microparticles transfer heme to endothelial cells and trigger vasoocclusions in sickle cell disease. Blood 2015, 125, 3805–3814. [Google Scholar] [CrossRef] [PubMed]
- Jana, S.; Meng, F.; Strader, M.B.; Hicks, W.; Kassa, T.; Tarandovskiy, I.; De Paoli, S.; Simak, J.; Miller, J.L.; Mendelsohn, L.; et al. Hemoglobin S oxidation promotes plasma-derived microparticle membrane alterations and toxicity. Blood 2016, 128, 856. [Google Scholar]
- Yabuki, A.; Matsushita, M.; Malchesky, P.S.; Iwasaki, K.; Iwashita, Y.; Nosé, Y. In vitro evaluation of a pyridoxalated hemoglobin polyoxyethylene conjugates in reversing cell sickling. ASAIO Trans. 1988, 34, 773–777. [Google Scholar] [PubMed]


{kind=link}
| Transient hypertension |
| Gastrointestinal symptoms |
| Pancreatic and liver enzyme elevation |
| Myocardial infarction; cardiac arrhythmias |
| Renal Injury |
| Mortality |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alayash, A.I. Hemoglobin-Based Blood Substitutes and the Treatment of Sickle Cell Disease: More Harm than Help? Biomolecules 2017, 7, 2. https://doi.org/10.3390/biom7010002
Alayash AI. Hemoglobin-Based Blood Substitutes and the Treatment of Sickle Cell Disease: More Harm than Help? Biomolecules. 2017; 7(1):2. https://doi.org/10.3390/biom7010002
Chicago/Turabian StyleAlayash, Abdu I. 2017. "Hemoglobin-Based Blood Substitutes and the Treatment of Sickle Cell Disease: More Harm than Help?" Biomolecules 7, no. 1: 2. https://doi.org/10.3390/biom7010002
APA StyleAlayash, A. I. (2017). Hemoglobin-Based Blood Substitutes and the Treatment of Sickle Cell Disease: More Harm than Help? Biomolecules, 7(1), 2. https://doi.org/10.3390/biom7010002