Dissection of Protein Kinase Pathways in Live Cells Using Photoluminescent Probes: Surveillance or Interrogation?


Abstract
1. Introduction
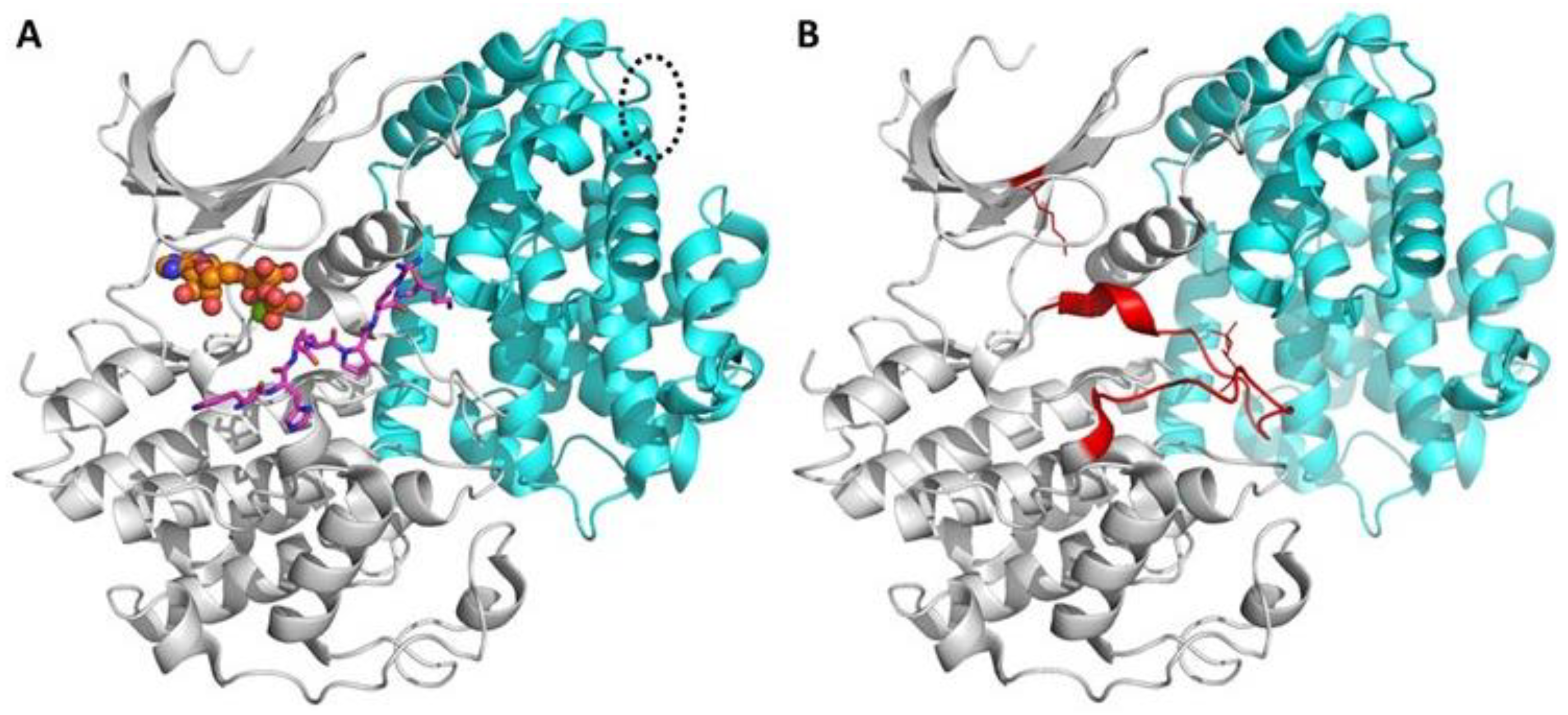
1.1. Brief Insight into Structure and Function of Protein Kinases
1.2. Brief Overview of Photo- and Bioluminescence Phenomena and Applications
1.2.1. Physical Background and Definitions
1.2.2. Main Classes of Fluorophores
- fluorescent protein tags (FPs);
- organic dyes; and
- fluorescent nanoparticles (NPs).
1.2.3. Photoluminescence Measurement Techniques
1.2.4. Bioluminescence: Mechanism, Molecules and Advantages
2. Probes Targeting PK Pathways: Classification and Main Features
- Probes reporting phosphorylation event by changes in photoluminescent characteristics
- genetically encoded tagged substrates of protein kinases
- labeled synthetic and semisynthetic substrate peptides
- Probes reporting phosphorylation event by changes in the intracellular localization (also known as translocation probes)
- Probes reporting binding event by local enrichment of fluorescence intensity, or by changes in photoluminescent characteristics
- labeled small- or medium-molecular-weight synthetic inhibitors
- labeled PK-targeting antibodies or their analogs; labeled endogenous substrates or ligands of PKs
- tagged protein kinases constituting FRET pairs with other tagged proteins or probes from classes 3A, 3B
- Probes reporting events upstream or downstream of signaling of the PK of interest
- tagged protein kinases reporting presence of activating molecules
- probes reporting physiological outcome of signaling pathway of target PK (gene expression, activation of metabolic processes, changes in cellular phenotype and cell viability, etc.)
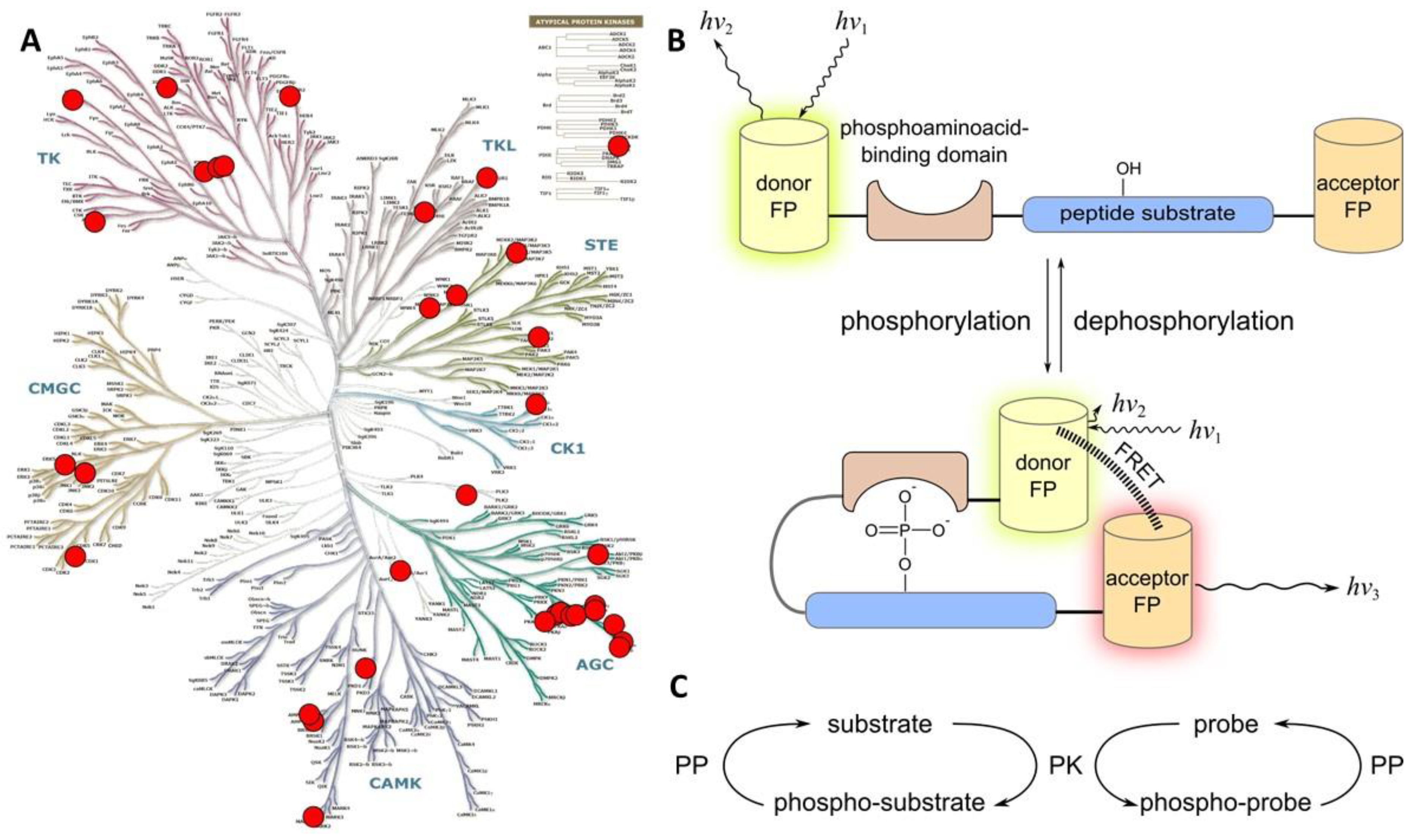
2.1. Class 1
2.1.1. Class 1A
- a consensus sequence that can be recognized and phosphorylated by the target PK;
- a phospho-amino acid recognizing domain (PAAD—such as SH2 that recognizes phospho-Tyr, or 14-3-3, FHA1, WW, and polo-box domain that recognize phospho-Ser/phospho-Thr);
- a FRET pair of fluorescent proteins.
2.1.2. Class 1B
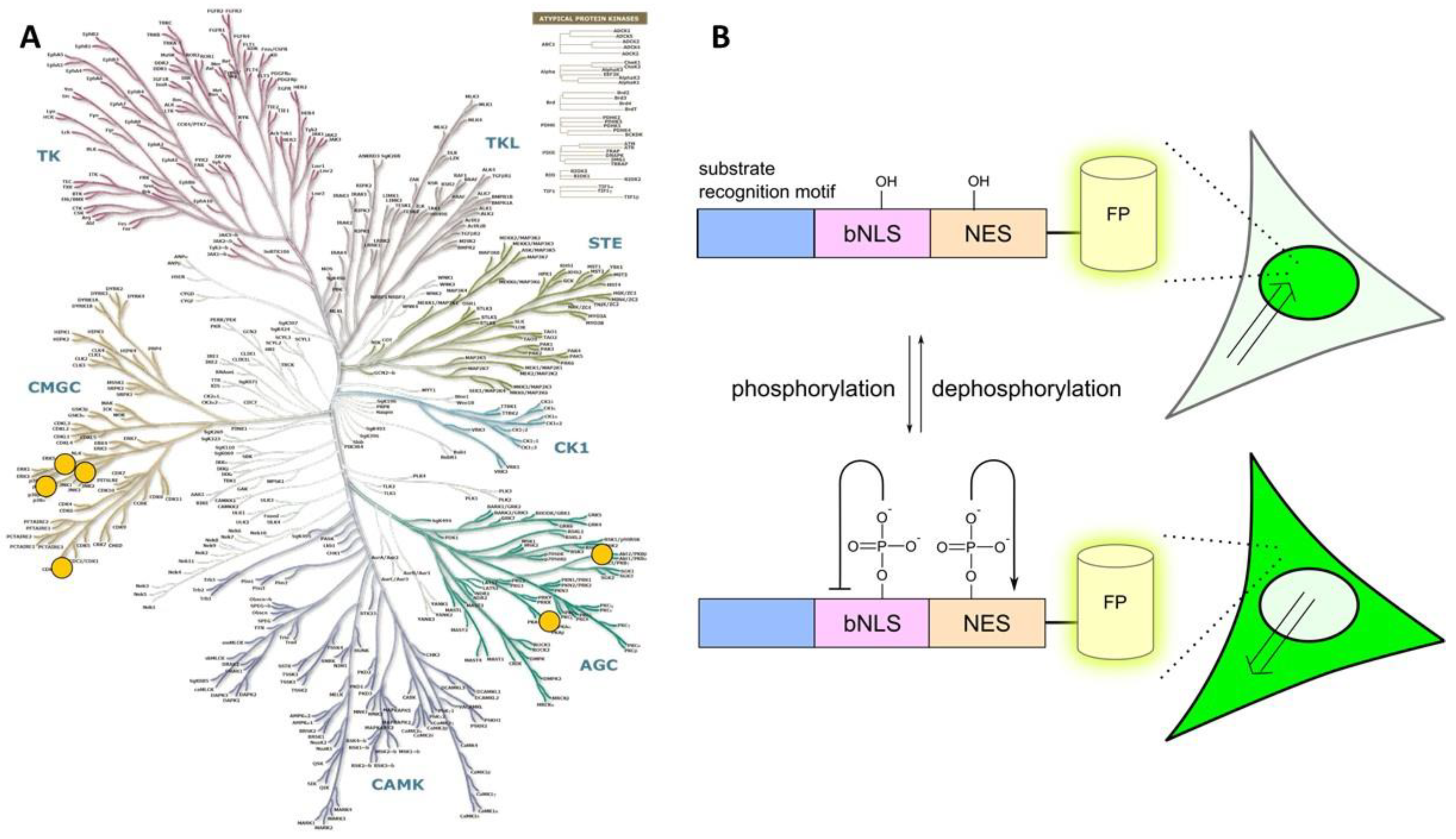
2.2. Class 2
- a consensus sequence that can be recognized and phosphorylated by the target PK;
- a nuclear localization sequence (NLS);
- a nuclear exclusion sequence (NES); and
- a fluorescent protein tag.
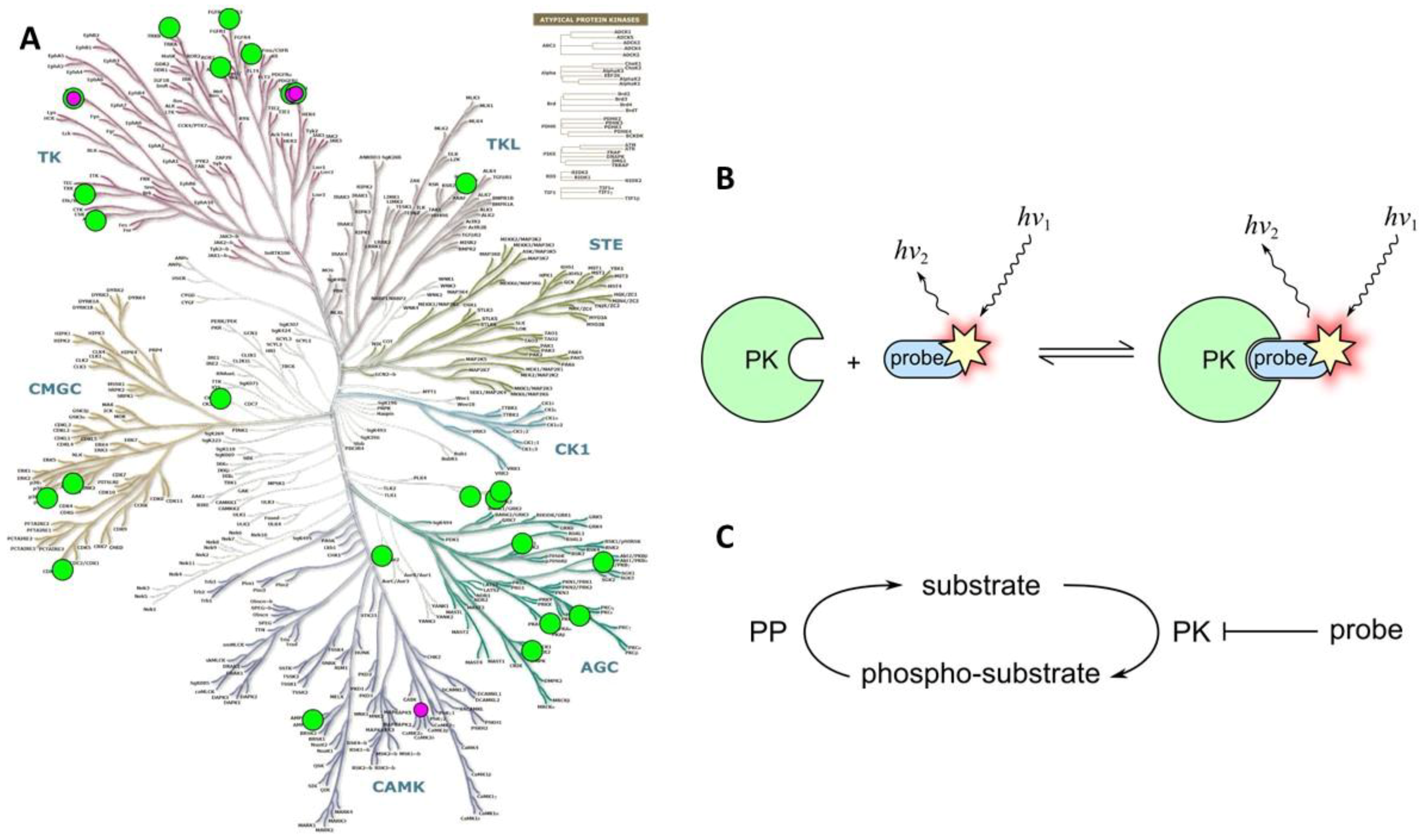
2.3. Class 3
2.3.1. Class 3A
2.3.2. Class 3B
2.3.3. Class 3C
2.4. Class 4
2.4.1. Class 4A
- a domain responsible for binding of the activating molecule—e.g., a secondary messenger (such as diacylglycerol (DAG) or phosphatidylinositol (3,4,5)-trisphosphate (PIP3)), or a regulatory protein (such as calmodulin or EGF);
- a catalytic core of the target PK, including autophosphorylatable region;
- a phospho-amino acid recognizing domain (PAAD); and
- a FRET pair of fluorescent proteins.
2.4.2. Class 4B
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABPP | activity-based protein profiling |
| CPP | cell-penetrating peptide |
| DAG | diacylglycerol |
| FLIM | fluorescence lifetime imaging |
| FP | fluorescent protein |
| KAR | kinase activity reporter |
| NES | nuclear export signal |
| NLS | nuclear localization signa |
| NP | nanoparticle |
| PAAD | phospho-amino acid binding domain |
| PIP3 | phosphatidylinositol(3,4,5)-trisphosphate |
| PK | protein kinase |
| PKAc | catalytic subunit of cAMP-dependent protein kinase |
| PKAr | regulatory subunit of cAMP-dependent protein kinase |
| QD | quantum dot |
| QY | quantum yield |
References
- Mildvan, A.S. Mechanisms of signaling and related enzymes. Proteins 1997, 29, 401–416. [Google Scholar] [CrossRef]
- Knight, J.D.R.; Qian, B.; Baker, D.; Kothary, R. Conservation, Variability and the Modeling of Active Protein Kinases. PLoS ONE 2007, 2, e982. [Google Scholar] [CrossRef] [PubMed]
- Kung, J.E.; Jura, N. Structural Basis for the Non-catalytic Functions of Protein Kinases. Structure 2016, 24, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.J.; Turk, B.E. Homing in: Mechanisms of Substrate Targeting by Protein Kinases. Trends Biochem. Sci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Jia, P.; Wang, Q.; Zhao, Z. Quantitative network mapping of the human kinome interactome reveals new clues for rational kinase inhibitor discovery and individualized cancer therapy. Oncotarget 2014, 5, 3697–3710. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Serine/Threonine Phosphatases: Mechanism through Structure. Cell 2009, 139, 468–484. [Google Scholar] [CrossRef] [PubMed]
- Lochhead, P.A. Protein Kinase Activation Loop Autophosphorylation in Cis: Overcoming a Catch-22 Situation. Biochemistry 2009, 2, pe4. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, S.R.; Mohammadi, M.; Schlessinger, J. Autoregulatory Mechanisms in Protein-tyrosine Kinases. J. Biol. Chem. 1998, 273, 11987–11990. [Google Scholar] [CrossRef] [PubMed]
- Carmena, M.; Ruchaud, S.; Earnshaw, W.C. Making the Auroras glow: Regulation of Aurora A and B kinase function by interacting proteins. Curr. Opin. Cell Biol. 2009, 21, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Hindriksen, S.; Lens, S.M.A.; Hadders, M.A. The Ins and Outs of Aurora B Inner Centromere Localization. Front. Cell Dev. Biol. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Corradini, E.; Burgers, P.P.; Plank, M.; Heck, A.J.R.; Scholten, A. Huntingtin-associated Protein 1 (HAP1) Is a cGMP-dependent Kinase Anchoring Protein (GKAP) Specific for the cGMP-dependent Protein Kinase Iβ Isoform. J. Biol. Chem. 2015, 290, 7887–7896. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.R.; Ron, D.; Kiely, P.A. RACK1, A multifaceted scaffolding protein: Structure and function. Cell Commun. Signal. 2011, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Bayer, K.-U.; Harbers, K.; Schulman, H. aKAP is an anchoring protein for a novel CaM kinase II isoform in skeletal muscle. EMBO J. 1998, 17, 5598–5605. [Google Scholar] [CrossRef] [PubMed]
- Eigenthaler, M.; Nolte, C.; Halbrugge, M.; Walter, U. Concentration and regulation of cyclic nucleotides, cyclic-nucleotide-dependent protein kinases and one of their major substrates in human platelets. Estimating the rate of cAMP-regulated and cGMP-regulated protein phosphorylation in intact cells. Eur. J. Biochem. 1992, 205, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Torkamani, A.; Verkhivker, G.; Schork, N.J. Cancer driver mutations in protein kinase genes. Cancer Lett. 2009, 281, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Quan, C.; Xiao, J.; Liu, L.; Duan, Q.; Yuan, P.; Zhu, F. Protein Kinases as Tumor Biomarkers and Therapeutic Targets. Curr. Pharm. Des. 2017, 23. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.-R.; Issaq, H.J.; Veenstra, T.D. Phosphoproteomics for the discovery of kinases as cancer biomarkers and drug targets. Proteom. Clin. Appl. 2007, 1, 1042–1057. [Google Scholar] [CrossRef] [PubMed]
- Nesterova, M.V.; Johnson, N.; Cheadle, C.; Bates, S.E.; Mani, S.; Stratakis, C.A.; Kahn, I.; Gupta, R.K.; Cho-Chung, Y.S. Autoantibody Cancer Biomarker: Extracellular Protein Kinase A. Cancer Res. 2006, 66, 8971–8974. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.-H.; Yu, J.-K.; Yang, F.-Q.; Zheng, S. Identification of a new protein biomarker for colorectal cancer diagnosis. Mol. Med. Rep. 2012, 6, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Chen, L.; Deng, H.; Zou, Y.; Liu, J.; Shi, H.; Xu, B.; Lu, M.; Li, C.; Jiang, J.; et al. Serum lemur tyrosine kinase-3: A novel biomarker for screening primary non-small cell lung cancer and predicting cancer progression. Int. J. Clin. Exp. Pathol. 2015, 8, 629–635. [Google Scholar] [PubMed]
- Hon, K.W.; Abu, N.; Ab Mutalib, N.-S.; Jamal, R. Exosomes As Potential Biomarkers and Targeted Therapy in Colorectal Cancer: A Mini-Review. Front. Pharmacol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, C.; Zhou, T.; Liu, X.; Liu, X.; Li, X.; Chen, D. Role of exosomal proteins in cancer diagnosis. Mol. Cancer 2017, 16. [Google Scholar] [CrossRef] [PubMed]
- Sandfeld-Paulsen, B.; Jakobsen, K.R.; Bæk, R.; Folkersen, B.H.; Rasmussen, T.R.; Meldgaard, P.; Varming, K.; Jørgensen, M.M.; Sorensen, B.S. Exosomal Proteins as Diagnostic Biomarkers in Lung Cancer. J. Thorac. Oncol. 2016, 11, 1701–1710. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Alessi, D.R. Kinase Drug Discovery—What’s Next in the Field? ACS Chem. Biol. 2013, 8, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Berndt, N.; Karim, R.M.; Schönbrunn, E. Advances of small molecule targeting of kinases. Curr. Opin. Chem. Biol. 2017, 39, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shyy, J.Y.; Chien, S. Fluorescence proteins, live-cell imaging, and mechanobiology: Seeing is believing. Annu. Rev. Biomed. Eng. 2008, 10, 1–38. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.X.; Lin, M.Z. Photoswitchable fluorescent proteins: Ten years of colorful chemistry and exciting applications. Curr. Opin. Chem. Biol. 2013, 17, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Lippincott-Schwartz, J.; Patterson, G.H. Photoactivatable fluorescent proteins for diffraction-limited and super-resolution imaging. Trends Cell Biol. 2009, 19, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, M. GFP: From jellyfish to the Nobel prize and beyond. Chem. Soc. Rev. 2009, 38, 3823–3832. [Google Scholar] [CrossRef] [PubMed]
- Cody, C.W.; Prasher, D.C.; Westler, W.M.; Prendergast, F.G.; Ward, W.W. Chemical structure of the hexapeptide chromophore of the Aequorea green-fluorescent protein. Biochemistry 1993, 32, 1212–1218. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Moss, L.G.; Phillips, G.N., Jr. The molecular structure of green fluorescent protein. Nat. Biotechnol. 1996, 14, 1246–1251. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.-T.; Cheng, L.; Kain, S.R. Optimized Codon Usage and Chromophore Mutations Provide Enhanced Sensitivity with the Green Fluorescent Protein. Nucleic Acids Res. 1996, 24, 4592–4593. [Google Scholar] [CrossRef] [PubMed]
- Subach, F.V.; Verkhusha, V.V. Chromophore Transformations in Red Fluorescent Proteins. Chem. Rev. 2012, 112, 4308–4327. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, A.; Shcherbakova, D.M.; Verkhusha, V.V. Red fluorescent proteins: Chromophore formation and cellular applications. Curr. Opin. Struct. Biol. 2012, 22, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, P.J.; Chen, Y.; Mueller, J.D. Chromophore maturation and fluorescence fluctuation spectroscopy of fluorescent proteins in a cell-free expression system. Anal. Biochem. 2012, 421, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Craggs, T.D. Green fluorescent protein: Structure, folding and chromophore maturation. Chem. Soc. Rev. 2009, 38, 2865–2875. [Google Scholar] [CrossRef] [PubMed]
- University of California Davis. Single Molecule Tracking, Figure 1. Available online: Https://phys.libretexts.org/LibreTexts/University_of_California_Davis/UCD%3A_Biophysics_241/Experimental_Characterization/Single_Molecule_Tracking (accessed on 24 April 2018).
- Chudakov, D.M.; Matz, M.V.; Lukyanov, S.; Lukyanov, K.A. Fluorescent Proteins and Their Applications in Imaging Living Cells and Tissues. Physiol. Rev. 2010, 90, 1103–1163. [Google Scholar] [CrossRef] [PubMed]
- Bulina, M.E.; Chudakov, D.M.; Mudrik, N.N.; Lukyanov, K.A. Interconversion of Anthozoa GFP-like fluorescent and non- fluorescent proteins by mutagenesis. BMC Biochem. 2002, 3, 7. [Google Scholar] [CrossRef]
- Zacharias, D.A. Partitioning of Lipid-Modified Monomeric GFPs into Membrane Microdomains of Live Cells. Science 2002, 296, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Yanushevich, Y.G.; Staroverov, D.B.; Savitsky, A.P.; Fradkov, A.F.; Gurskaya, N.G.; Bulina, M.E.; Lukyanov, K.A.; Lukyanov, S.A. A strategy for the generation of non-aggregating mutants of Anthozoa fluorescent proteins. FEBS Lett. 2002, 511, 11–14. [Google Scholar] [CrossRef]
- Chen, M.; Yin, M. Design and development of fluorescent nanostructures for bioimaging. Progr. Polym. Sci. 2014, 39, 365–395. [Google Scholar] [CrossRef]
- Wolfbeis, O.S. An overview of nanoparticles commonly used in fluorescent bioimaging. Chem. Soc. Rev. 2015, 44, 4743–4768. [Google Scholar] [CrossRef] [PubMed]
- Medintz, I.L.; Clapp, A.R.; Mattoussi, H.; Goldman, E.R.; Fisher, B.; Mauro, J.M. Self-assembled nanoscale biosensors based on quantum dot FRET donors. Nat. Mater. 2003, 2, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Wegner, K.D.; Hildebrandt, N. Quantum dots: Bright and versatile in vitro and in vivo fluorescence imaging biosensors. Chem. Soc. Rev. 2015, 44, 4792–4834. [Google Scholar] [CrossRef] [PubMed]
- Biju, V. Chemical modifications and bioconjugate reactions of nanomaterials for sensing, imaging, drug delivery and therapy. Chem. Soc. Rev. 2014, 43, 744–764. [Google Scholar] [CrossRef] [PubMed]
- Delehanty, J.B.; Mattoussi, H.; Medintz, I.L. Delivering quantum dots into cells: Strategies, progress and remaining issues. Anal. Bioanal. Chem. 2009, 393, 1091–1105. [Google Scholar] [CrossRef] [PubMed]
- McNamara, George GEOMCNAMARA. Available online: http://www.geomcnamara.com/fluorophore-table (accessed on 24 April 2018).
- McNamara, George GEOMCNAMARA. Available online: http://www.geomcnamara.com/fluorescent-proteins-photophysics-data (accessed on 24 April 2018).
- Cserép, G.B.; Herner, A.; Kele, P. Bioorthogonal fluorescent labels: A review on combined forces. Methods Appl. Fluoresc. 2015, 3, 042001. [Google Scholar] [CrossRef] [PubMed]
- Geertsema, H.J.; Schulte, A.C.; Spenkelink, L.M.; McGrath, W.J.; Morrone, S.R.; Sohn, J.; Mangel, W.F.; Robinson, A.; van Oijen, A.M. Single-Molecule Imaging at High Fluorophore Concentrations by Local Activation of Dye. Biophys. J. 2015, 108, 949–956. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fink, C.; Morgan, F.; Loew, L.M. Intracellular Fluorescent Probe Concentrations by Confocal Microscopy. Biophys. J. 1998, 75, 1648–1658. [Google Scholar] [CrossRef]
- Rahnel, H.; Viht, K.; Lavogina, D.; Mazina, O.; Haljasorg, T.; Enkvist, E.; Uri, A. A Selective Biligand Inhibitor of CK2 Increases Caspase-3 Activity in Cancer Cells and Inhibits Platelet Aggregation. ChemMedChem 2017, 12, 1723–1736. [Google Scholar] [CrossRef] [PubMed]
- Kriisa, M.; Sinijärv, H.; Vaasa, A.; Enkvist, E.; Kostenko, S.; Moens, U.; Uri, A. Inhibition of CREB Phosphorylation by Conjugates of Adenosine Analogues and Arginine-Rich Peptides, Inhibitors of PKA Catalytic Subunit. ChemBioChem 2015, 16, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Felber, L.M.; Cloutier, S.M.; Kündig, C.; Kishi, T.; Brossard, V.; Jichlinski, P.; Leisinger, H.-J.; Deperthes, D. Evaluation of the CFP-substrate-YFP system for protease studies: Advantages and limitations. Proteom. Technol. 2004, 36, 878–885. [Google Scholar]
- Morikawa, T.J.; Fujita, H.; Kitamura, A.; Horio, T.; Yamamoto, J.; Kinjo, M.; Sasaki, A.; Machiyama, H.; Yoshizawa, K.; Ichimura, T.; et al. Dependence of fluorescent protein brightness on protein concentration in solution and enhancement of it. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: Berlin, Germany, 2006; ISBN 978-0-387-46312-4. [Google Scholar]
- Barbero, N.; Barni, E.; Barolo, C.; Quagliotto, P.; Viscardi, G.; Napione, L.; Pavan, S.; Bussolino, F. A study of the interaction between fluorescein sodium salt and bovine serum albumin by steady-state fluorescence. Dyes Pigments 2009, 80, 307–313. [Google Scholar] [CrossRef]
- Hitosugi, T.; Sato, M.; Sasaki, K.; Umezawa, Y. Lipid Raft Specific Knockdown of Src Family Kinase Activity Inhibits Cell Adhesion and Cell Cycle Progression of Breast Cancer Cells. Cancer Res. 2007, 67, 8139–8148. [Google Scholar] [CrossRef] [PubMed]
- Axelrod, D. Total Internal Reflection Fluorescence Microscopy in Cell Biology: Total Internal Reflection Fluorescence. Traffic 2001, 2, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Becker, W. Fluorescence lifetime imaging—Techniques and applications: Fluorescence Lifetime Imaging. J. Microsc. 2012, 247, 119–136. [Google Scholar] [CrossRef] [PubMed]
- Mérola, F.; Fredj, A.; Betolngar, D.-B.; Ziegler, C.; Erard, M.; Pasquier, H. Newly engineered cyan fluorescent proteins with enhanced performances for live cell FRET imaging. Biotechnol. J. 2014, 9, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Nie, H.; Yang, W.; Yang, M.; Jing, J.; Zhang, X. Highly specific and ratiometric fluorescent probe for ozone assay in indoor air and living cells. Dyes Pigments 2016, 127, 67–72. [Google Scholar] [CrossRef]
- Bajar, B.; Wang, E.; Zhang, S.; Lin, M.; Chu, J. A Guide to Fluorescent Protein FRET Pairs. Sensors 2016, 16, 1488. [Google Scholar] [CrossRef] [PubMed]
- Zaccolo, M.; De Giorgi, F.; Cho, C.Y.; Feng, L.; Knapp, T.; Negulescu, P.A.; Taylor, S.S.; Tsien, R.Y.; Pozzan, T. A genetically encoded, fluorescent indicator for cyclic AMP in living cells. Nat. Cell Biol. 2000, 2, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Biener-Ramanujan, E.; Ramanujan, V.K.; Herman, B.; Gertler, A. Spatio-temporal kinetics of growth hormone receptor signaling in single cells using FRET microscopy. Growth Horm. IGF Res. 2006, 16, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Koschut, D.; Richert, L.; Pace, G.; Niemann, H.H.; Mély, Y.; Orian-Rousseau, V. Live cell imaging shows hepatocyte growth factor-induced Met dimerization. Biochim. Biophys. Acta 2016, 1863, 1552–1558. [Google Scholar] [CrossRef] [PubMed]
- Herbst, K.J.; Allen, M.D.; Zhang, J. Luminescent Kinase Activity Biosensors Based on a Versatile Bimolecular Switch. J. Am. Chem. Soc. 2011, 133, 5676–5679. [Google Scholar] [CrossRef] [PubMed]
- Goedhart, J.; van Weeren, L.; Adjobo-Hermans, M.J.W.; Elzenaar, I.; Hink, M.A.; Gadella, T.W.J. Quantitative Co-Expression of Proteins at the Single Cell Level—Application to a Multimeric FRET Sensor. PLoS ONE 2011, 6, e27321. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, S.; Ameer-beg, S.M.; Ng, T.T.C.; Vojnovic, B.; Wouters, F.S. A dark yellow fluorescent protein (YFP)-based Resonance Energy-Accepting Chromoprotein (REACh) for Forster resonance energy transfer with GFP. Proc. Natl. Acad. Sci. USA 2006, 103, 4089–4094. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-J.R.; Escobedo-Lozoya, Y.; Szatmari, E.M.; Yasuda, R. Activation of CaMKII in single dendritic spines during long-term potentiation. Nature 2009, 458, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Vaasa, A.; Ligi, K.; Mohandessi, S.; Enkvist, E.; Uri, A.; Miller, L.W. Time-gated luminescence microscopy with responsive nonmetal probes for mapping activity of protein kinases in living cells. Chem. Commun. 2012, 48, 8595. [Google Scholar] [CrossRef] [PubMed]
- Dickson, R.M.; Cubitt, A.B.; Tsien, R.Y.; Moerner, W.E. On/off blinking and switching behaviour of single molecules of green fluorescent protein. Nature 1997, 388, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois, D.; Adam, V. Reversible photoswitching in fluorescent proteins: A mechanistic view. IUBMB Life 2012, 64, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Mo, G.C.H.; Ross, B.; Hertel, F.; Manna, P.; Yang, X.; Greenwald, E.; Booth, C.; Plummer, A.M.; Tenner, B.; Chen, Z.; et al. Genetically encoded biosensors for visualizing live-cell biochemical activity at super-resolution. Nat. Methods 2017, 14, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Sahl, S.J.; Hell, S.W.; Jakobs, S. Fluorescence nanoscopy in cell biology. Nat. Rev. Mol. Cell Biol. 2017, 18, 685–701. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Shim, S.-H.; He, J.; Zhuang, X. Fast, three-dimensional super-resolution imaging of live cells. Nat. Methods 2011, 8, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Winckler, P.; Lartigue, L.; Giannone, G.; De Giorgi, F.; Ichas, F.; Sibarita, J.-B.; Lounis, B.; Cognet, L. Identification and super-resolution imaging of ligand-activated receptor dimers in live cells. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Agnes, R.S.; Broome, A.-M.; Wang, J.; Verma, A.; Lavik, K.; Basilion, J.P. An Optical Probe for Noninvasive Molecular Imaging of Orthotopic Brain Tumors Overexpressing Epidermal Growth Factor Receptor. Mol. Cancer Ther. 2012, 11, 2202–2211. [Google Scholar] [CrossRef] [PubMed]
- Ardeshirpour, Y.; Chernomordik, V.; Zielinski, R.; Capala, J.; Griffiths, G.; Vasalatiy, O.; Smirnov, A.V.; Knutson, J.R.; Lyakhov, I.; Achilefu, S.; et al. In Vivo Fluorescence Lifetime Imaging Monitors Binding of Specific Probes to Cancer Biomarkers. PLoS ONE 2012, 7, e31881. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.; Rouhi, S.; Taniguchi, S.; Yang, S.Y.; Green, M.; Keshtgar, M.; Seifalian, A. Near-infrared quantum dots for HER2 localization and imaging of cancer cells. Int. J. Nanomed. 2014. [Google Scholar] [CrossRef]
- Mahalingam, S.M.; Dudkin, V.Y.; Goldberg, S.; Klein, D.; Yi, F.; Singhal, S.; O’Neil, K.T.; Low, P.S. Evaluation of a Centyrin-Based Near-Infrared Probe for Fluorescence-Guided Surgery of Epidermal Growth Factor Receptor Positive Tumors. Bioconjug. Chem. 2017, 28, 2865–2873. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.-T.; Ko, K.-P.; Shi, H.; Ren, W.X.; Verwilst, P.; Koo, S.; Lee, J.Y.; Chi, S.-G.; Kim, J.S. PLK1-Targeted Fluorescent Tumor Imaging with High Signal-to-Background Ratio. ACS Sens. 2017, 2, 1512–1516. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Kim, E.; Cuccarese, M.F.; Plotkin, A.L.; Prytyskach, M.; Kohler, R.H.; Pittet, M.J.; Weissleder, R. Near infrared imaging of Mer tyrosine kinase (MERTK) using MERi-SiR reveals tumor associated macrophage uptake in metastatic disease. Chem. Commun. 2018, 54, 42–45. [Google Scholar] [CrossRef] [PubMed]
- Helmchen, F.; Denk, W. Deep tissue two-photon microscopy. Nat. Methods 2005, 2, 932–940. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.R.; Jarrett, J.W.; Hassan, A.M.; Dunn, A.K. Deep tissue imaging with multiphoton fluorescence microscopy. Curr. Opin. Biomed. Eng. 2017, 4, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Thorne, N.; Inglese, J.; Auld, D.S. Illuminating Insights into Firefly Luciferase and Other Bioluminescent Reporters Used in Chemical Biology. Chem. Biol. 2010, 17, 646–657. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Rowe, L.; Dikici, E.; Ensor, M.; Daunert, S. Aequorin mutants with increased thermostability. Anal. Bioanal. Chem. 2014, 406, 5639–5643. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Inouye, S.; Shimomura, O. The use of Renilla luciferase, Oplophorus luciferase, and apoaequorin as bioluminescent reporter protein in the presence of coelenterazine analogues as substrate. Biochem. Biophys. Res. Commun. 1997, 233, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Nyati, S.; Schinske, K.; Ray, D.; Nyati, M.; Ross, B.D.; Rehemtulla, A. Molecular Imaging of TGF -Induced Smad2/3 Phosphorylation Reveals a Role for Receptor Tyrosine Kinases in Modulating TGF Signaling. Clin. Cancer Res. 2011, 17, 7424–7439. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.P.; Unch, J.; Binkowski, B.F.; Valley, M.P.; Butler, B.L.; Wood, M.G.; Otto, P.; Zimmerman, K.; Vidugiris, G.; Machleidt, T.; et al. Engineered Luciferase Reporter from a Deep Sea Shrimp Utilizing a Novel Imidazopyrazinone Substrate. ACS Chem. Biol. 2012, 7, 1848–1857. [Google Scholar] [CrossRef] [PubMed]
- Robers, M.B.; Dart, M.L.; Woodroofe, C.C.; Zimprich, C.A.; Kirkland, T.A.; Machleidt, T.; Kupcho, K.R.; Levin, S.; Hartnett, J.R.; Zimmerman, K.; et al. Target engagement and drug residence time can be observed in living cells with BRET. Nat. Comm. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Vasta, J.D.; Corona, C.R.; Wilkinson, J.; Zimprich, C.A.; Hartnett, J.R.; Ingold, M.R.; Zimmerman, K.; Machleidt, T.; Kirkland, T.A.; Huwiler, K.G.; et al. Quantitative, Wide-Spectrum Kinase Profiling in Live Cells for Assessing the Effect of Cellular ATP on Target Engagement. Cell Chem. Biol. 2018, 25. [Google Scholar] [CrossRef] [PubMed]
- González-Vera, J.; Morris, M. Fluorescent Reporters and Biosensors for Probing the Dynamic Behavior of Protein Kinases. Proteomes 2015, 3, 369–410. [Google Scholar] [CrossRef] [PubMed]
- Oldach, L.; Zhang, J. Genetically Encoded Fluorescent Biosensors for Live-Cell Visualization of Protein Phosphorylation. Chem. Biol. 2014, 21, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Tarrant, M.K.; Cole, P.A. The Chemical Biology of Protein Phosphorylation. Annu. Rev. Biochem. 2009, 78, 797–825. [Google Scholar] [CrossRef] [PubMed]
- Ni, Q.; Titov, D.; Zhang, J. Analyzing protein kinase dynamics in living cells with FRET reporters. Methods 2006, 40, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Antal, C.E.; Newton, A.C. Spatiotemporal Dynamics of Phosphorylation in Lipid Second Messenger Signaling. Mol. Cell. Proteom. 2013, 12, 3498–3508. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Allen, M.D. FRET-based biosensors for protein kinases: Illuminating the kinome. Mol. BioSyst. 2007, 3, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Aoki, K.; Kiyokawa, E.; Nakamura, T.; Matsuda, M. Visualization of growth signal transduction cascades in living cells with genetically encoded probes based on Forster resonance energy transfer. Philos. Trans. R. Soc. B 2008, 363, 2143–2151. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, D.S.; Wang, Q. Seeing Is Believing: Peptide-Based Fluorescent Sensors of Protein Tyrosine Kinase Activity. ChemBioChem 2007, 8, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Miyazaki, M.; Aoki, R.; Zama, T.; Inouye, S.; Hirose, K.; Iino, M.; Hagiwara, M. A fluorescent indicator for visualizing cAMP-induced phosphorylation in vivo. Nat. Biotechnol. 2000, 18, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ma, Y.; Taylor, S.S.; Tsien, R.Y. Genetically encoded reporters of protein kinase A activity reveal impact of substrate tethering. Proc. Natl. Acad. Sci. USA 2001, 98, 14997–15002. [Google Scholar] [CrossRef] [PubMed]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed]
- Eid, S.; Fulle, S. KinMap. Available online: http://www.kinhub.org/kinmap/index.html (accessed on 24 April 2018).
- Romoser, V.A.; Hinkle, P.M.; Persechini, A. Detection in Living Cells of Ca2+-dependent Changes in the Fluorescence Emission of an Indicator Composed of Two Green Fluorescent Protein Variants Linked by a Calmodulin-binding Sequence. J. Biol. Chem. 1997, 272, 13270–13274. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, A.; Llopis, J.; Heim, R.; McCaffery, J.M.; Adams, J.A.; Ikura, M.; Tsien, R.Y. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature 1997, 388, 882–887. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, K.; Mochizuki, N.; Ohba, Y.; Mizuno, H.; Miyawaki, A.; Matsuda, M. A Pair of Fluorescent Resonance Energy Transfer-based Probes for Tyrosine Phosphorylation of the CrkII Adaptor Protein in Vivo. J. Biol. Chem. 2001, 276, 31305–31310. [Google Scholar] [CrossRef] [PubMed]
- VanEngelenburg, S.B.; Palmer, A.E. Fluorescent biosensors of protein function. Curr. Opin. Chem. Biol. 2008, 12, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Ting, A.Y.; Kain, K.H.; Klemke, R.L.; Tsien, R.Y. Genetically encoded fluorescent reporters of protein tyrosine kinase activities in living cells. Proc. Natl. Acad. Sci. USA 2001, 98, 15003–15008. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Ozawa, T.; Inukai, K.; Asano, T.; Umezawa, Y. Fluorescent Indicators for Imaging Protein Phosphorylation in Single Living Cells. Nat. Biotechnol. 2002, 20, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Seong, J.; Ouyang, M.; Kim, T.; Sun, J.; Wen, P.-C.; Lu, S.; Zhuo, Y.; Llewellyn, N.M.; Schlaepfer, D.D.; Guan, J.-L.; et al. Detection of focal adhesion kinase activation at membrane microdomains by fluorescence resonance energy transfer. Nat. Commun. 2011, 2, 406. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Sun, J.; Wu, J.; He, H.-T.; Wang, Y.; Zhu, C. A FRET-Based Biosensor for Imaging SYK Activities in Living Cells. Cell. Mol. Bioeng. 2011, 4, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Randriamampita, C.; Mouchacca, P.; Malissen, B.; Marguet, D.; Trautmann, A.; Lellouch, A.C. A Novel ZAP-70 Dependent FRET Based Biosensor Reveals Kinase Activity at both the Immunological Synapse and the Antisynapse. PLoS ONE 2008, 3, e1521. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.D.; Zhang, J. Subcellular dynamics of protein kinase a activity visualized by FRET-based reporters. Biochem. Biophys. Res. Commun. 2006, 348, 716–721. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, M.T.; Ni, Q.; Tsien, R.Y.; Zhang, J.; Newton, A.C. Spatio-temporal Dynamics of Protein Kinase B/Akt Signaling Revealed by a Genetically Encoded Fluorescent Reporter. J. Biol. Chem. 2005, 280, 5581–5587. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhang, J. Spatiotemporal Analysis of Differential Akt Regulation in Plasma Membrane Microdomains. Mol. Biol. Cell 2008, 19, 4366–4373. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Sato, M.; Umezawa, Y. Fluorescent Indicators for Akt/Protein Kinase B and Dynamics of Akt Activity Visualized in Living Cells. J. Biol. Chem. 2003, 278, 30945–30951. [Google Scholar] [CrossRef] [PubMed]
- Tsou, P.; Zheng, B.; Hsu, C.-H.; Sasaki, A.T.; Cantley, L.C. A Fluorescent Reporter of AMPK Activity and Cellular Energy Stress. Cell Metab. 2011, 13, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Konagaya, Y.; Terai, K.; Hirao, Y.; Takakura, K.; Imajo, M.; Kamioka, Y.; Sasaoka, N.; Kakizuka, A.; Sumiyama, K.; Asano, T.; et al. A Highly Sensitive FRET Biosensor for AMPK Exhibits Heterogeneous AMPK Responses among Cells and Organs. Cell Rep. 2017, 21, 2628–2638. [Google Scholar] [CrossRef] [PubMed]
- Fosbrink, M.; Aye-Han, N.-N.; Cheong, R.; Levchenko, A.; Zhang, J. Visualization of JNK activity dynamics with a genetically encoded fluorescent biosensor. Proc. Natl. Acad. Sci. USA 2010, 107, 5459–5464. [Google Scholar] [CrossRef] [PubMed]
- Gallegos, L.L.; Kunkel, M.T.; Newton, A.C. Targeting Protein Kinase C Activity Reporter to Discrete Intracellular Regions Reveals Spatiotemporal Differences in Agonist-dependent Signaling. J. Biol. Chem. 2006, 281, 30947–30956. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, M.T.; Newton, A.C. Imaging Kinase Activity at Protein Scaffolds. Methods Mol. Biol. 2014, 1071, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Harvey, C.D.; Ehrhardt, A.G.; Cellurale, C.; Zhong, H.; Yasuda, R.; Davis, R.J.; Svoboda, K. A genetically encoded fluorescent sensor of ERK activity. Proc. Natl. Acad. Sci. USA 2008, 105, 19264–19269. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Kawai, Y.; Umezawa, Y. Genetically Encoded Fluorescent Indicators To Visualize Protein Phosphorylation by Extracellular Signal-Regulated Kinase in Single Living Cells. Anal. Chem. 2007, 79, 2570–2575. [Google Scholar] [CrossRef] [PubMed]
- Hukasova, E.; Silva Cascales, H.; Kumar, S.R.; Lindqvist, A. Monitoring Kinase and Phosphatase Activities Through the Cell Cycle by Ratiometric FRET. J. Vis. Exp. 2012. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.A.; You, Z.; Hunter, T. Monitoring ATM kinase activity in living cells. DNA Repair 2007, 6, 1277–1284. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-W.; Ting, A.Y. A Genetically Encoded Fluorescent Reporter of Histone Phosphorylation in Living Cells. Angew. Chem. Int. Ed. 2004, 43, 2940–2943. [Google Scholar] [CrossRef] [PubMed]
- Gavet, O.; Pines, J. Progressive Activation of CyclinB1-Cdk1 Coordinates Entry to Mitosis. Dev. Cell 2010, 18, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Fuller, B.G.; Lampson, M.A.; Foley, E.A.; Rosasco-Nitcher, S.; Le, K.V.; Tobelmann, P.; Brautigan, D.L.; Stukenberg, P.T.; Kapoor, T.M. Midzone activation of aurora B in anaphase produces an intracellular phosphorylation gradient. Nature 2008, 453, 1132–1136. [Google Scholar] [CrossRef] [PubMed]
- Macůrek, L.; Lindqvist, A.; Lim, D.; Lampson, M.A.; Klompmaker, R.; Freire, R.; Clouin, C.; Taylor, S.S.; Yaffe, M.B.; Medema, R.H. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature 2008, 455, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Tomida, T.; Takekawa, M.; O’Grady, P.; Saito, H. Stimulus-Specific Distinctions in Spatial and Temporal Dynamics of Stress-Activated Protein Kinase Kinase Kinases Revealed by a Fluorescence Resonance Energy Transfer Biosensor. Mol. Cell. Biol. 2009, 29, 6117–6127. [Google Scholar] [CrossRef] [PubMed]
- Timm, T.; von Kries, J.P.; Li, X.; Zempel, H.; Mandelkow, E.; Mandelkow, E.-M. Microtubule Affinity Regulating Kinase Activity in Living Neurons Was Examined by a Genetically Encoded Fluorescence Resonance Energy Transfer/Fluorescence Lifetime Imaging-based Biosensor. J. Biol. Chem. 2011, 286, 41711–41722. [Google Scholar] [CrossRef] [PubMed]
- Kamioka, Y.; Sumiyama, K.; Mizuno, R.; Matsuda, M. Live imaging of transgenic mice expressing FRET biosensors. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2013, 2013, 125–128. [Google Scholar] [PubMed]
- Hiratsuka, T.; Fujita, Y.; Naoki, H.; Aoki, K.; Kamioka, Y.; Matsuda, M. Intercellular propagation of extracellular signal-regulated kinase activation revealed by in vivo imaging of mouse skin. eLife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.M.; Nyati, S.; Ross, B.D.; Rehemtulla, A. Molecular Imaging of the ATM Kinase Activity. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lee, K.C.; Bhojani, M.S.; Khan, A.P.; Shilman, A.; Holland, E.C.; Ross, B.D.; Rehemtulla, A. Molecular imaging of Akt kinase activity. Nat. Med. 2007, 13, 1114–1119. [Google Scholar] [CrossRef] [PubMed]
- Post, P.L.; DeBiasio, R.L.; Taylor, D.L. A fluorescent protein biosensor of myosin II regulatory light chain phosphorylation reports a gradient of phosphorylated myosin II in migrating cells. Mol. Biol. Cell 1995, 6, 1755–1768. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Van, T.N.N.; Pellerano, M.; Lykaso, S.; Morris, M.C. Fluorescent Protein Biosensor for Probing CDK/Cyclin Activity in vitro and in Living Cells. ChemBioChem 2014, 15, 2298–2305. [Google Scholar] [CrossRef] [PubMed]
- Damayanti, N.P.; Parker, L.L.; Irudayaraj, J.M.K. Fluorescence Lifetime Imaging of Biosensor Peptide Phosphorylation in Single Live Cells. Angew. Chem. Int. Ed. 2013, 52, 3931–3934. [Google Scholar] [CrossRef] [PubMed]
- Damayanti, N.P.; Buno, K.; Cui, Y.; Voytik-Harbin, S.L.; Pili, R.; Freeman, J.; Irudayaraj, J.M.K. Real-Time Multiplex Kinase Phosphorylation Sensors in Living Cells. ACS Sens. 2017, 2, 1225–1230. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, M.E.; Hällbrink, M.M.; Elmquist, A.M.; Langel, Ü. Passage of cell-penetrating peptides across a human epithelial cell layer in vitro. Biochem. J. 2004, 377, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Higashi, H.; Sato, K.; Ohtake, A.; Omori, A.; Yoshida, S.; Kudo, Y. Imaging of cAMP-dependent protein kinase activity in living neural cells using a novel fluorescent substrate. FEBS Lett. 1997, 414, 55–60. [Google Scholar] [CrossRef]
- Yeh, R.H.; Yan, X.; Cammer, M.; Bresnick, A.R.; Lawrence, D.S. Real Time Visualization of Protein Kinase Activity in Living Cells. J. Biol. Chem. 2002, 277, 11527–11532. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Dulyaninova, N.G.; Kumar, S.; Bresnick, A.R.; Lawrence, D.S. Visual Snapshots of Intracellular Kinase Activity at the Onset of Mitosis. Chem. Biol. 2007, 14, 1254–1260. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Dai, Z.; Cahill, S.M.; Blumenstein, M.; Lawrence, D.S. Light-Regulated Sampling of Protein Tyrosine Kinase Activity. J. Am. Chem. Soc. 2006, 128, 14016–14017. [Google Scholar] [CrossRef] [PubMed]
- Oien, N.P.; Nguyen, L.T.; Jernigan, F.E.; Priestman, M.A.; Lawrence, D.S. Long-Wavelength Fluorescent Reporters for Monitoring Protein Kinase Activity. Angew. Chem. Int. Ed. 2014, 53, 3975–3978. [Google Scholar] [CrossRef] [PubMed]
- Veldhuyzen, W.F.; Nguyen, Q.; McMaster, G.; Lawrence, D.S. A Light-Activated Probe of Intracellular Protein Kinase Activity. J. Am. Chem. Soc. 2003, 125, 13358–13359. [Google Scholar] [CrossRef] [PubMed]
- Aujard, I.; Benbrahim, C.; Gouget, M.; Ruel, O.; Baudin, J.B.; Neveu, P.; Jullien, L. o-nitrobenzyl photolabile protecting groups with red-shifted absorption: Syntheses and uncaging cross-sections for one- and two-photon excitation. Chemistry 2006, 12, 6865–6879. [Google Scholar] [CrossRef] [PubMed]
- Hammer, C.A.; Falahati, K.; Jakob, A.; Klimek, R.; Burghardt, I.; Heckel, A.; Wachtveitl, J. Sensitized Two-Photon Activation of Coumarin Photocages. J. Phys. Chem. Lett. 2018, 9, 1448–1453. [Google Scholar] [CrossRef] [PubMed]
- Regot, S.; Hughey, J.J.; Bajar, B.T.; Carrasco, S.; Covert, M.W. High-Sensitivity Measurements of Multiple Kinase Activities in Live Single Cells. Cell 2014, 157, 1724–1734. [Google Scholar] [CrossRef] [PubMed]
- Kudo, T.; Jeknić, S.; Macklin, D.N.; Akhter, S.; Hughey, J.J.; Regot, S.; Covert, M.W. Live-cell measurements of kinase activity in single cells using translocation reporters. Nat. Protoc. 2017, 13, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt Promotes Cell Survival by Phosphorylating and Inhibiting a Forkhead Transcription Factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Hung, Y.P. Single Cell Imaging of Metabolism with Fluorescent Biosensors; Harvard University: Cambridge, MA, USA, 2012. [Google Scholar]
- Gross, S.M.; Rotwein, P. Akt signaling dynamics in individual cells. J. Cell Sci. 2015, 128, 2509–2519. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.M.; Rotwein, P. Mapping growth-factor-modulated Akt signaling dynamics. J. Cell Sci. 2016, 129, 2052–2063. [Google Scholar] [CrossRef] [PubMed]
- Maryu, G.; Matsuda, M.; Aoki, K. Multiplexed Fluorescence Imaging of ERK and Akt Activities and Cell-cycle Progression. Cell Struct. Funct. 2016, 41, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, K.A.; Chen, Y.-T.; Haskins, J.R. Fluorescent protein biosensors. Mod. Drug Discov. 2003, 6, 33–37. [Google Scholar]
- Hahn, A.T.; Jones, J.T.; Meyer, T. Quantitative analysis of cell cycle phase durations and PC12 differentiation using fluorescent biosensors. Cell Cycle 2009, 8, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Spencer, S.L.; Cappell, S.D.; Tsai, F.-C.; Overton, K.W.; Wang, C.L.; Meyer, T. The Proliferation-Quiescence Decision Is Controlled by a Bifurcation in CDK2 Activity at Mitotic Exit. Cell 2013, 155, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Sample, V.; Ni, Q.; Mehta, S.; Inoue, T.; Zhang, J. Controlling Enzymatic Action in Living Cells with a Kinase-Inducible Bimolecular Switch. ACS Chem. Biol. 2013, 8, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Milanole, G.; Gao, B.; Paoletti, A.; Pieters, G.; Dugave, C.; Deutsch, E.; Rivera, S.; Law, F.; Perfettini, J.-L.; Mari, E.; et al. Bimodal fluorescence/ 129 Xe NMR probe for molecular imaging and biological inhibition of EGFR in Non-Small Cell Lung Cancer. Bioorg. Med. Chem. 2017, 25, 6653–6660. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Yin, J.; Haiyang, H.; Peng, X.; Gao, Q.; Duan, C. Conformationally Induced Off−On Cell Membrane Chemosensor Targeting Receptor Protein-Tyrosine Kinases for in Vivo and in Vitro Fluorescence Imaging of Cancers. J. Am. Chem. Soc. 2018. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. 2016, 103, 26–48. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Ivan, T.; Enkvist, E.; Sinijarv, H.; Uri, A. Competitive ligands facilitate dissociation of the complex of bifunctional inhibitor and protein kinase. Biophys. Chem. 2017, 228, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Kestav, K.; Viht, K.; Konovalov, A.; Enkvist, E.; Uri, A.; Lavogina, D. Slowly on, Slowly off: Bisubstrate-Analogue Conjugates of 5-Iodotubercidin and Histone H3 Peptide Targeting Protein Kinase Haspin. ChemBioChem 2017, 18, 790–798. [Google Scholar] [CrossRef] [PubMed]
- Vaasa, A.; Lust, M.; Terrin, A.; Uri, A.; Zaccolo, M. Small-molecule FRET probes for protein kinase activity monitoring in living cells. Biochem. Biophys. Res. Commun. 2010, 397, 750–755. [Google Scholar] [CrossRef] [PubMed]
- Kamkaew, A.; Burgess, K. Double-Targeting Using a TrkC Ligand Conjugated to Dipyrrometheneboron Difluoride (BODIPY) Based Photodynamic Therapy (PDT) Agent. J. Med. Chem. 2013, 56, 7608–7614. [Google Scholar] [CrossRef] [PubMed]
- Ko, E.; Kamkaew, A.; Burgess, K. Small Molecule Ligands for Active Targeting of TrkC-Expressing Tumor Cells. ACS Med. Chem. Lett. 2012, 3, 1008–1012. [Google Scholar] [CrossRef] [PubMed]
- Kurzawa, L.; Pellerano, M.; Coppolani, J.B.; Morris, M.C. Fluorescent Peptide Biosensor for Probing the Relative Abundance of Cyclin-Dependent Kinases in Living Cells. PLoS ONE 2011, 6, e26555. [Google Scholar] [CrossRef] [PubMed]
- Viht, K.; Saaver, S.; Vahter, J.; Enkvist, E.; Lavogina, D.; Sinijärv, H.; Raidaru, G.; Guerra, B.; Issinger, O.-G.; Uri, A. Acetoxymethyl Ester of Tetrabromobenzimidazole–Peptoid Conjugate for Inhibition of Protein Kinase CK2 in Living Cells. Bioconjug. Chem. 2015, 26, 2324–2335. [Google Scholar] [CrossRef] [PubMed]
- Vetter, M.L.; Zhang, Z.; Liu, S.; Wang, J.; Cho, H.; Zhang, J.; Zhang, W.; Gray, N.S.; Yang, P.L. Fluorescent Visualization of Src by Using Dasatinib-BODIPY. ChemBioChem 2014, 15, 1317–1324. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Budin, G.; Yang, K.S.; Reiner, T.; Weissleder, R. Bioorthogonal Probes for Polo-like Kinase 1 Imaging and Quantification. Angew. Chem. Int. Ed. 2011, 50, 9378–9381. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.S.; Budin, G.; Reiner, T.; Vinegoni, C.; Weissleder, R. Bioorthogonal Imaging of Aurora Kinase A in Live Cells. Angew. Chem. Int. Ed. 2012, 51, 6598–6603. [Google Scholar] [CrossRef] [PubMed]
- Mikula, H.; Stapleton, S.; Kohler, R.H.; Vinegoni, C.; Weissleder, R. Design and Development of Fluorescent Vemurafenib Analogs for In Vivo Imaging. Theranostics 2017, 7, 1257–1265. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Kwiatkowski, N.; Zeng, H.; Lim, S.M.; Gray, N.S.; Zhang, W.; Yang, P.L. Leveraging kinase inhibitors to develop small molecule tools for imaging kinases by fluorescence microscopy. Mol. BioSyst. 2012, 8, 2523. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Hao, P.; Li, L.; Tan, C.Y.J.; Cheng, X.; Chen, G.Y.J.; Sze, S.K.; Shen, H.-M.; Yao, S.Q. Design and Synthesis of Minimalist Terminal Alkyne-Containing Diazirine Photo-Crosslinkers and Their Incorporation into Kinase Inhibitors for Cell- and Tissue-Based Proteome Profiling. Angew. Chem. Int. Ed. 2013, 52, 8551–8556. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Cheng, X.; Sze, S.K.; Yao, S.Q. Proteome profiling reveals potential cellular targets of staurosporine using a clickable cell-permeable probe. Chem. Commun. 2011, 47, 11306. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Qian, L.; Li, L.; Bernhammer, J.C.; Huynh, H.V.; Lee, J.-S.; Yao, S.Q. Tetrazole Photoclick Chemistry: Reinvestigating Its Suitability as a Bioorthogonal Reaction and Potential Applications. Angew. Chem. Int. Ed. 2016, 55, 2002–2006. [Google Scholar] [CrossRef] [PubMed]
- Hwang, G.; Kim, H.; Yoon, H.; Song, C.; Lim, D.-K.; Sim, T.; Lee, J. In situ imaging of quantum dot-AZD4547 conjugates for tracking the dynamic behavior of fibroblast growth factor receptor 3. Int. J. Nanomed. 2017, 12, 5345–5357. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liu, H.; Pan, Z. A general approach for the development of fluorogenic probes suitable for no-wash imaging of kinases in live cells. Chem. Commun. 2014, 50, 15319–15322. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Liu, W.; Brown, A.S.; Landgraf, R.; Wilson, J.N. Fluorescent Kinase Probes Enabling Identification and Dynamic Imaging of HER2(+) Cells. Anal. Chem. 2016, 88, 11310–11313. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Landgraf, R.; Wilson, J.N. Synthesis and photophysical properties of a fluorescent cyanoquinoline probe for profiling ERBB2 kinase inhibitor response. Bioorg. Med. Chem. 2017, 25, 6016–6023. [Google Scholar] [CrossRef] [PubMed]
- Zambaldo, C.; Sadhu, K.K.; Karthikeyan, G.; Barluenga, S.; Daguer, J.-P.; Winssinger, N. Selective affinity-based probe for oncogenic kinases suitable for live cell imaging. Chem. Sci. 2013, 4, 2088. [Google Scholar] [CrossRef]
- Turetsky, A.; Kim, E.; Kohler, R.H.; Miller, M.A.; Weissleder, R. Single cell imaging of Bruton’s Tyrosine Kinase using an irreversible inhibitor. Sci. Rep. 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Zhang, C.-J.; Chen, G.Y.J.; Yao, S.Q. Cell-Based Proteome Profiling of Potential Dasatinib Targets by Use of Affinity-Based Probes. J. Am. Chem. Soc. 2012, 134, 3001–3014. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Pan, S.; Li, Z.; Li, L.; Wu, X.; Hao, P.; Sze, S.K.; Yao, S.Q. Multiplex Imaging and Cellular Target Identification of Kinase Inhibitors via an Affinity-Based Proteome Profiling Approach. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Barcomb, K.; Goodell, D.J.; Arnold, D.B.; Bayer, K.U. Live imaging of endogenous Ca2+/calmodulin-dependent protein kinase II in neurons reveals that ischemia-related aggregation does not require kinase activity. J. Neurochem. 2015, 135, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Kantelhardt, S.R.; Caarls, W.; de Vries, A.H.B.; Hagen, G.M.; Jovin, T.M.; Schulz-Schaeffer, W.; Rohde, V.; Giese, A.; Arndt-Jovin, D.J. Specific Visualization of Glioma Cells in Living Low-Grade Tumor Tissue. PLoS ONE 2010, 5, e11323. [Google Scholar] [CrossRef] [PubMed]
- Lyakhov, I.; Zielinski, R.; Kuban, M.; Kramer-Marek, G.; Fisher, R.; Chertov, O.; Bindu, L.; Capala, J. HER2- and EGFR-Specific Affiprobes: Novel Recombinant Optical Probes for Cell Imaging. ChemBioChem 2010, 11, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Telmer, C.A.; Schmidt, B.F.; Franke, J.D.; Ort, S.; Arndt-Jovin, D.J.; Bruchez, M.P. Fluorogen Activating Protein–Affibody Probes: Modular, No-Wash Measurement of Epidermal Growth Factor Receptors. Bioconjug. Chem. 2015, 26, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Gulyani, A.; Vitriol, E.; Allen, R.; Wu, J.; Gremyachinskiy, D.; Lewis, S.; Dewar, B.; Graves, L.M.; Kay, B.K.; Kuhlman, B.; et al. A biosensor generated via high-throughput screening quantifies cell edge Src dynamics. Nat. Chem. Biol. 2011, 7, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Malkov, D.; Fetter, J.; Zenser, N.; Song, K. A Novel Biosensor Assay for Detecting Activation of Endogenous EGFR in Living Cells; Sigma® Life Science: St. Louis, MO, USA, 2011. [Google Scholar]
- Hayashi-Takanaka, Y.; Yamagata, K.; Nozaki, N.; Kimura, H. Visualizing histone modifications in living cells: Spatiotemporal dynamics of H3 phosphorylation during interphase. J. Cell Biol. 2009, 187, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Giannone, G.; Hosy, E.; Sibarita, J.-B.; Choquet, D.; Cognet, L. High-Content Super-Resolution Imaging of Live Cell by uPAINT. In Nanoimaging; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2013; Volume 950, pp. 95–110. ISBN 978-1-62703-137-0. [Google Scholar]
- Ng, T.; Squire, A.; Hansra, G.; Bornancin, F.; Prevostel, C.; Hanby, A.; Harris, W.; Barnes, D.; Schmidt, S.; Mellor, H.; et al. Imaging Protein Kinase Calpha Activation in Cells. Science 1999, 283, 2085–2089. [Google Scholar] [CrossRef] [PubMed]
- Chew, T.-L.; Wolf, W.A.; Gallagher, P.J.; Matsumura, F.; Chisholm, R.L. A fluorescent resonant energy transfer–based biosensor reveals transient and regional myosin light chain kinase activation in lamella and cleavage furrows. J. Cell Biol. 2002, 156, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Takao, K. Visualization of Synaptic Ca2+/Calmodulin-Dependent Protein Kinase II Activity in Living Neurons. J. Neurosci. 2005, 25, 3107–3112. [Google Scholar] [CrossRef] [PubMed]
- Bertolin, G.; Sizaire, F.; Herbomel, G.; Reboutier, D.; Prigent, C.; Tramier, M. A FRET biosensor reveals spatiotemporal activation and functions of aurora kinase A in living cells. Nat. Commun. 2016, 7, 12674. [Google Scholar] [CrossRef] [PubMed]
- Broussard, J.A.; Rappaz, B.; Webb, D.J.; Brown, C.M. Fluorescence resonance energy transfer microscopy as demonstrated by measuring the activation of the serine/threonine kinase Akt. Nat. Protoc. 2013, 8, 265–281. [Google Scholar] [CrossRef] [PubMed]
- Offterdinger, M.; Georget, V.; Girod, A.; Bastiaens, P.I.H. Imaging Phosphorylation Dynamics of the Epidermal Growth Factor Receptor. J. Biol. Chem. 2004, 279, 36972–36981. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.C.E.; Maebashi, H.K.; Nakahata, Y.; Nabekura, J.; Murakoshi, H. Development of a Molecularly Evolved, Highly Sensitive CaMKII FRET Sensor with Improved Expression Pattern. PLoS ONE 2015, 10, e0121109. [Google Scholar] [CrossRef] [PubMed]
- Bayliss, R.; Sardon, T.; Vernos, I.; Conti, E. Structural Basis of Aurora-A Activation by TPX2 at the Mitotic Spindle. Mol. Cell 2003, 12, 851–862. [Google Scholar] [CrossRef]
- Ohashi, S.; Sakashita, G.; Ban, R.; Nagasawa, M.; Matsuzaki, H.; Murata, Y.; Taniguchi, H.; Shima, H.; Furukawa, K.; Urano, T. Phospho-regulation of human protein kinase Aurora-A: Analysis using anti-phospho-Thr288 monoclonal antibodies. Oncogene 2006, 25, 7691–7702. [Google Scholar] [CrossRef] [PubMed]
- Paster, W.; Paar, C.; Eckerstorfer, P.; Jakober, A.; Drbal, K.; Schutz, G.J.; Sonnleitner, A.; Stockinger, H. Genetically Encoded Forster Resonance Energy Transfer Sensors for the Conformation of the Src Family Kinase Lck. J. Immunol. 2009, 182, 2160–2167. [Google Scholar] [CrossRef] [PubMed]
- Tewson, P.; Westenberg, M.; Zhao, Y.; Campbell, R.E.; Quinn, A.M.; Hughes, T.E. Simultaneous Detection of Ca2+ and Diacylglycerol Signaling in Living Cells. PLoS ONE 2012, 7, e42791. [Google Scholar] [CrossRef] [PubMed]
- Ritt, M.; Guan, J.L.; Sivaramakrishnan, S. Visualizing and Manipulating Focal Adhesion Kinase Regulation in Live Cells. J. Biol. Chem. 2013, 288, 8875–8886. [Google Scholar] [CrossRef] [PubMed]
- Goni, G.M.; Epifano, C.; Boskovic, J.; Camacho-Artacho, M.; Zhou, J.; Bronowska, A.; Martin, M.T.; Eck, M.J.; Kremer, L.; Grater, F.; et al. Phosphatidylinositol 4,5-bisphosphate triggers activation of focal adhesion kinase by inducing clustering and conformational changes. Proc. Natl. Acad. Sci. USA 2014, 111, E3177–E3186. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.R.; Harootunian, A.T.; Buechler, Y.J.; Taylor, S.S.; Tsien, R.Y. Fluorescence ratio imaging of cyclic AMP in single cells. Nature 1991, 349, 694–697. [Google Scholar] [CrossRef] [PubMed]
- Goaillard, J.-M.; Vincent, P.; Fischmeister, R. Simultaneous measurements of intracellular cAMP and l-type Ca2+ current in single frog ventricular myocytes. J. Physiol. 2001, 530, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Schleicher, K.; Zaccolo, M. Using cAMP Sensors to Study Cardiac Nanodomains. J. Cardiovasc. Dev. Dis. 2018, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Honda, A.; Adams, S.R.; Sawyer, C.L.; Lev-Ram, V.; Tsien, R.Y.; Dostmann, W.R.G. Spatiotemporal dynamics of guanosine 3’,5’-cyclic monophosphate revealed by a genetically encoded, fluorescent indicator. Proc. Natl. Acad. Sci. USA 2001, 98, 2437–2442. [Google Scholar] [CrossRef] [PubMed]
- Hodneland Nilsson, L.I.; Nitschke Pettersen, I.K.; Nikolaisen, J.; Micklem, D.; Avsnes Dale, H.; Vatne Røsland, G.; Lorens, J.; Tronstad, K.J. A new live-cell reporter strategy to simultaneously monitor mitochondrial biogenesis and morphology. Sci. Rep. 2015, 5, 17217. [Google Scholar] [CrossRef] [PubMed]
- Balboula, A.Z.; Nguyen, A.L.; Gentilello, A.S.; Quartuccio, S.M.; Drutovic, D.; Solc, P.; Schindler, K. Haspin kinase regulates microtubule-organizing center clustering and stability through Aurora kinase C in mouse oocytes. J. Cell Sci. 2016, 129, 3648–3660. [Google Scholar] [CrossRef] [PubMed]
- Verma, D.; Bajpai, V.K.; Ye, N.; Maneshi, M.M.; Jetta, D.; Andreadis, S.T.; Sachs, F.; Hua, S.Z. Flow induced adherens junction remodeling driven by cytoskeletal forces. Exp. Cell Res. 2017, 359, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Nahar, K.; Goto, T.; Kaida, A.; Deguchi, S.; Miura, M. Effects of Chk1 inhibition on the temporal duration of radiation-induced G2 arrest in HeLa cells. J. Radiat. Res. 2014, 55, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Rosado, A.; Zanella, F.; Garcia, B.; Carnero, A.; Link, W. A Dual-Color Fluorescence-Based Platform to Identify Selective Inhibitors of Akt Signaling. PLoS ONE 2008, 3, e1823. [Google Scholar] [CrossRef] [PubMed]
- Shcherbo, D.; Souslova, E.A.; Goedhart, J.; Chepurnykh, T.V.; Gaintzeva, A.; Shemiakina, I.I.; Gadella, T.W.; Lukyanov, S.; Chudakov, D.M. Practical and reliable FRET/FLIM pair of fluorescent proteins. BMC Biotechnol. 2009, 9, 24. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
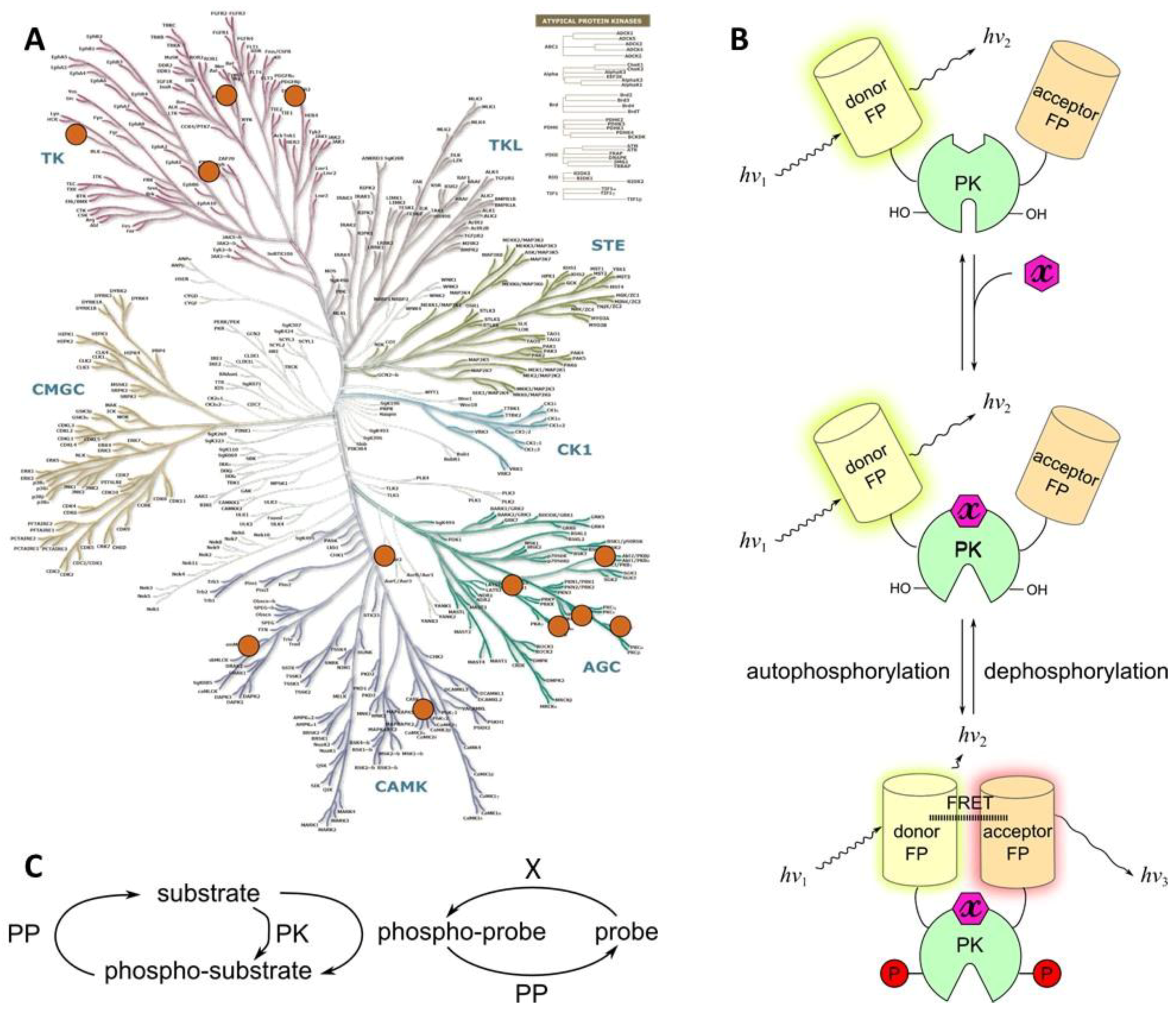{kind=link}
| Biochemical Assays | In cellulo Assays |
|---|---|
| Phosphorylation assay in the presence/absence of dilution series of target PK | Utilization of probe in a cell line with low endogenous expression of target PK, in the presence/absence of the overexpressed target PK |
| Phosphorylation assay in the presence of target PK and in the presence/absence of a dephosphorylating phosphatase 1 | Utilization of probe in conditions where the target PK knocked down/out |
| For probes utilizing FRET phenomenon, phosphorylation assay in the presence of target PK and in the presence/absence of a protease that does not degrade fluorescent proteins but cuts the probe (to establish minimal level of FRET) | Utilization of probe in conditions where the kinase-dead mutant of the target PK is overexpressed |
| Application of selective activators and inhibitors of target PK, and corresponding compounds that should affect closely related PKs yet not the target PK | |
| Use of a point-mutated probe variant as a negative control where the non-phosphorylatable amino acid is substituted for the Ser, Thr or Tyr contained in the consensus sequence. | |
| For probes that contain PAAD, introduction of point-mutations to the crucial positions of PAAD domain should also be considered to provide another set of negative controls that can be phosphorylated by target PK, yet recognition of the phosphorylated sequence by PAAD is disrupted. | |
| Use of a truncated probe variant as a negative control where one of the fluorescent proteins is omitted | |
| For probes utilizing FRET phenomenon, acceptor photobleaching experiment to monitor presence/absence of increase of fluorescence intensity of donor fluorophore dependent on the assay conditions used | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lavogina, D.; Kopanchuk, S.; Viht, K. Dissection of Protein Kinase Pathways in Live Cells Using Photoluminescent Probes: Surveillance or Interrogation? Chemosensors 2018, 6, 19. https://doi.org/10.3390/chemosensors6020019
Lavogina D, Kopanchuk S, Viht K. Dissection of Protein Kinase Pathways in Live Cells Using Photoluminescent Probes: Surveillance or Interrogation? Chemosensors. 2018; 6(2):19. https://doi.org/10.3390/chemosensors6020019
Chicago/Turabian StyleLavogina, Darja, Sergei Kopanchuk, and Kaido Viht. 2018. "Dissection of Protein Kinase Pathways in Live Cells Using Photoluminescent Probes: Surveillance or Interrogation?" Chemosensors 6, no. 2: 19. https://doi.org/10.3390/chemosensors6020019
APA StyleLavogina, D., Kopanchuk, S., & Viht, K. (2018). Dissection of Protein Kinase Pathways in Live Cells Using Photoluminescent Probes: Surveillance or Interrogation? Chemosensors, 6(2), 19. https://doi.org/10.3390/chemosensors6020019