Deciphering microRNAs and Their Associated Hairpin Precursors in a Non-Model Plant, Abelmoschus esculentus


Abstract
:1. Introduction
2. Results
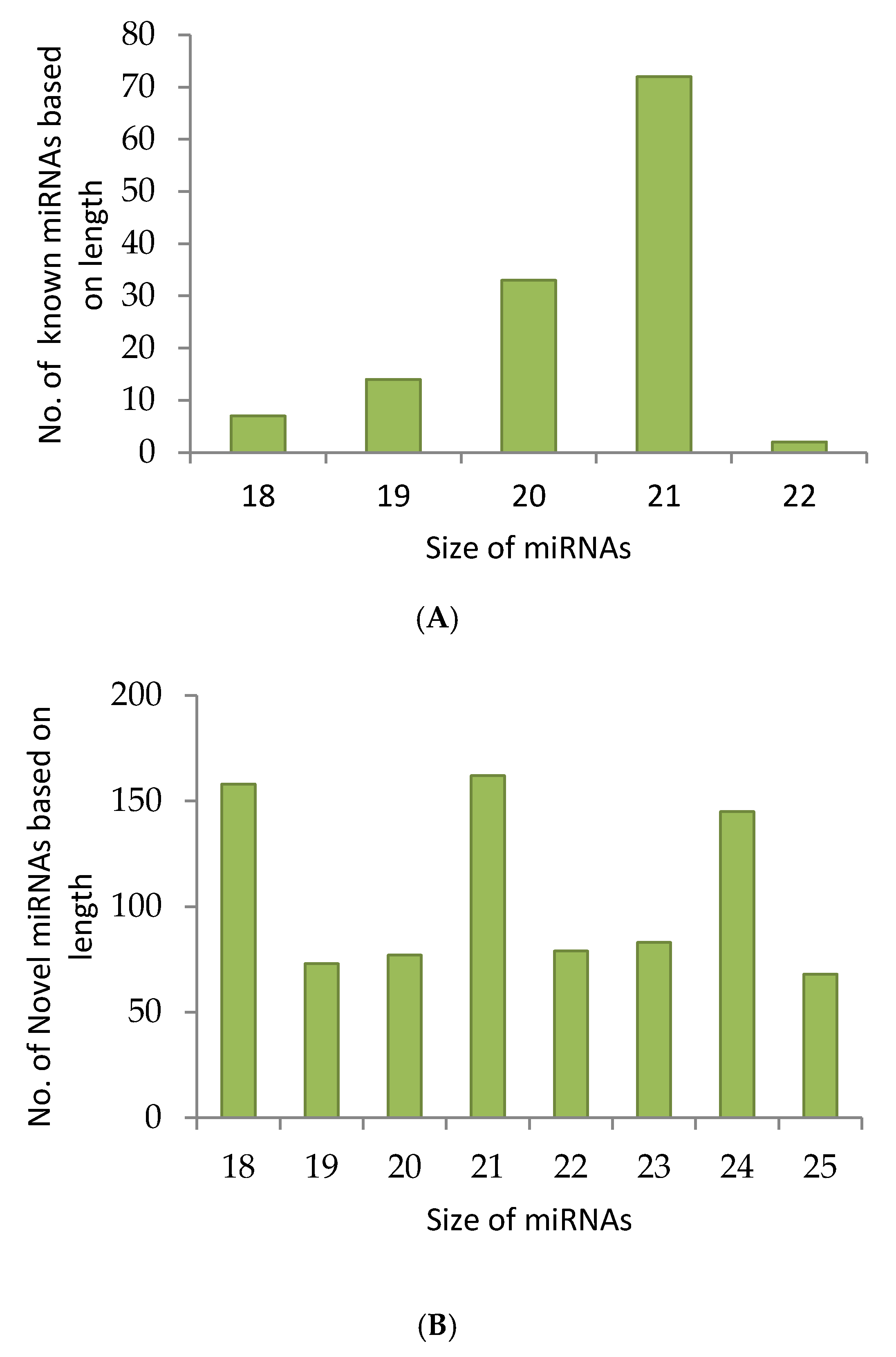
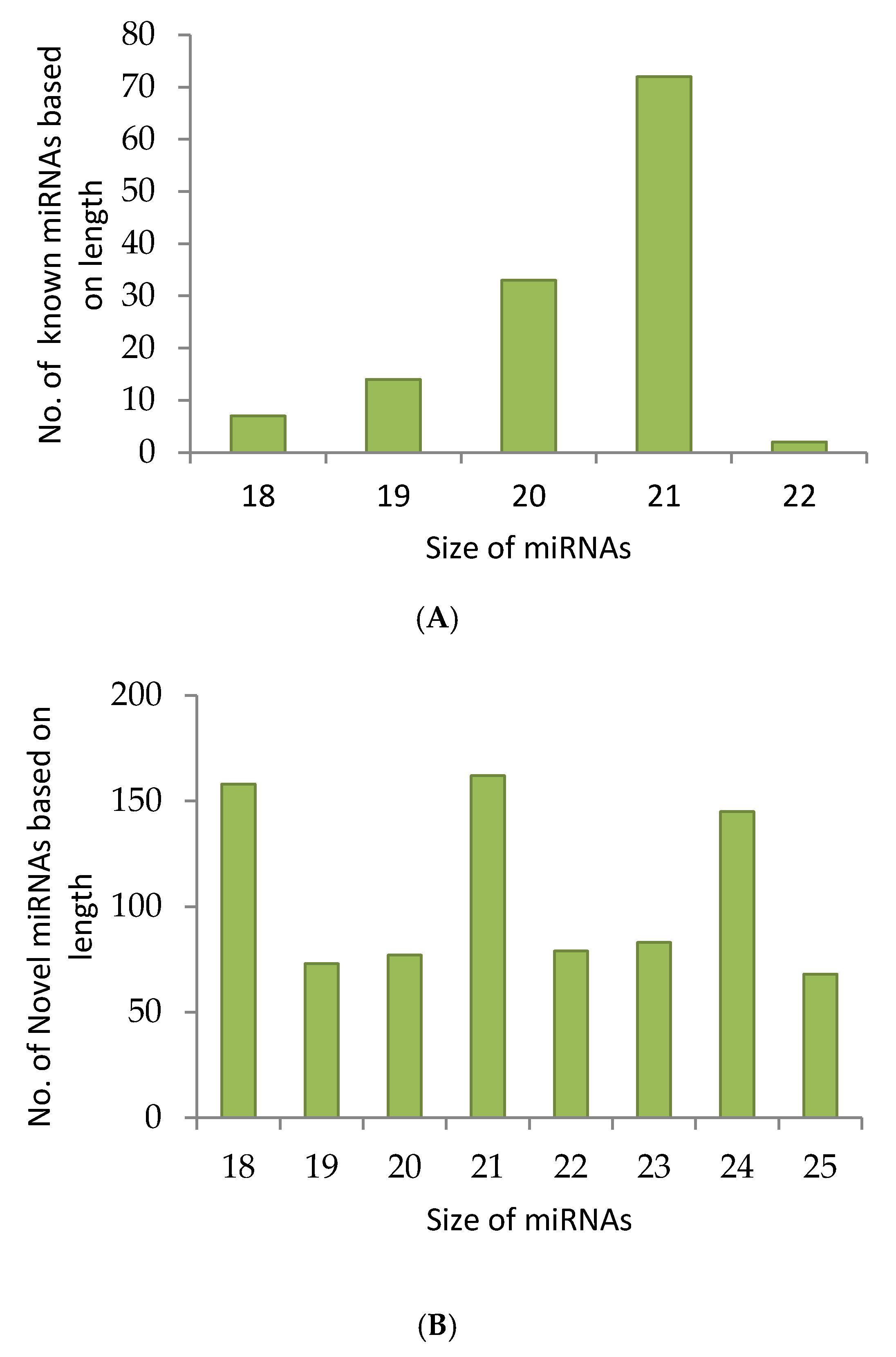
2.1. sRNA Sequencing Reveals Known as Well as Novel miRNA Candidates
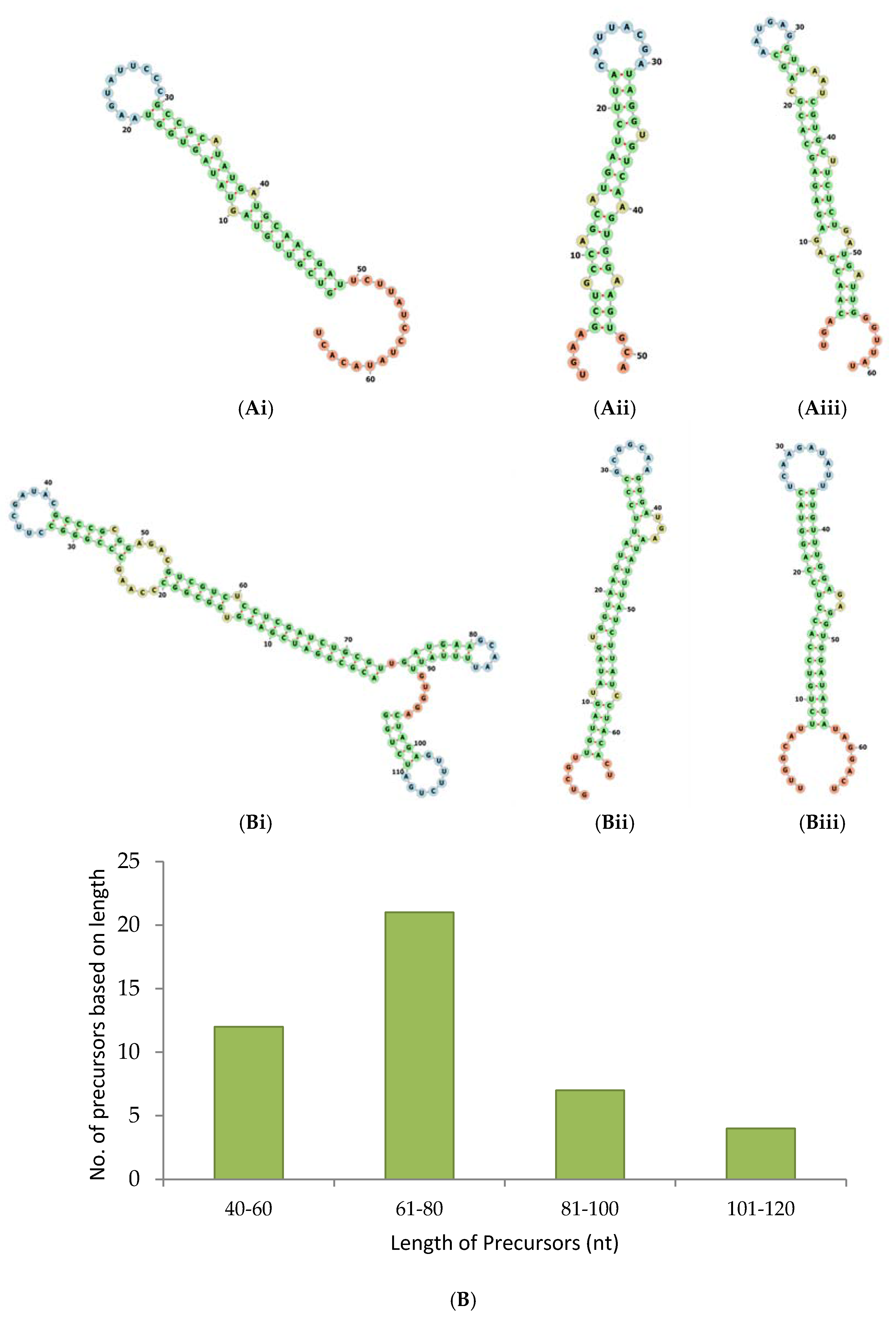
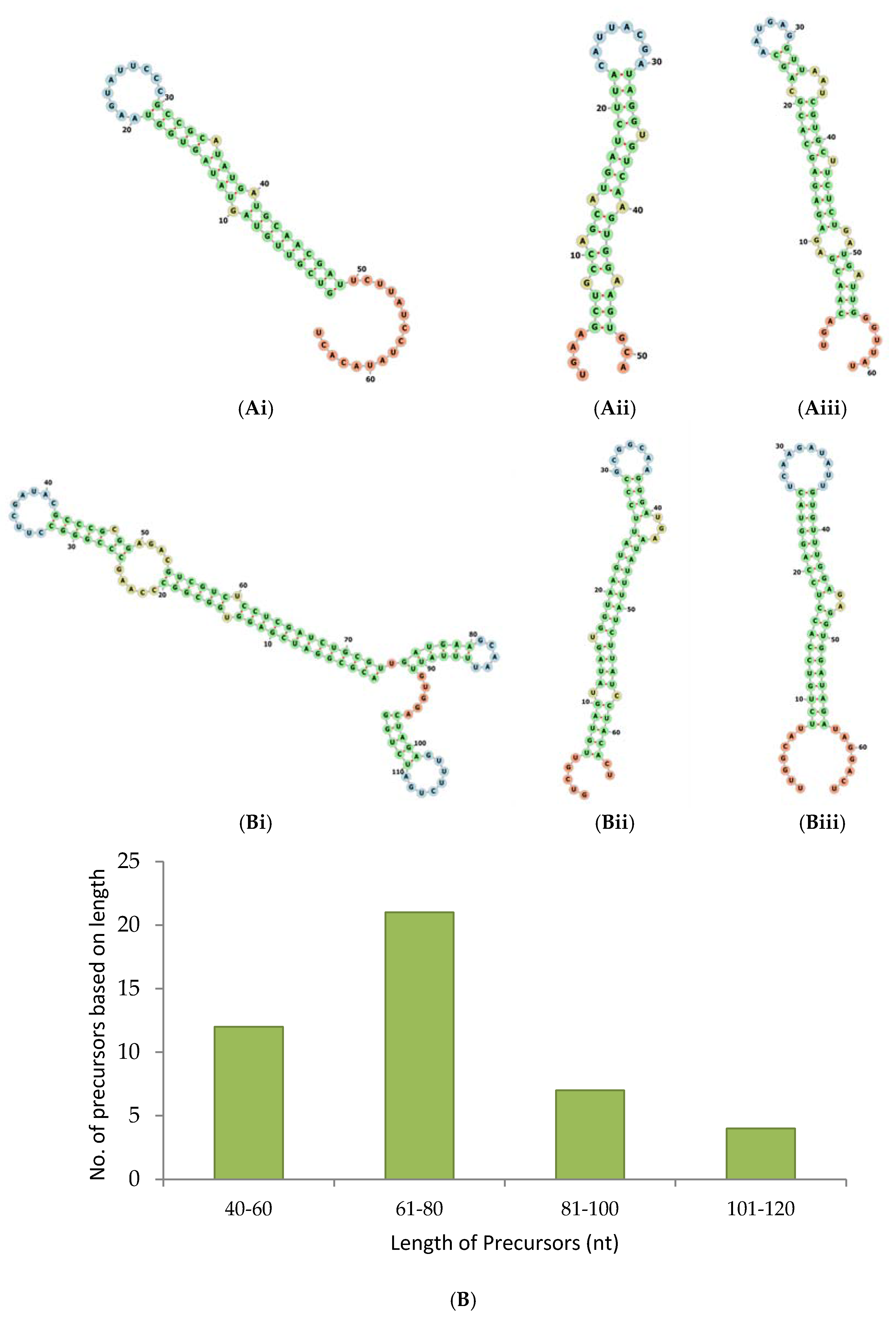
2.2. Pre-miRNAs Profiling and Determination of Pre-miRNAs for Known and Novel miRNAs
- (I)
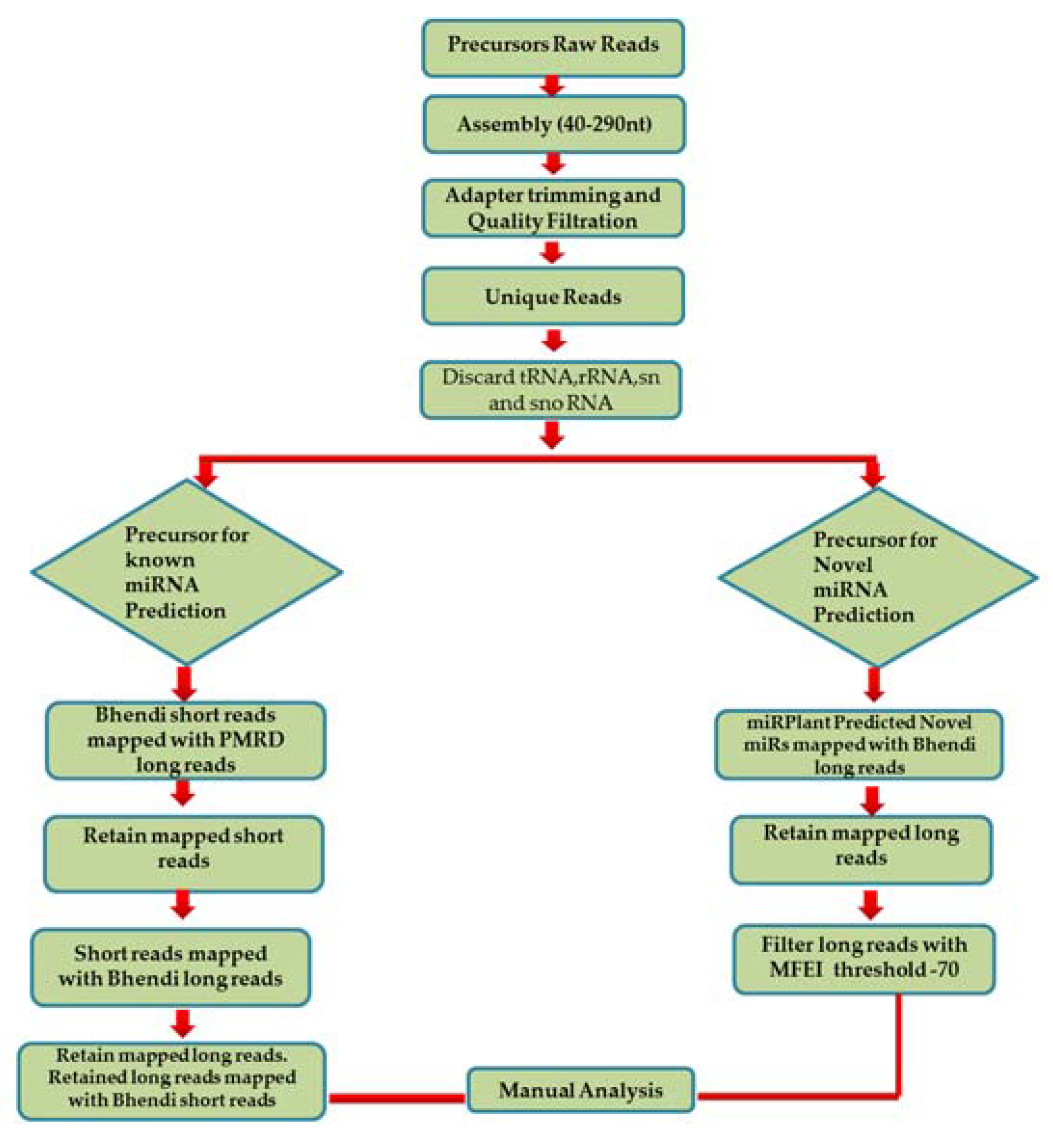
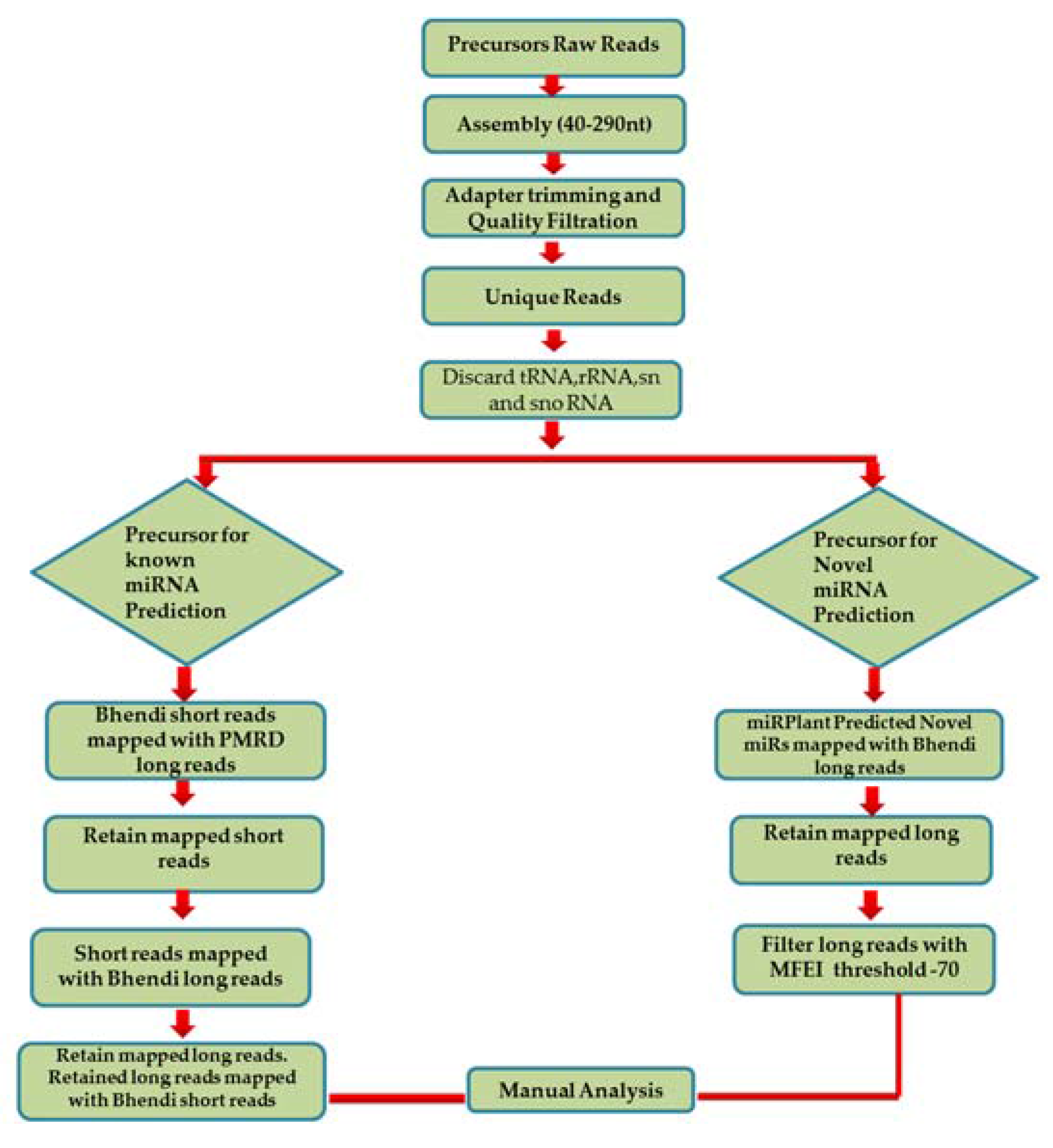
- In order to predict the precursors for conserved miRNAs, initially, 15,041 known pre-miRNA sequences retrieved from PMRD were considered as long reads. The short reads of A. esculentus were mapped with the long reads from PMRD to analyse the overlapping position by not allowing any mismatches; it was speculated that only conserved miRNA: miRNA* would be aligned with the PMRD long reads. A total of 5069 short reads showing alignment were extracted and mapped to A. esculentus pre-miRNA data to determine the pre-miRNA reads precisely. A total of 549 mapped long reads were retrieved and remapped with short reads for complete cluster formation. The predicted precursors were subjected to manual analysis to filter out the incomplete precursors and precursors whose mature miRNA falls in the loop region. Manual analysis retained only 25 complete precursors with perfect stem-loop structure, with the mature miRNA held in their stem region. The mapping summary for the three small RNA datasets is shown in the Table 1 and the mapping pattern of miRNA 156 with its precursor is shown in Figure 2B.
- (II)
- To predict the novel pre-miRNA candidates, the pre-miRNA reads mapped with the 845 novel miRNAs, predicted by miRPlant—were retrieved and aligned with the A. esculentus small RNA dataset to see the mature miRNA and its complimentary sequence clusters. The final results were subjected to manual analysis to reveal the mapping pattern of the novel miRNAs as depicted in Figure 2C.
- The long reads which showed unambiguous secondary structure are considered as possible pre-miRNA candidates.
- The minimum length of the long reads chosen was 40 nt.
- We calculated the minimal fold energy (MFE) for the reads using RNA Fold and Randfold. The minimum fold energy index (MFEI) was calculated [22] as follows.
- The base pairing between the mature miRNA and its complimentary sequence includes no more than four mismatches.
- The asymmetric bulges in between the duplex region are less frequent and should contain less than four bases. However, the total number of mismatches in the duplex region is no more than four.
- The precursors which carry the mature miRNA in its loop region are discarded. Also, the precursors with bigger loops in between the duplex region are also discarded.
- For novel precursors, we preceded with sequences holding the MFEI at −70 as their minimum threshold to discard other RNA species contamination such as rRNAs and tRNAs. Moreover, we manually checked the individual precursors by similarity searching in National Center for Biotechnology Information (NCBI) [23] to ensure that the sequences are devoid of these contaminating RNAs.
- In the case of novel precursors, some novel miRNAs are predicted based on their expression as well as their mapping pattern with the precursor.
2.3. Precursors of Conserved miRNAs and Novel miRNAs
2.4. Precursors of miRNAs from PMRD Not in miRBase
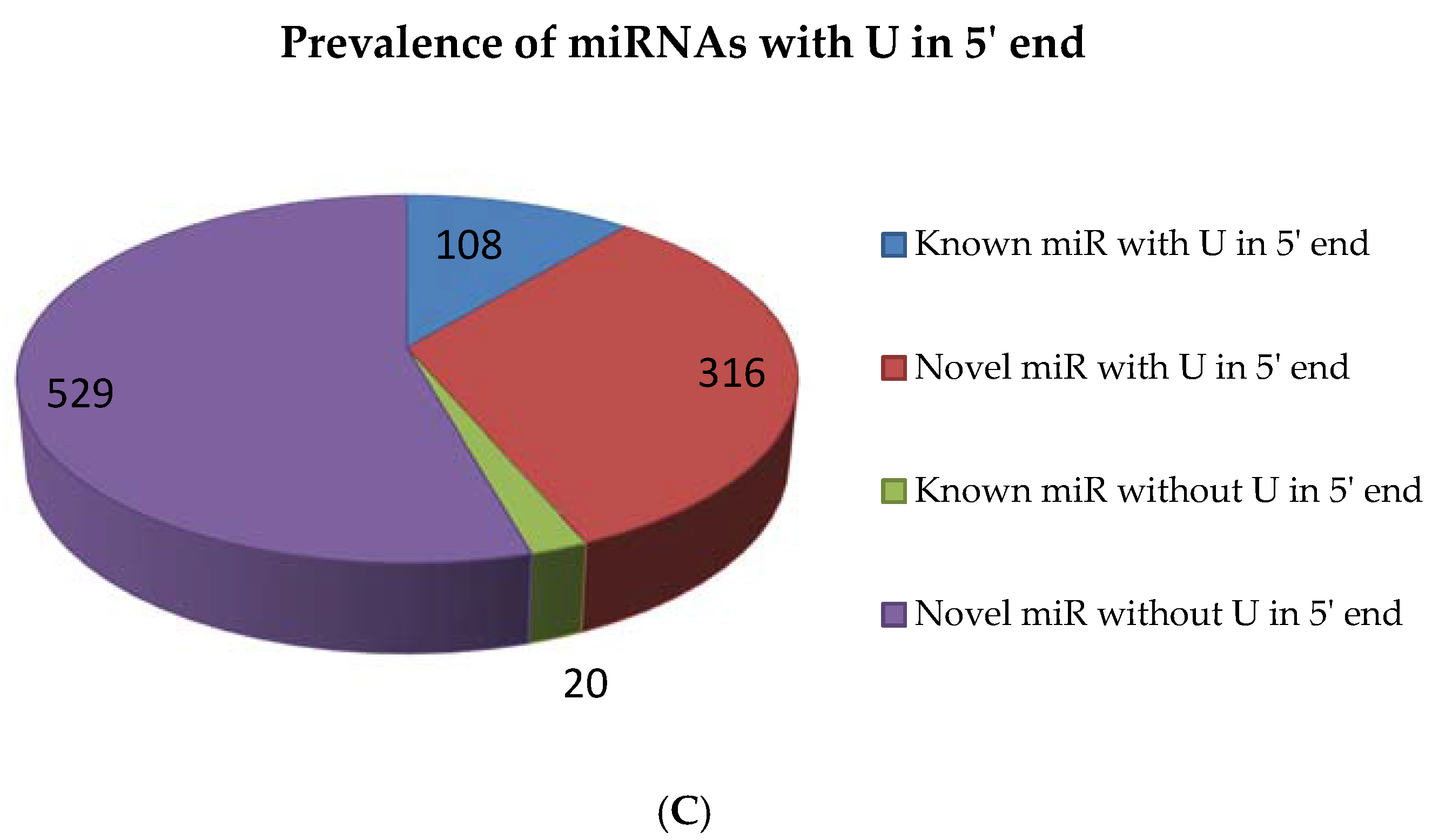
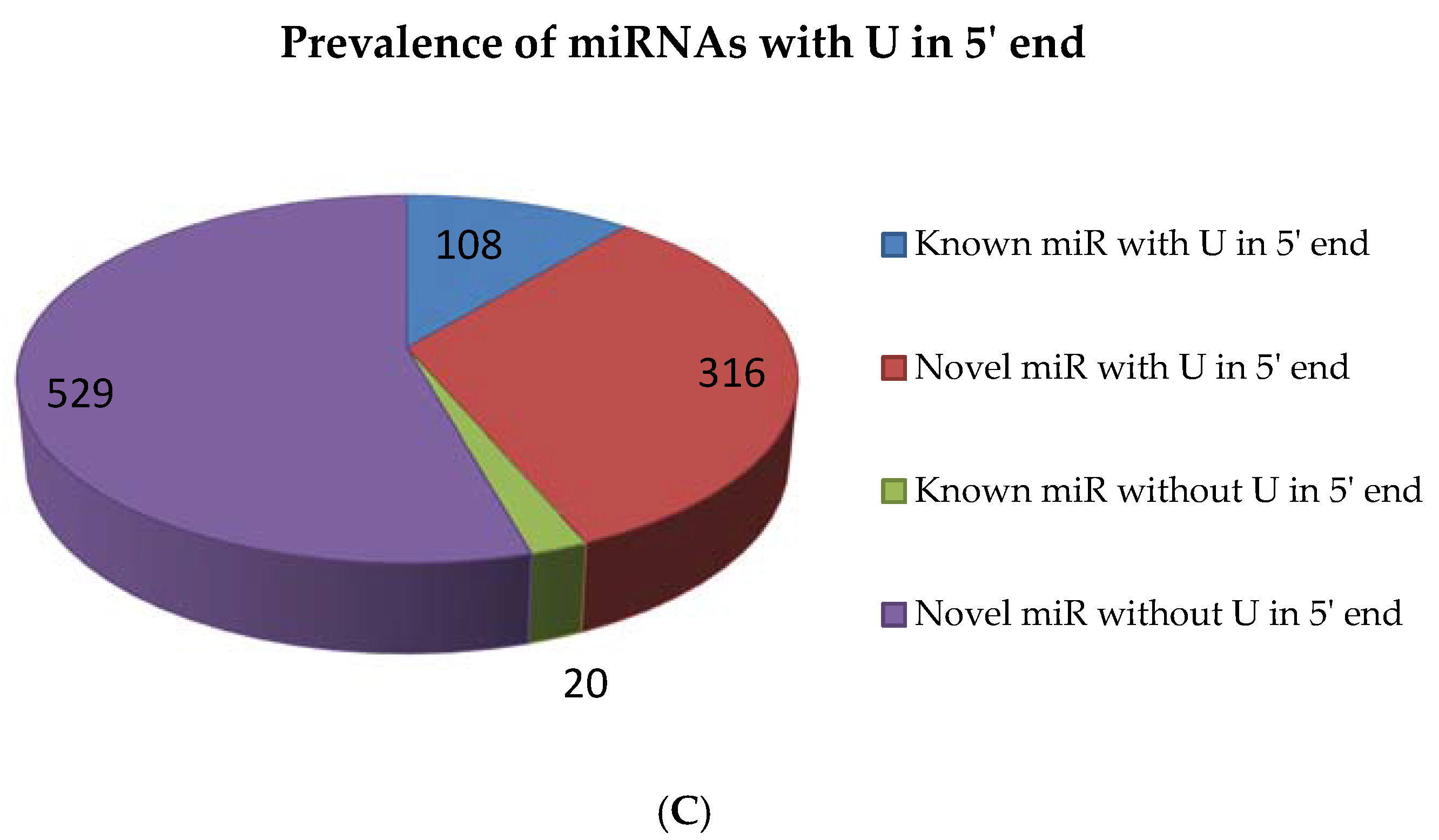
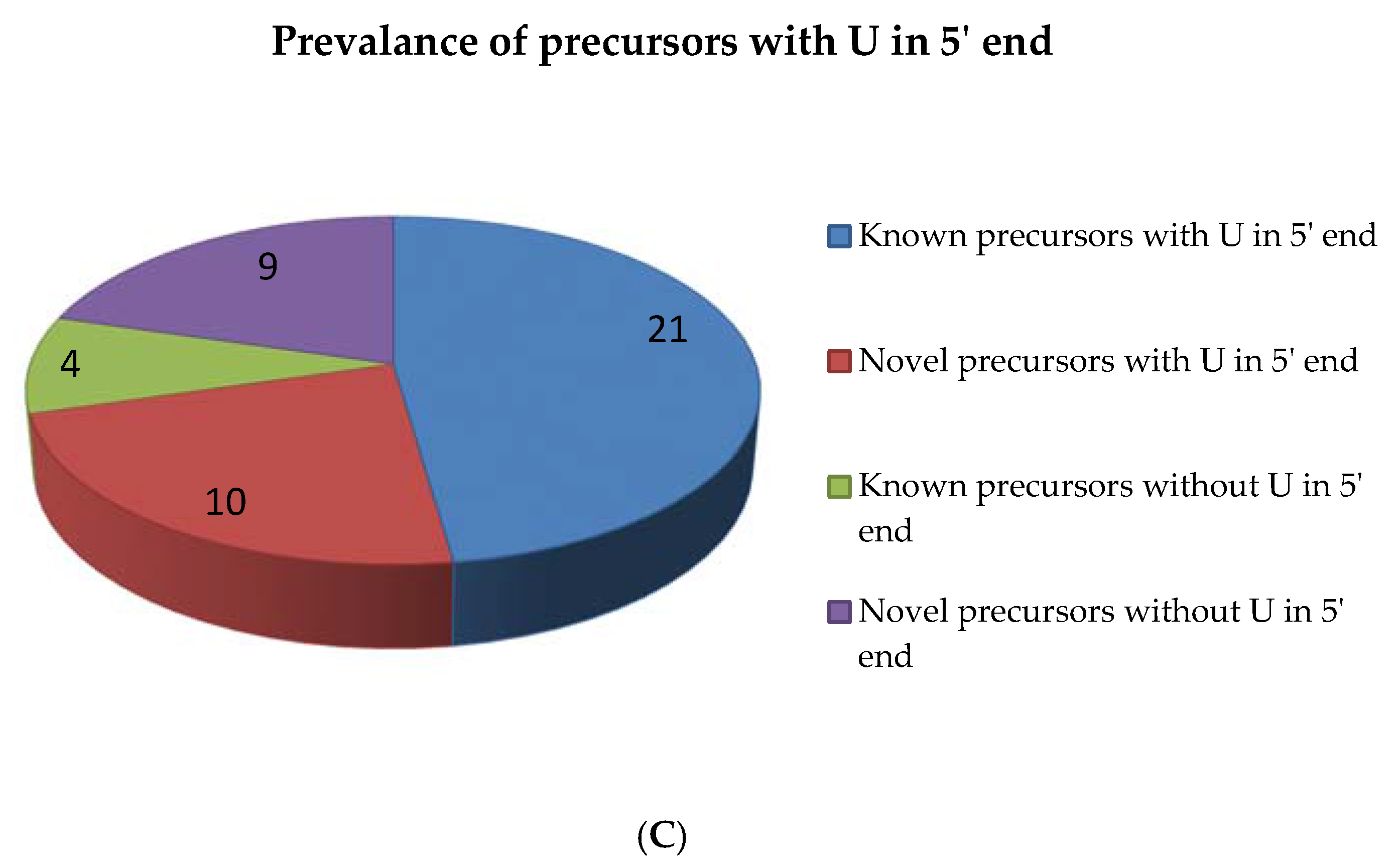
2.5. miRNAs from 5′ and 3′ Positions of Precursors
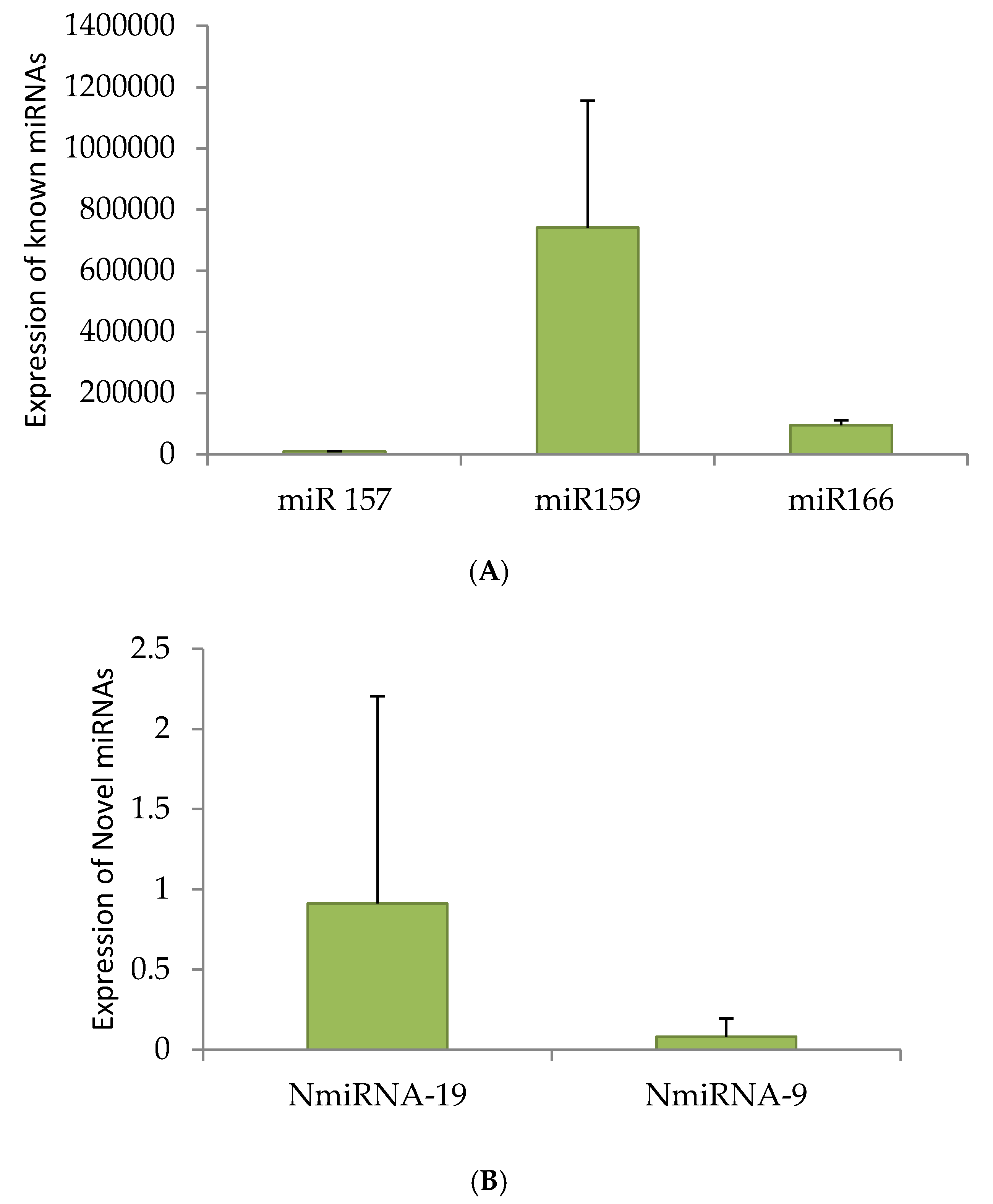
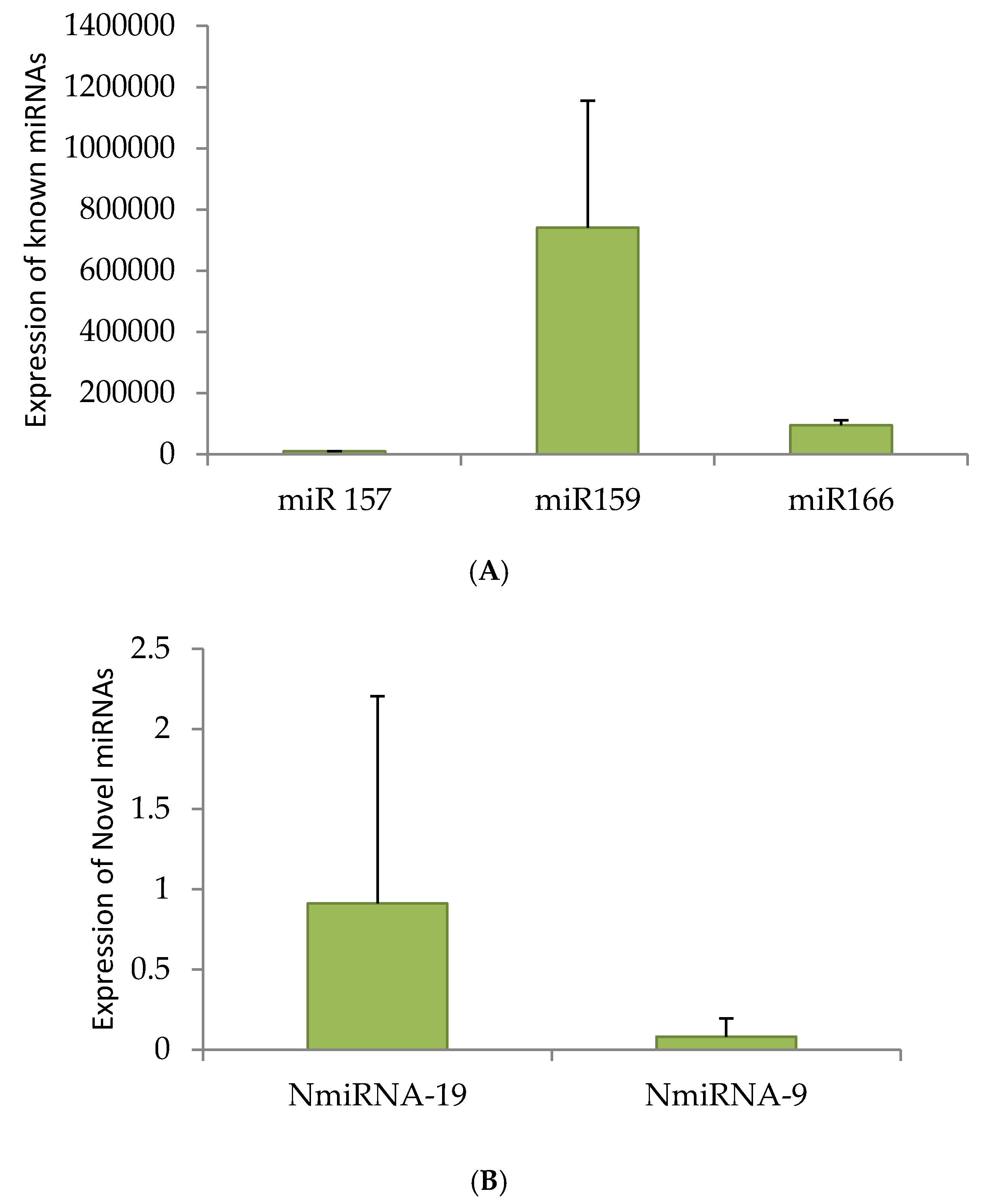
2.6. qRT-PCR
2.7. Target Prediction:
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. RNA Extraction
5.2. sRNA Sequencing Libraries Preparation
5.3. sRNA Sequence Read Mapping
5.4. Precursor miRNA Library Preparation
5.5. Data Analysis
5.6. qRT PCR
5.7. Prediction of miRNA Target Genes
5.8. Accession Numbers
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Mateos, J.L.; Bologna, N.G.; Chorostecki, U.; Palatnik, J.F. Identification of MicroRNA Processing Determinants by Random Mutagenesis of Arabidopsis MIR172a Precursor. Curr. Biol. 2010, 20, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Werner, S.; Wollmann, H.; Schneeberger, K.; Weigel, D. Structure Determinants for Accurate Processing of miR172a in Arabidopsis thaliana. Curr. Biol. 2010, 20, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Czech, B.; Hannon, G.J. Small RNA sorting: Matchmaking for Argonautes. Nat. Rev. Genet. 2011, 12, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, Z.; Yu, B.; Liu, J.; Chen, X. Methylation Protects miRNAs and siRNAs from a 3′-End Uridylation Activity in Arabidopsis. Curr. Biol. 2005, 15, 1501–1507. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Yang, Z.; Li, J.; Minakhina, S.; Yang, M.; Padgett, R.W.; Steward, R.; Chen, X. Methylation as a crucial step in plant microRNA biogenesis. Science 2005, 307, 932–935. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Mo, B.; Chen, X. Mechanisms that impact microRNA stability in plants. RNA Biol. 2012, 9, 1218–1223. [Google Scholar] [CrossRef] [PubMed]
- Mallory, A.; Vaucheret, H. Form, Function, and Regulation of ARGONAUTE Proteins. Plant Cell. 2010, 22, 3879–3889. [Google Scholar] [CrossRef] [PubMed]
- Voinnet, O. Origin, Biogenesis, and Activity of Plant MicroRNAs. Cell 2009, 136, 669–687. [Google Scholar] [CrossRef] [PubMed]
- Fahlgren, N.; Howell, M.D.; Kasschau, K.D.; Chapman, E.J.; Sullivan, C.M.; Cumbie, J.S.; Givan, S.A.; Law, T.F.; Grant, S.R.; Dangl, J.L.; et al. High-Throughput Sequencing of Arabidopsis microRNAs: Evidence for Frequent Birth and Death of MIRNA Genes. PLoS ONE 2007, 2, e219. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Marshall, D.; Bryan, G.J.; Hornyik, C. Identification and Characterization of miRNATranscriptome in Potato by High-Throughput Sequencing. PLoS ONE 2013, 8, e57233. [Google Scholar]
- Hsieh, L.C.; Lin, S.I.; Shih, A.C.C.; Chen, J.W.; Lin, W.Y.; Tseng, C.Y.; Li, W.H.; Chiou, T.J. Uncovering Small RNA-Mediated Responses to Phosphate Deficiency in Arabidopsis by Deep Sequencing. Plant Physiol. 2009, 151, 2120–2132. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.H.; Spriggs, A.; Matthew, L.; Fan, L.; Kennedy, G.; Gubler, F.; Helliwell, C. A diverse set of microRNAs and microRNA-like small RNAs in developing rice grains. Genome Res. 2008, 18, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; You, X.; Chen, T.; Mackowiak, S.D.; Friedlander, M.R.; Weigt, M.; Du, H.; Gogol-Doring, A.; Chang, Z.; Dieterich, C.; et al. Global profiling of miRNAs and the hairpin precursors: Insights into miRNA processing and novel miRNA discovery. Nucleic Acids Res. 2013, 41, 3619–3634. [Google Scholar] [CrossRef] [PubMed]
- Rhoades, M.W.J.; Bartel, D.P.; Bartel, B. Micro RNAs and Their Regulatory roles in Plants. Annu. Rev. Plant Biol. 2006, 57, 19–53. [Google Scholar] [CrossRef] [PubMed]
- Priyavathi, P.; Kavitha, V.; Gopal, P. Complex nature of infection associated with yellow vein mosaic disease in Bhendi (Abelmoschus esculentus). Curr. Sci. 2016, 111, 1511–1515. [Google Scholar] [CrossRef]
- An, J.; Lai, J.; Sajjanhar, A.; Lehman, M.L.; Nelson, C.C. miRPlant: An integrated tool for identification of plant miRNA from RNA sequencing data. BMC Bioinform. 2014, 15, 275. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D732. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yu, J.; Li, D.; Zhang, Z.; Liu, F.; Zhou, X.; Wang, T.; Ling, Y.; Su, Z. PMRD: plant microRNA database. Nucleic Acids Res. 2010, 38, D806–D813. [Google Scholar] [CrossRef] [PubMed]
- Hofacker, I.L. Vienna RNA secondary structure server. Nucleic Acids Res. 2003, 31, 3429–3431. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, E.; Wuyts, J.; Rouzé, P.; Peer, Y.V. Evidence that microRNA precursors, unlike other non-coding RNAs, have lower folding free energies than random sequences. Bioinformatics 2004, 20, 2911–2917. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.H.; Pan, X.P.; Cox, S.B.; Cobb, G.P.; Anderson, T.A. Evidence that miRNAs are different from other RNAs. Cell. Mol. Life. Sci. 2006, 63, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Moazed, D. Small RNAs in transcriptional gene silencing and genome defence. Nature 2009, 457, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Aukerman, M.J.; Sakai, H. Regulation of Flowering Time and Floral Organ Identity by a MicroRNA and Its APETALA2 like Target Genes. Plant Cell 2003, 15, 2730–2741. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Ye, J.; Fang, R. Artificial Micro-RNA mediated Virus Resistance in Plants. J. Virol. 2007, 81, 6690–6699. [Google Scholar] [CrossRef] [PubMed]
- Moxon, S.; Jing, R.; Szittya, G.; Schwach, F.; Pilcher, R.L.R.; Moulton, V.; Dalmay, T. Deep sequencing of tomato short RNAs identifies microRNAs targeting genes involved in fruit ripening. Genome Res. 2008, 18, 1602–1609. [Google Scholar] [CrossRef] [PubMed]
- Martinez, G.; Forment, J.; Llave, C.; Pallas, V.; Gomez, G. High-Throughput Sequencing, Characterization and Detection of New and Conserved Cucumber miRNAs. PLoS ONE 2011, 6, e19523. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, H.; Li, D.; Chen, H. Identification and characterization of maize microRNAs involved in the very early stage of seed germination. BMC Genom. 2011, 12, 154. [Google Scholar] [CrossRef] [PubMed]
- Briere, C.L.; Naya, L.; Sallet, E.; Calenge, F.; Frugier, F.; Hartmann, C.; Gouzy, J.; Crespi, M. Genome-Wide Medicago truncatula Small RNA Analysis Revealed Novel MicroRNAs and Isoforms Differentially Regulated in Roots and Nodules. Plant Cell 2009, 21, 2780–2796. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Wang, C.; Zhang, C.; Korir, N.K.; Yu, H.; Ma, Z.; Fang, J. Deep sequencing discovery of novel and conserved microRNAs in trifoliate orange (Citrus trifoliata). BMC Genom. 2010, 11, 43. [Google Scholar] [CrossRef] [PubMed]
- Gebelin, V.; Argout, X.; Engchuan, W.; Pitollat, B.; Duan, C.; Montoro, P.; Leclercq, J. Identification of novel microRNAs in Hevea brasiliensis and computational prediction of their targets. BMC Plant Biol. 2012, 12, 18. [Google Scholar] [CrossRef] [PubMed]
- Naoumkina, M.; Thyssen, G.N.; Fang, D.D.; Hinchliffe, D.J.; Florane, C.B.; Jenkins, J.N. Small RNA sequencing and degradome analysis of developing fibers of short fiber mutants Ligon-lintles-1 (Li1) and −2 (Li2) revealed a role for miRNAs and their targets in cotton fiber elongation. BMC Genom. 2016, 17, 360. [Google Scholar] [CrossRef] [PubMed]
- Burroughs, A.M.; Kawano, M.; Ando, Y.; Daub, C.O.; Hayashizaki, Y. pre-miRNA profiles obtained through application of locked nucleic acids and deep sequencing reveals complex 5′/3′ arm variation including concomitant cleavage and polyuridylation patterns. Nucleic Acids Res. 2011, 40, 1424–1437. [Google Scholar] [CrossRef] [PubMed]
- Newman, M.A.; Mani, V.; Hammond, S.M. Deep sequencing of microRNA precursors reveals extensive 3′ end modification. RNA 2011, 17, 1795–1803. [Google Scholar] [CrossRef] [PubMed]
- Xuan, P.; Guo, M.Z.; Wang, J.; Wang, C.Y.; Liu, X.Y.; Liu, Y. Genetic algorithm-based efficient feature selection for classification of pre-miRNAs. Genet. Mol. Res. 2011, 10, 588–603. [Google Scholar] [CrossRef] [PubMed]
- Lacombe, S.; Nagasaki, H.; Santi, C.; Duval, D.; Piegu, B.; Bangratz, M.; Breitler, J.; Guiderdoni, E.; Brugidou, C.; Hirsch, J.; et al. Identification of precursor transcripts for 6 novel miRNAs expands the diversity on the genomic organisation and expression of miRNA genes in rice. BMC Plant Biol. 2008, 8, 123. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, Q.; Wang, B. Identification and Characterization of MicroRNAs in Asiatic Cotton (Gossypium arboreum L.). PLoS ONE 2012, 7, e33696. [Google Scholar] [CrossRef] [PubMed]
- Rogans, S.J.; Rey, C. Unveiling the Micronome of Cassava (Manihot esculenta Crantz). PLoS ONE 2016, 11, e0147251. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk?/projects/fastqc/.2010 (accessed on 13 July 2015).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Fast X Artefacts Filter. Available online: http://hannonlab.cshl.edu/fastx_toolkit/index.html (accessed on 13 July 2015).
- Fqtrim. Available online: http://ccb.jhu.edu/software/fqtrim/.
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina Paired-End reAd merger. Bioinformatics 2013, 30, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2013, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Balcells, I.; Cirera, S.; Busk, P.K. Specific and sensitive quantitative RT-PCR of miRNAs with DNA primers. BMC Biotechnol. 2011, 11, 70. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using RealTime Quantitative PCR and the 2−ΔΔcT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server. Nucleic Acids Res. 2011, 39, W155–W159. [Google Scholar] [CrossRef] [PubMed]
- Schafleitner, R.; Kumar, S.; Lin, C.; Hegde, S.G.; Ebert, A. The okra (Abelmoschus esculentus) transcriptome as a source for gene sequence information and molecular markers for diversity analysis. Gene 2013, 517, 27–36. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No | Precursors | Small RNA Dataset 1 | Small RNA Dataset 2 | Small RNA Dataset 3 |
|---|---|---|---|---|
| 1 | No of precursors for known miRNAs | 309 | 305 | 241 |
| 2 | No of common precursors (for Known miRNAs) among the three datasets | 219 | ||
| 3 | No of unique precursors (for known miRNAs) among the three datasets | 330 | ||
| 4 | No of precursors for novel miRNAs | 4863 | 4115 | 3509 |
| 5 | No of common precursors (for novel miRNAs) among the three datasets | 2003 | ||
| 6 | No of unique precursors (for novel miRNAs) among the three datasets | 6793 | ||
| miRNA ID | miRNA Sequence | Precursor Sequence | MFEI |
|---|---|---|---|
| miR156 | TTGACAGAAGATAGAGAG | TTGACAGAAGATAGAGAGCACCCTCTCTCTCTCTCCCTGTCTGCCTTTCTGTCTGTCTTATTATTGACACGGCTGATGACTTGTAAATTCTCCATGAGAATCAGTT | −0.696 |
| miR482a | TCTTTCCTACTCCTCCCA | TCTTTCCTACTCCTCCCATTCCAGGGACGGAGGAGGCTAGGT | −0.750 |
| miR6300 | GTCGTTGTAGTATAGTGGT | GTCGTTGTAGTATAGTGGTAAGTATTCCCGCCGCATATGATGCAACGATTCTTATCCTATACACT | −0.659 |
| miR157 | TTGACAGAAGATAGAGAGCA | TTGACAGAAGATAGAGAGCACCCTCTCTCTCTCTCCCTGTCTGCCTTTCTGTCTGTCTTATTATTGACACGGCTGATGACTTGTAAATTCTCCATGAGAATCAGTT | −0.696 |
| miR159 | GGATTGAAGGGAGCTCTA | TTTGGATTGAAGGGAGCTCTATTCTGTGATGAAGCAATTTTATTGTGGACTAGAGTTTCTGATCTGG | −0.769 |
| miR396 | CACAGCTTTCTTGAACTT | TTCCACAGCTTTCTTGAACTTGCGAGTCAACGGGTTAGCAAACCCGCAAGGCGCAAGGAAGCTGATTGGCGGGATCCCTCGCGGGTGCACCGCCGA | −0.750 |
| miR6424 | TGGTGCCACGCTGTGTGCG | AGAGCTGAATGTGGTGTTTTGGGCTCATGCTTGCGTGGTGCCATCAAAACTTGGCATTGGAGAAGCGGTGATGGTGCCACGCTGTGTGCGACTGA | −0.696 |
| miR168 | TCGCTTGGTGCAGGTCGG | TCGCTTGGTGCAGGTCGGGAAATTACGATAGGTGTCAAGTGGAAGTGCA | −0.604 |
| miR160 | TGCCTGGCTCCCTGTATG | TGCCTGGCTCCCTGTATGCCACAATGTAGGCAAGGGAAGTCGGCAAAATGG | −0.775 |
| miR530 | TGCATTTGCACCTGCACC | TGCATTTGCACCTGCACCTTCTCATTACGATAGGTGTCAAGTGGAAGTGCA | −0.878 |
| miR166 | TCTCGGACCAGGCTTCAT | TCTCGGACCAGGCTTCATTCCCGAAGCCTGCCCAGCAGAACGACCCGCGAACGTGTTATCGAAAAAC | −0.463 |
| miR535 | CAACGAGAGAGAGCACGC | TGACAACGAGAGAGAGCACGCAGCAATGAGGTTAATCGTGCTTCTCTGATGATTGGGTTAT | −0.571 |
| miR162 | TCGATAAACCTCTGCATC | TCGATAAACCTCTGCATCCAGGAGCAATGAGGATAATCTGCTCTTGTGATGATAGGGTTATC | −0.881 |
| miR408 | CACTGCCTCTTCCCTGGCT | TGCACTGCCTCTTCCCTGGCTTTCAGGTCTCCAAGGTGAACAGCCTCTGGTCGATGGAACAATGTAGGCAAGGGAAGTCGGCAAAATG | −0.646 |
| miR167 | GCTGCCAGCATGATCTTA | TGAAGCTGCCAGCATGATCTTACATTACGATAGGTGTCAAGTGGAAGTGCA | −0.339 |
| S. No | miRNA | Target |
|---|---|---|
| 1 | miR-169 |
|
| 2 | miR-166 |
|
| 3 | miR-157 |
|
| 4 | miR-159 |
|
| 5 | NmiRNA-4 |
|
| 6 | NmiRNA-1 |
|
| 7 | NmiRNA-7 |
|
| 8 | NmiRNA-18 |
|
| S. No | miRNA | Forward Primer | Reverse Primer |
|---|---|---|---|
| 1 | miRNA159a | GCAGTTTGGATTGAAGGGA | AGTCCAGTTTTTTTTTTTTTTTAGAGC |
| 2 | miRNA 166a | GTCGGACCAGGCTTCAT | CCAGTTTTTTTTTTTTTTTGGGGA |
| 3 | miRNA 157a | CGCAGTTGACAGAAGATAGAG | TCCAGTTTTTTTTTTTTTTTGTGCT |
| 4 | NmiRNA 19 | GGCGCAGAGTTACTAATTCATGA | GTCCAGTTTTTTTTTTTTTTTCAGAT |
| 5 | NmiRNA 9 | CGCAGGGTGGCTGTAGTTTA | GTCCAGTTTTTTTTTTTTTTTACCAC |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Velayudha Vimala Kumar, K.; Srikakulam, N.; Padbhanabhan, P.; Pandi, G. Deciphering microRNAs and Their Associated Hairpin Precursors in a Non-Model Plant, Abelmoschus esculentus. Non-Coding RNA 2017, 3, 19. https://doi.org/10.3390/ncrna3020019
Velayudha Vimala Kumar K, Srikakulam N, Padbhanabhan P, Pandi G. Deciphering microRNAs and Their Associated Hairpin Precursors in a Non-Model Plant, Abelmoschus esculentus. Non-Coding RNA. 2017; 3(2):19. https://doi.org/10.3390/ncrna3020019
Chicago/Turabian StyleVelayudha Vimala Kumar, Kavitha, Nagesh Srikakulam, Priyavathi Padbhanabhan, and Gopal Pandi. 2017. "Deciphering microRNAs and Their Associated Hairpin Precursors in a Non-Model Plant, Abelmoschus esculentus" Non-Coding RNA 3, no. 2: 19. https://doi.org/10.3390/ncrna3020019