
Phosphoproteome Discovery in Human Biological Fluids

Abstract
:1. Introduction
2. Bioanalytical Strategies
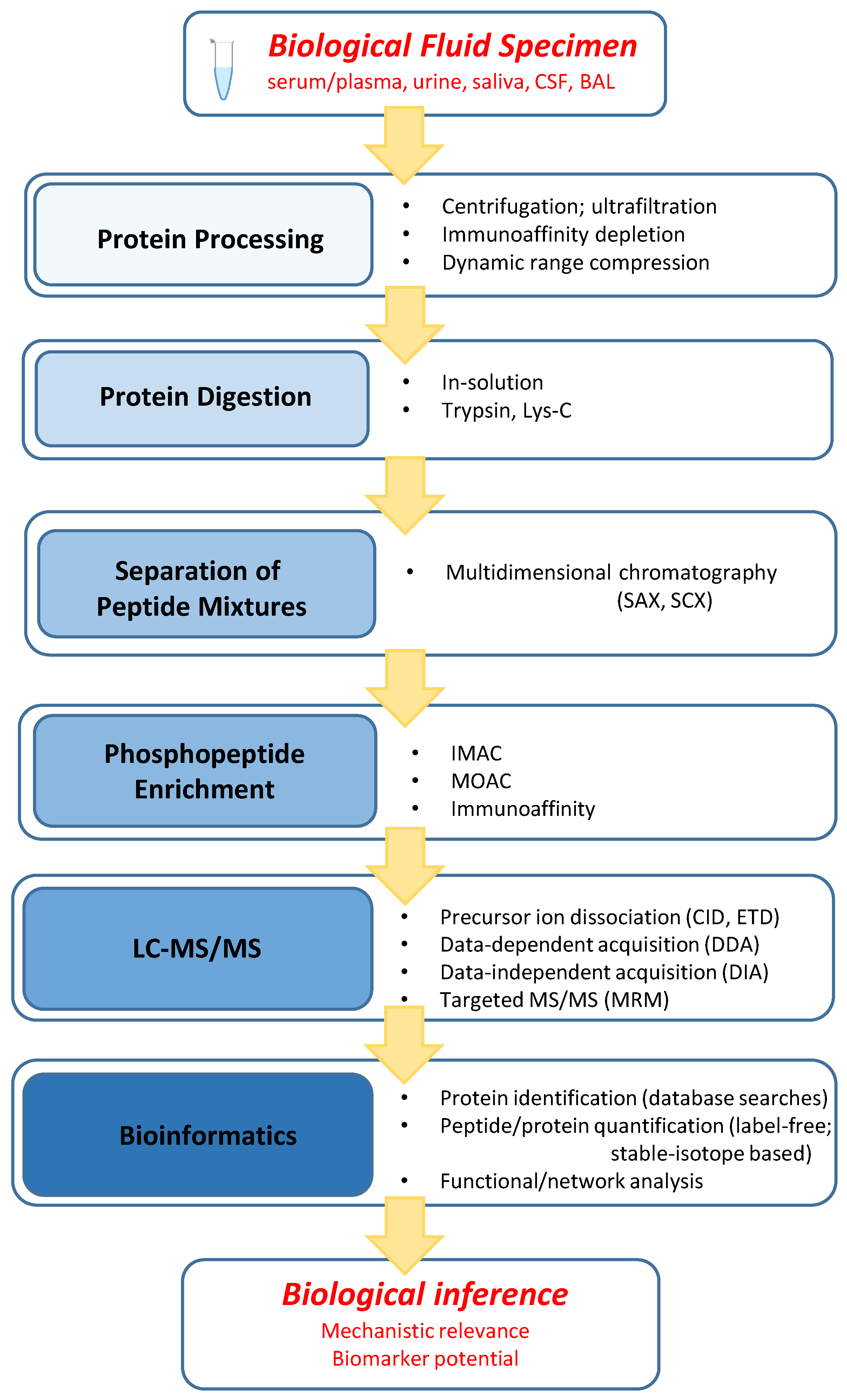
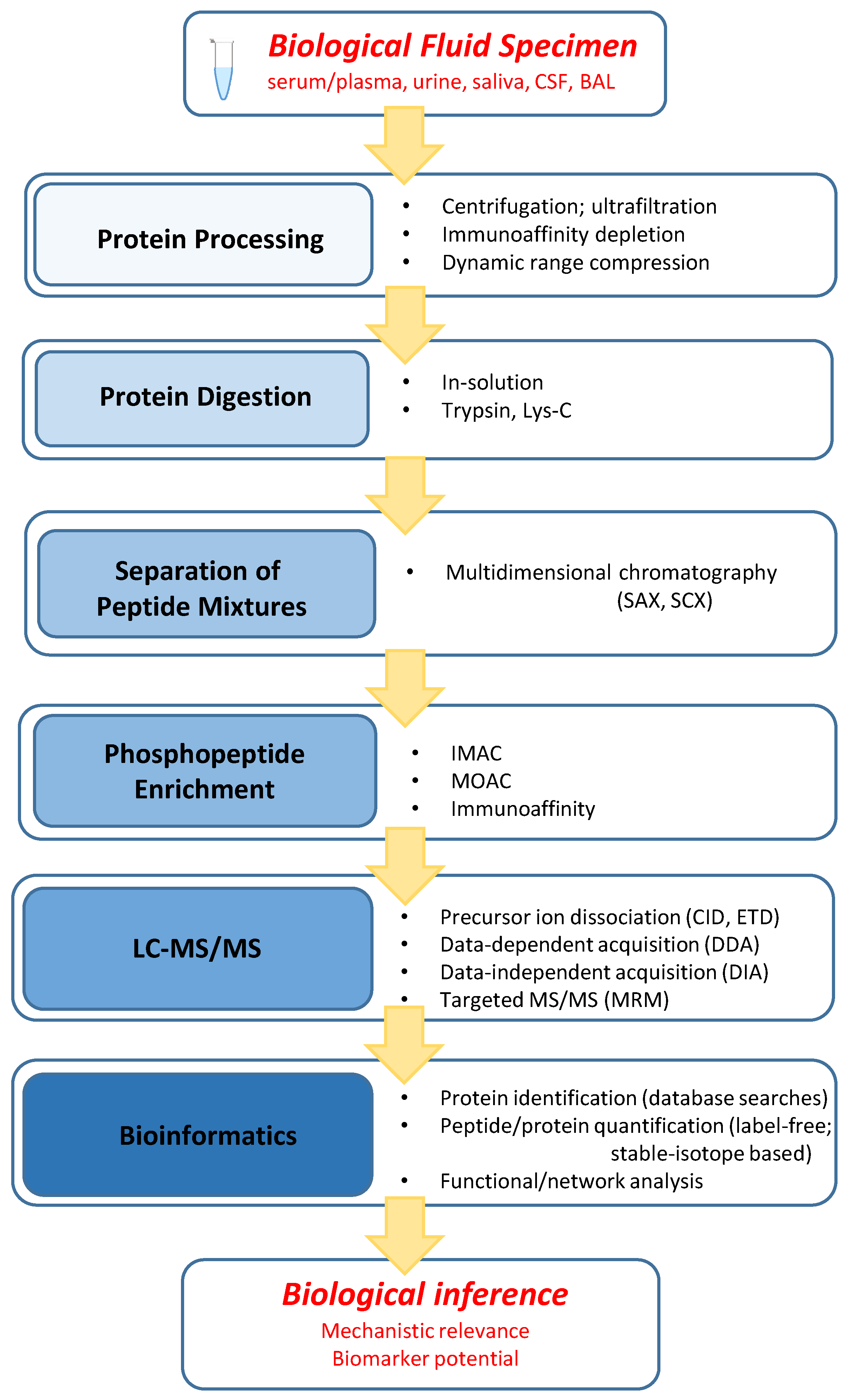
2.1. General Workflow Characteristics
2.2. Protein Processing
2.3. Protein Digestion
2.4. Separation of Peptide Mixtures
2.5. Phosphopeptide Enrichment
2.6. LC–MS/MS
2.7. Bioinformatics
3. Applications to Biofluid Phosphoproteome Characterization
3.1. Serum/Plasma
3.2. Urine
3.3. Cerebrospinal Fluid
3.4. Saliva
3.5. Bronchoalveolar Lavage Fluid
4. Concluding Remarks
Acknowledgments
Conflicts of Interest
Abbreviations
| BAL | bronchoalveolar lavage fluid |
| CID | collision-induced dissociation |
| CSF | cerebrospinal fluid |
| DIA | data-independent acquisition |
| DDA | data-dependent acquisition |
| ETD | electron-transfer dissociation |
| IMAC | immobilized metal ion affinity chromatography |
| iTRAQ | isobaric tag for relative and absolute quantification |
| MARS | Multiple Affinity Removal System |
| MDLC | multidimensional liquid chromatography |
| MRM | multiple reaction monitoring |
| MOAC | metal oxide affinity chromatography |
| mTRAQ | mass differential tag for relative and absolute quantification |
| RP | reversed-phase |
| SAX | strong anion exchange |
| SCX | strong cation exchange |
| TMT | tandem mass tag |
| TOF | time-of-flight |
| UPLC | ultra-high pressure chromatography |
References
- Hunter, T. Tyrosine phosphorylation: Thirty years and counting. Curr. Opin. Cell Biol. 2009, 21, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P. The regulation of protein function by multisite phosphorylation—A 25 year update. Trends Biochem. Sci. 2000, 25, 596–601. [Google Scholar] [CrossRef]
- Marmiroli, S.; Fabbro, D.; Miyata, Y.; Pierobon, M.; Ruzzene, M. Phosphorylation, Signaling, and Cancer: Targets and Targeting. BioMed Res. Int. 2015, 2015, 601543. [Google Scholar] [CrossRef] [PubMed]
- Sever, R.; Brugge, J.S. Signal transduction in cancer. Cold Spring Harb. Perspect. Med. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Rosca, M.; Minkler, P.; Hoppel, C.L. Cardiac mitochondria in heart failure: Normal cardiolipin profile and increased threonine phosphorylation of complex IV. Biochim. Biophys. Acta (BBA) Bioenerg. 2011, 1807, 1373–1382. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.A.; Fullerton, D.A.; Buttrick, P.M. Contractile protein phosphorylation predicts human heart disease phenotypes. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1644–H1650. [Google Scholar] [CrossRef] [PubMed]
- Asfa, A.S.; Qiu, B.; Wee, S.; Choi, H.; Gunaratne, J.; Tergaonkar, V. Phosphoprotein network analysis of white adipose tissues unveils deregulated pathways in response to high-fat diet. Sci. Rep. 2016, 6, 25844. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Banks, A.S.; Estall, J.L.; Kajimura, S.; Bostrom, P.; Laznik, D.; Ruas, J.L.; Chalmers, M.J.; Kamenecka, T.M.; Bluher, M.; et al. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature 2010, 466, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Tenreiro, S.; Eckermann, K.; Outeiro, T.F. Protein phosphorylation in neurodegeneration: Friend or foe? Front. Mol. Neurosci. 2014, 7, 42. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, A.; Diamandis, E.P.; Rodriguez, H.; Hoofnagle, A.N.; Ioannidis, J.; Lopez, M. What is wrong with clinical proteomics? Clin. Chem. 2014, 60, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Zhang, H. Glycoproteomics and clinical applications. Proteom. Clin. Appl. 2010, 4, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Tagliabracci, V.S.; Pinna, L.A.; Dixon, J.E. Secreted protein kinases. Trends Biochem. Sci. 2013, 38, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Yalak, G.; Vogel, V. Extracellular phosphorylation and phosphorylated proteins: Not just curiosities but physiologically important. Sci. Signal. 2012, 5, re7. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chance, M.R. Integrating phosphoproteomics in systems biology. Comput. Struct. Biotechnol. J. 2014, 10, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Jaros, J.A.; Guest, P.C.; Ramoune, H.; Rothermundt, M.; Leweke, F.M.; Martins-de-Souza, D.; Bahn, S. Clinical use of phosphorylated proteins in blood serum analysed by immobilised metal ion affinity chromatography and mass spectrometry. J. Proteom. 2012, 76, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Jaros, J.A.; Martins-de-Souza, D.; Rahmoune, H.; Rothermundt, M.; Leweke, F.M.; Guest, P.C.; Bahn, S. Protein phosphorylation patterns in serum from schizophrenia patients and healthy controls. J. Proteom. 2012, 76, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Liu, L.; Wang, J.; Jin, Q. Urinary proteomic and non-prefractionation quantitative phosphoproteomic analysis during pregnancy and non-pregnancy. BMC Genom. 2013, 14, 777. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.L.; Anderson, N.G. The human plasma proteome: History, character, and diagnostic prospects. Mol. Cell. Proteom. 2002, 1, 845–867. [Google Scholar] [CrossRef]
- Hortin, G.L.; Sviridov, D.; Anderson, N.L. High-abundance polypeptides of the human plasma proteome comprising the top 4 logs of polypeptide abundance. Clin. Chem. 2008, 54, 1608–1616. [Google Scholar] [CrossRef] [PubMed]
- Mrozinski, P.; Zolotarjova, N.; Chen, H. Human Serum and Plasma Protein Depletion–Novel High-Capacity Affinity Column for the Removal of the “Top 14” Abundant Proteins Application; Agilent Technologies, Inc.: Santa Clara, CA, USA, 2008. [Google Scholar]
- Yocum, A.K.; Yu, K.; Oe, T.; Blair, I.A. Effect of immunoaffinity depletion of human serum during proteomic investigations. J. Proteome Res. 2005, 4, 1722–1731. [Google Scholar] [CrossRef] [PubMed]
- Righetti, P.G.; Boschetti, E. The ProteoMiner and the FortyNiners: Searching for gold nuggets in the proteomic arena. Mass Spectrom. Rev. 2008, 27, 596–608. [Google Scholar] [CrossRef] [PubMed]
- Dickhut, C.; Feldmann, I.; Lambert, J.; Zahedi, R.P. Impact of digestion conditions on phosphoproteomics. J. Proteome Res. 2014, 13, 2761–2770. [Google Scholar] [CrossRef] [PubMed]
- Bahl, J.M.; Jensen, S.S.; Larsen, M.R.; Heegaard, N.H. Characterization of the human cerebrospinal fluid phosphoproteome by titanium dioxide affinity chromatography and mass spectrometry. Anal. Chem. 2008, 80, 6308–6316. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R. Quantitative Evaluation of Filter Aided Sample Preparation (FASP) and Multienzyme Digestion FASP Protocols. Anal. Chem. 2016, 88, 5438–5443. [Google Scholar] [CrossRef] [PubMed]
- Batth, T.S.; Francavilla, C.; Olsen, J.V. Off-line high-pH reversed-phase fractionation for in-depth phosphoproteomics. J. Proteome Res. 2014, 13, 6176–6186. [Google Scholar] [CrossRef] [PubMed]
- Kocher, T.; Pichler, P.; Swart, R.; Mechtler, K. Analysis of protein mixtures from whole-cell extracts by single-run nanoLC-MS/MS using ultralong gradients. Nat. Protoc. 2012, 7, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Dayon, L.; Kussmann, M. Proteomics of human plasma: A critical comparison of analytical workflows in terms of effort, throughput and outcome. EuPA Open Proteom. 2013, 1, 8–16. [Google Scholar] [CrossRef]
- Marcantonio, M.; Trost, M.; Courcelles, M.; Desjardins, M.; Thibault, P. Combined enzymatic and data mining approaches for comprehensive phosphoproteome analyses: Application to cell signaling events of interferon-gamma-stimulated macrophages. Mol. Cell. Proteom. 2008, 7, 645–660. [Google Scholar] [CrossRef] [PubMed]
- Fila, J.; Honys, D. Enrichment techniques employed in phosphoproteomics. Amino Acids 2012, 43, 1025–1047. [Google Scholar] [CrossRef] [PubMed]
- Neville, D.C.; Rozanas, C.R.; Price, E.M.; Gruis, D.B.; Verkman, A.S.; Townsend, R.R. Evidence for phosphorylation of serine 753 in CFTR using a novel metal-ion affinity resin and matrix-assisted laser desorption mass spectrometry. Protein Sci. 1997, 6, 2436–2445. [Google Scholar] [CrossRef] [PubMed]
- Posewitz, M.C.; Tempst, P. Immobilized gallium(III) affinity chromatography of phosphopeptides. Anal. Chem. 1999, 71, 2883–2892. [Google Scholar] [CrossRef] [PubMed]
- Thingholm, T.E.; Larsen, M.R. Sequential Elution from IMAC (SIMAC): An Efficient Method for Enrichment and Separation of Mono- and Multi-phosphorylated Peptides. Methods Mol. Biol. 2016, 1355, 147–160. [Google Scholar] [PubMed]
- Iliuk, A.B.; Martin, V.A.; Alicie, B.M.; Geahlen, R.L.; Tao, W.A. In-depth analyses of kinase-dependent tyrosine phosphoproteomes based on metal ion-functionalized soluble nanopolymers. Mol. Cell. Proteom. 2010, 9, 2162–2172. [Google Scholar] [CrossRef] [PubMed]
- Zoumaro-Djayoon, A.D.; Heck, A.J.; Munoz, J. Targeted analysis of tyrosine phosphorylation by immuno-affinity enrichment of tyrosine phosphorylated peptides prior to mass spectrometric analysis. Methods 2012, 56, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Salih, E.; Siqueira, W.L.; Helmerhorst, E.J.; Oppenheim, F.G. Large-scale phosphoproteome of human whole saliva using disulfide-thiol interchange covalent chromatography and mass spectrometry. Anal. Biochem. 2010, 407, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Hebert, A.S.; Richards, A.L.; Bailey, D.J.; Ulbrich, A.; Coughlin, E.E.; Westphall, M.S.; Coon, J.J. The one hour yeast proteome. Mol. Cell. Proteom. 2014, 13, 339–347. [Google Scholar] [CrossRef] [PubMed]
- DeGnore, J.P.; Qin, J. Fragmentation of phosphopeptides in an ion trap mass spectrometer. J. Am. Soc. Mass Spectrom. 1998, 9, 1175–1188. [Google Scholar] [CrossRef]
- Villen, J.; Beausoleil, S.A.; Gygi, S.P. Evaluation of the utility of neutral-loss-dependent MS3 strategies in large-scale phosphorylation analysis. Proteomics 2008, 8, 4444–4452. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, J.; Premsler, T.; Sickmann, A. Application of electron transfer dissociation (ETD) for the analysis of posttranslational modifications. Proteomics 2008, 8, 4466–4483. [Google Scholar] [CrossRef] [PubMed]
- Kalli, A.; Smith, G.T.; Sweredoski, M.J.; Hess, S. Evaluation and optimization of mass spectrometric settings during data-dependent acquisition mode: Focus on LTQ-Orbitrap mass analyzers. J. Proteome Res. 2013, 12, 3071–3086. [Google Scholar] [CrossRef] [PubMed]
- Bateman, R.H.; Carruthers, R.; Hoyes, J.B.; Jones, C.; Langridge, J.I.; Millar, A.; Vissers, J.P. A novel precursor ion discovery method on a hybrid quadrupole orthogonal acceleration time-of-flight (Q-TOF) mass spectrometer for studying protein phosphorylation. J. Am. Soc. Mass Spectrom. 2002, 13, 792–803. [Google Scholar] [CrossRef]
- Chapman, J.D.; Goodlett, D.R.; Masselon, C.D. Multiplexed and data-independent tandem mass spectrometry for global proteome profiling. Mass Spectrom. Rev. 2014, 33, 452–470. [Google Scholar] [CrossRef] [PubMed]
- Geromanos, S.J.; Vissers, J.P.; Silva, J.C.; Dorschel, C.A.; Li, G.Z.; Gorenstein, M.V.; Bateman, R.H.; Langridge, J.I. The detection, correlation, and comparison of peptide precursor and product ions from data independent LC-MS with data dependant LC-MS/MS. Proteomics 2009, 9, 1683–1695. [Google Scholar] [CrossRef] [PubMed]
- Gillet, L.C.; Navarro, P.; Tate, S.; Rost, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis. Mol. Cell. Proteom. 2012, 11, O111.016717. [Google Scholar] [CrossRef] [PubMed]
- Adachi, J.; Narumi, R.; Tomonaga, T. Targeted Phosphoproteome Analysis Using Selected/Multiple Reaction Monitoring (SRM/MRM). Methods Mol. Biol. 2016, 1394, 87–100. [Google Scholar] [PubMed]
- Yates, J.R., 3rd; Eng, J.K.; McCormack, A.L.; Schieltz, D. Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal. Chem. 1995, 67, 1426–1436. [Google Scholar] [CrossRef] [PubMed]
- Hoopmann, M.R.; Moritz, R.L. Current algorithmic solutions for peptide-based proteomics data generation and identification. Curr. Opin. Biotechnol. 2013, 24, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.C.; Jones, A.R.; Hubbard, S.J. Computational phosphoproteomics: From identification to localization. Proteomics 2015, 15, 950–963. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Schilling, B.; Rardin, M.J.; MacLean, B.X.; Zawadzka, A.M.; Frewen, B.E.; Cusack, M.P.; Sorensen, D.J.; Bereman, M.S.; Jing, E.; Wu, C.C.; et al. Platform-independent and label-free quantitation of proteomic data using MS1 extracted ion chromatograms in skyline: Application to protein acetylation and phosphorylation. Mol. Cell. Proteom. 2012, 11, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Laukens, K.; Naulaerts, S.; Berghe, W.V. Bioinformatics approaches for the functional interpretation of protein lists: From ontology term enrichment to network analysis. Proteomics 2015, 15, 981–996. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Kornhauser, J.M.; Tkachev, S.; Zhang, B.; Skrzypek, E.; Murray, B.; Latham, V.; Sullivan, M. PhosphoSitePlus: A comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012, 40, D261–D270. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Ross, M.M.; Tessitore, A.; Ornstein, D.; Vanmeter, A.; Liotta, L.A.; Petricoin, E.F., 3rd. An initial characterization of the serum phosphoproteome. J. Proteome Res. 2009, 8, 5523–5531. [Google Scholar] [CrossRef] [PubMed]
- Carrascal, M.; Gay, M.; Ovelleiro, D.; Casas, V.; Gelpi, E.; Abian, J. Characterization of the human plasma phosphoproteome using linear ion trap mass spectrometry and multiple search engines. J. Proteome Res. 2010, 9, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Garbis, S.D.; Roumeliotis, T.I.; Tyritzis, S.I.; Zorpas, K.M.; Pavlakis, K.; Constantinides, C.A. A novel multidimensional protein identification technology approach combining protein size exclusion prefractionation, peptide zwitterion-ion hydrophilic interaction chromatography, and nano-ultraperformance RP chromatography/nESI-MS2 for the in-depth analysis of the serum proteome and phosphoproteome: Application to clinical sera derived from humans with benign prostate hyperplasia. Anal. Chem. 2011, 83, 708–718. [Google Scholar] [PubMed]
- Li, Q.R.; Fan, K.X.; Li, R.X.; Dai, J.; Wu, C.C.; Zhao, S.L.; Wu, J.R.; Shieh, C.H.; Zeng, R. A comprehensive and non-prefractionation on the protein level approach for the human urinary proteome: Touching phosphorylation in urine. Rapid Commun. Mass Spectrom. 2010, 24, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Liu, K.; Gao, Y. Phosphoproteins with Stability Against All Urinary Phosphatases as Potential Biomarkers in Urine. Protein Pept. Lett. 2015, 22, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Stone, M.D.; Chen, X.; McGowan, T.; Bandhakavi, S.; Cheng, B.; Rhodus, N.L.; Griffin, T.J. Large-scale phosphoproteomics analysis of whole saliva reveals a distinct phosphorylation pattern. J. Proteome Res. 2011, 10, 1728–1736. [Google Scholar] [CrossRef] [PubMed]
- Giorgianni, F.; Mileo, V.; Desiderio, D.M.; Catinella, S.; Beranova-Giorgianni, S. Characterization of the phosphoproteome in human bronchoalveolar lavage fluid. Int. J. Proteom. 2012, 2012, 460261. [Google Scholar] [CrossRef] [PubMed]
- Total Plasma Protein. Available online: https://www.nlm.nih.gov/medlineplus/ency/article/003483.htm (accessed on 26 May 2016).
- Kalantari, S.; Jafari, A.; Moradpoor, R.; Ghasemi, E.; Khalkhal, E. Human Urine Proteomics: Analytical Techniques and Clinical Applications in Renal Diseases. Int. J. Proteom. 2015, 2015, 782798. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Jin, W.H.; Sheng, Q.H.; Shieh, C.H.; Wu, J.R.; Zeng, R. Protein phosphorylation and expression profiling by Yin-yang multidimensional liquid chromatography (Yin-yang MDLC) mass spectrometry. J. Proteome Res. 2007, 6, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Doherty, C.M.; Forbes, R.B. Diagnostic Lumbar Puncture. Ulster Med. J. 2014, 83, 93–102. [Google Scholar] [PubMed]
- Schutzer, S.E.; Liu, T.; Natelson, B.H.; Angel, T.E.; Schepmoes, A.A.; Purvine, S.O.; Hixson, K.K.; Lipton, M.S.; Camp, D.G.; Coyle, P.K.; et al. Establishing the proteome of normal human cerebrospinal fluid. PLoS ONE 2010, 5, e10980. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.R.; Jensen, S.S.; Jakobsen, L.A.; Heegaard, N.H. Exploring the sialiome using titanium dioxide chromatography and mass spectrometry. Mol. Cell. Proteom. 2007, 6, 1778–1787. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, S.P.; Williamson, R.T. A review of saliva: Normal composition, flow, and function. J. Prosthet. Dent. 2001, 85, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Ruhl, S. The scientific exploration of saliva in the post-proteomic era: From database back to basic function. Expert Rev. Proteom. 2012, 9, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Schipper, R.G.; Silletti, E.; Vingerhoeds, M.H. Saliva as research material: Biochemical, physicochemical and practical aspects. Arch. Oral Biol. 2007, 52, 1114–1135. [Google Scholar] [CrossRef] [PubMed]
- Basic Principles and Techniques of Bronchoalveolar Lavage. Available online: http://www.uptodate.com/contents/basic-principles-and-technique-of-bronchoalveolar-lavage (accessed on 26 May 2016).
- Nguyen, E.V.; Gharib, S.A.; Schnapp, L.M.; Goodlett, D.R. Shotgun MS proteomic analysis of bronchoalveolar lavage fluid in normal subjects. Proteom. Clin. Appl. 2014, 8, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Wattiez, R.; Falmagne, P. Proteomics of bronchoalveolar lavage fluid. J. Chromatogr. B 2005, 815, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Giorgianni, F.; Mileo, V.; Chen, L.; Desiderio, D.M.; Beranova-Giorgianni, S. Proteomics of human bronchoalveolar lavage fluid: Discovery of biomarkers of chronic obstructive pulmonary disease (COPD) with difference gel electrophoresis (DIGE) and mass spectrometry (MS). In Methods in Protein Biochemistry; Tschesche, H., Ed.; Walter de Gruyter: Berlin, Germany, 2012. [Google Scholar]

{kind=link}
| Fluid Studied | Disease or Condition | Protein Depletion | MDLC | Phospho Enrichment | CID/ETD | DDA/DIA | Phosphoproteome Panel Reported | Reference |
|---|---|---|---|---|---|---|---|---|
| Serum | Prostate Cancer | Y | N | Y | Y/Y | DDA (qual.) | ~100 phosphopeptides | [54] |
| Plasma | Normal | Y | Y | Y | Y/N | DDA (qual.) | 138 phosphopeptides/127 sites in 70 proteins | [55] |
| Serum | Benign Prostate Hyperplasia | N | Y | Y | Y/N | DDA (qual.) | 375 phosphopeptides in 375 proteins | [56] |
| Serum | N/A | Y | N | Y (at protein level) | Y/N | DIA (qual.) | 5800 phosphopeptides in 502 proteins | [15] |
| Serum | Schizophrenia vs. Control | Y | N | Y (at protein level) | Y/N | DIA (quant.) | 59 altered phosphoproteins | [16] |
| Urine | Normal | N | Y | N | Y/N | DDA (qual.) | 45 phosphopeptides/59 sites in 31 proteins | [57] |
| Urine | Pregnancy (Before/after delivery) | N | N | Y | Y/N | DDA (quant.) | 130 phosphopeptides/222 sites in 105 proteins; 16 altered phosphoproteins | [17] |
| Urine | Normal | N | N | Y | Y/N | DDA (qual.) | 106 phosphosites in 64 proteins | [58] |
| CSF | Suspected Neurological Disorder | N | N | Y | Y/N | DDA (qual.) | 44 phosphoproteins (include 56 novel sites) | [24] |
| Saliva | Normal | N | N | Y (derivatization) | Y/N | DDA (qual.) | 65 phosphoproteins | [36] |
| Saliva | Normal | Y | Y | Y | Y/Y | DDA (qual.) | 217 phosphopeptides in 85 phosphoproteins | [59] |
| BAL | N/A (not Lung Cancer or COPD) | Y | N | Y | Y/N | DDA (qual.) | 36 phosphopeptides/26 sites in 21 proteins | [60] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giorgianni, F.; Beranova-Giorgianni, S. Phosphoproteome Discovery in Human Biological Fluids. Proteomes 2016, 4, 37. https://doi.org/10.3390/proteomes4040037
Giorgianni F, Beranova-Giorgianni S. Phosphoproteome Discovery in Human Biological Fluids. Proteomes. 2016; 4(4):37. https://doi.org/10.3390/proteomes4040037
Chicago/Turabian StyleGiorgianni, Francesco, and Sarka Beranova-Giorgianni. 2016. "Phosphoproteome Discovery in Human Biological Fluids" Proteomes 4, no. 4: 37. https://doi.org/10.3390/proteomes4040037
APA StyleGiorgianni, F., & Beranova-Giorgianni, S. (2016). Phosphoproteome Discovery in Human Biological Fluids. Proteomes, 4(4), 37. https://doi.org/10.3390/proteomes4040037