Calcium-Dependent Interaction Occurs between Slow Skeletal Myosin Binding Protein C and Calmodulin


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
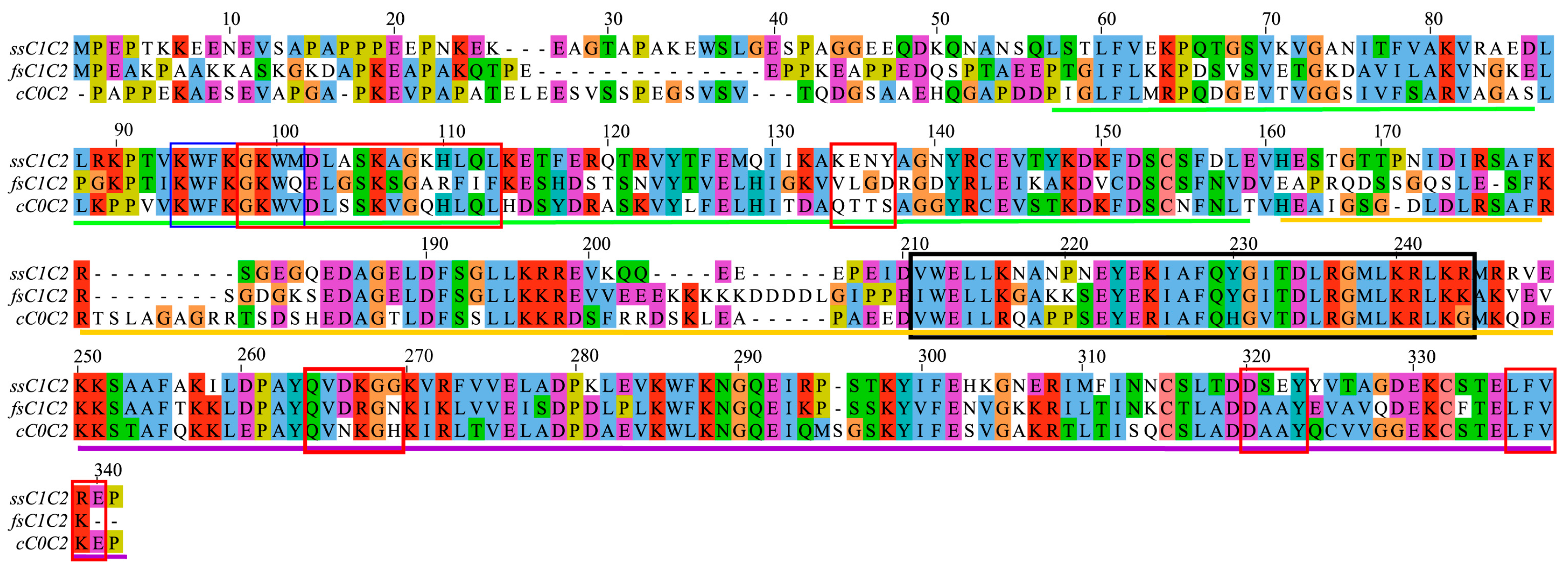
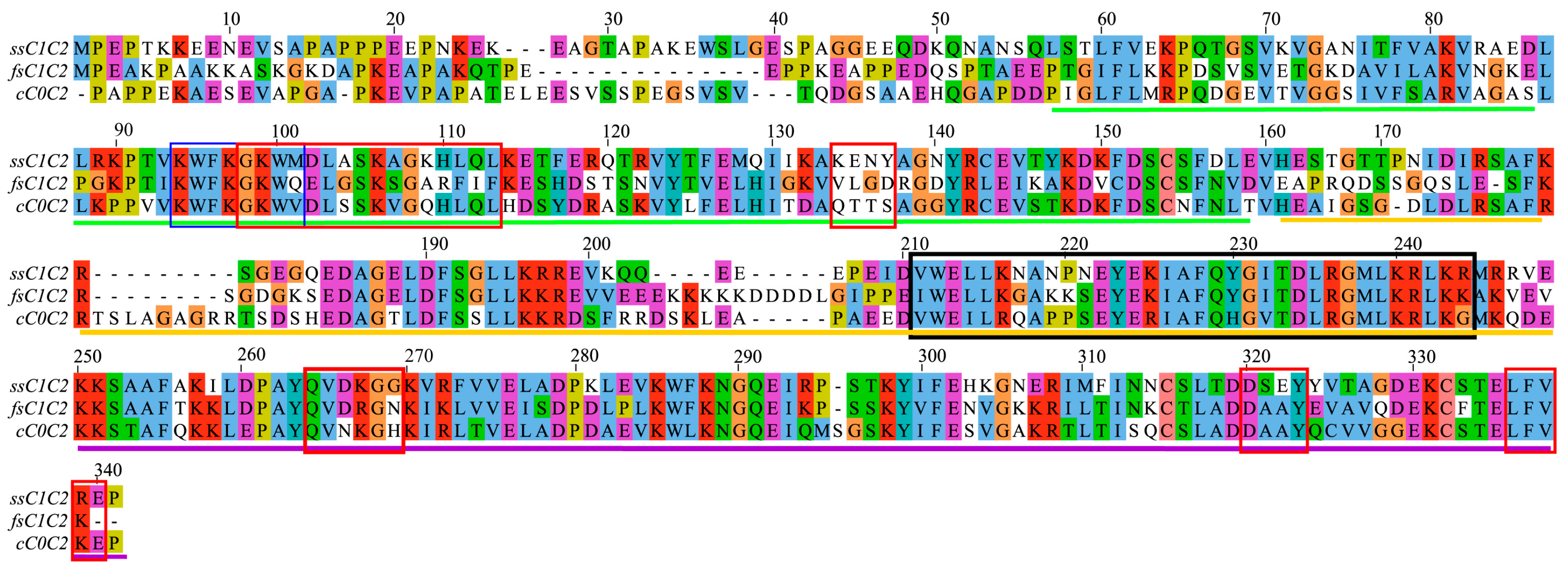
2.1. Analysis of Amino Acid Conservation Provides Clues about Regulatory Functions in MyBP-C Isoforms
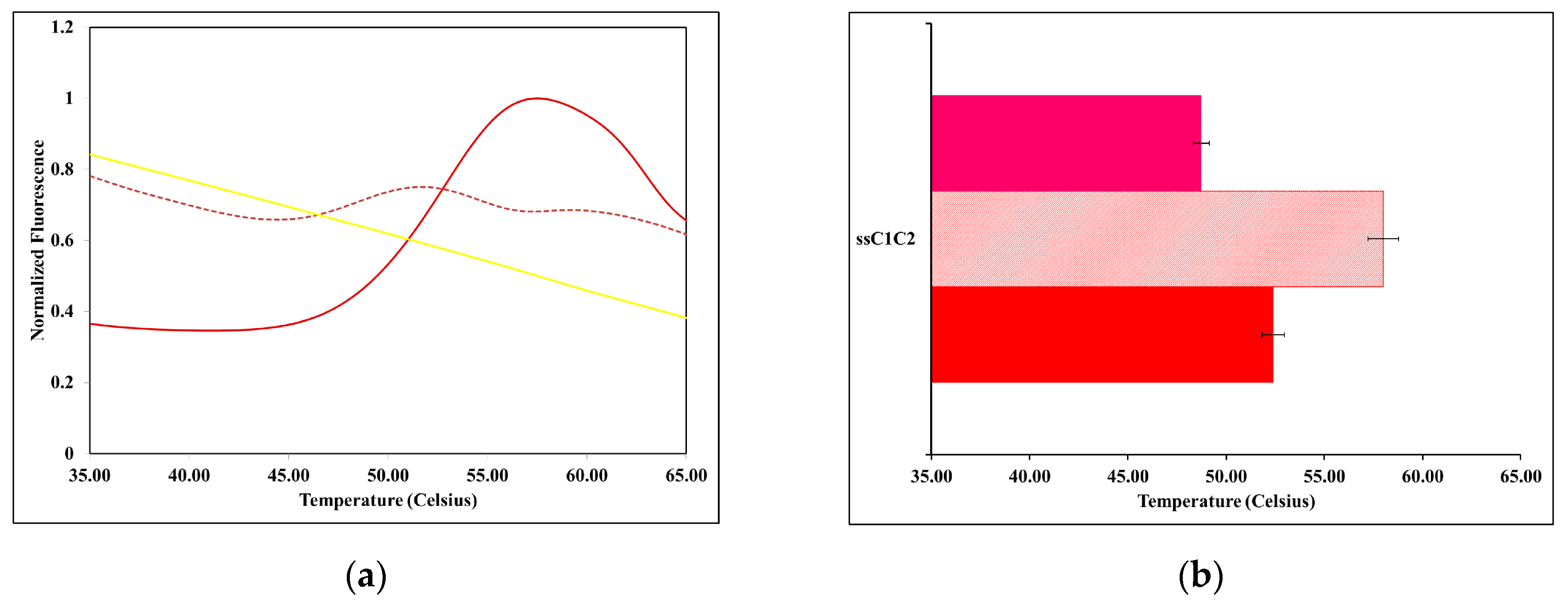
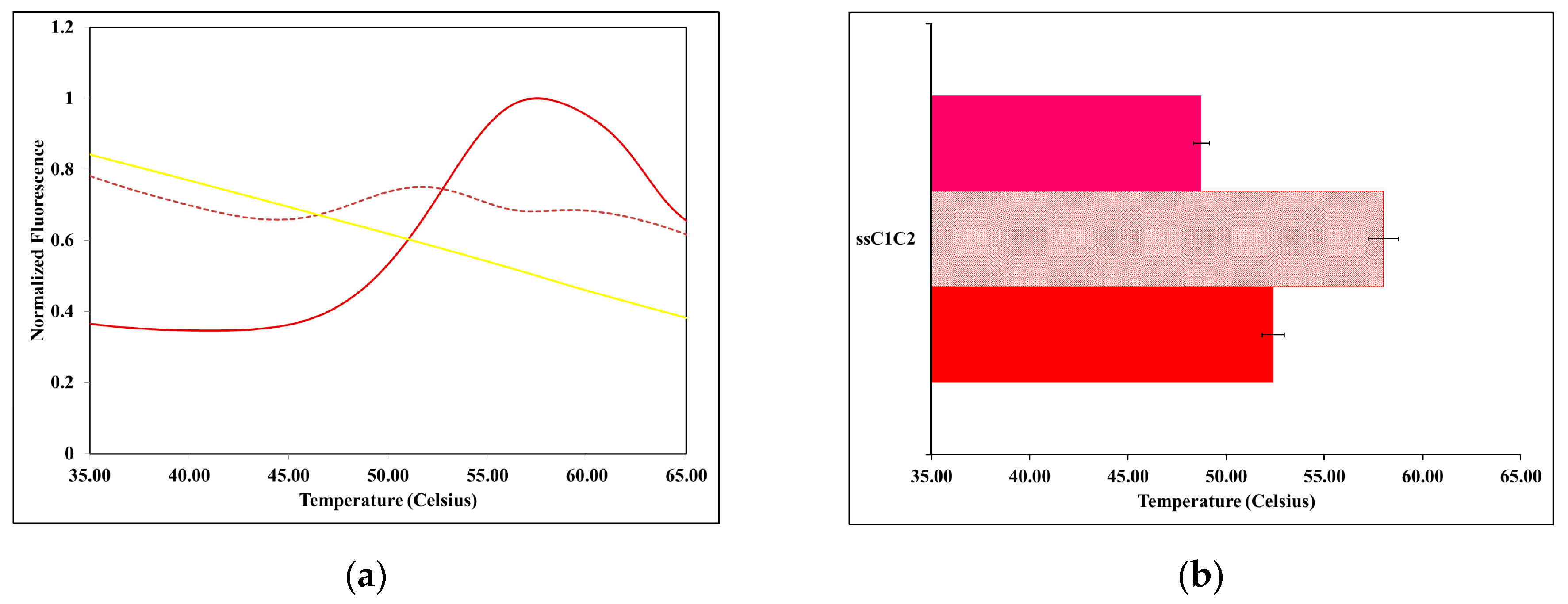
2.2. The Thermal Stability of ssMyBP-C Is Impacted by Ca2+-CaM
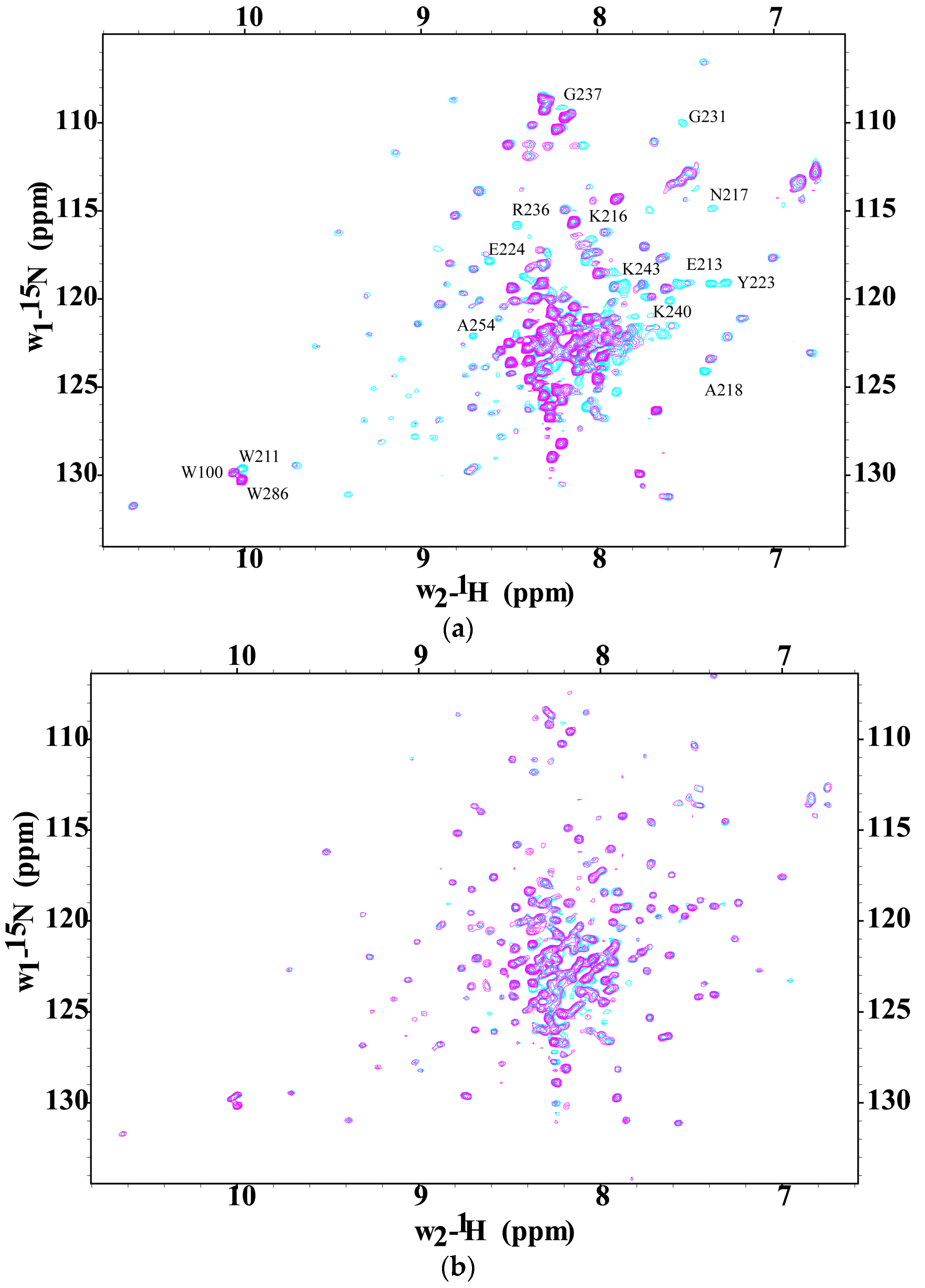
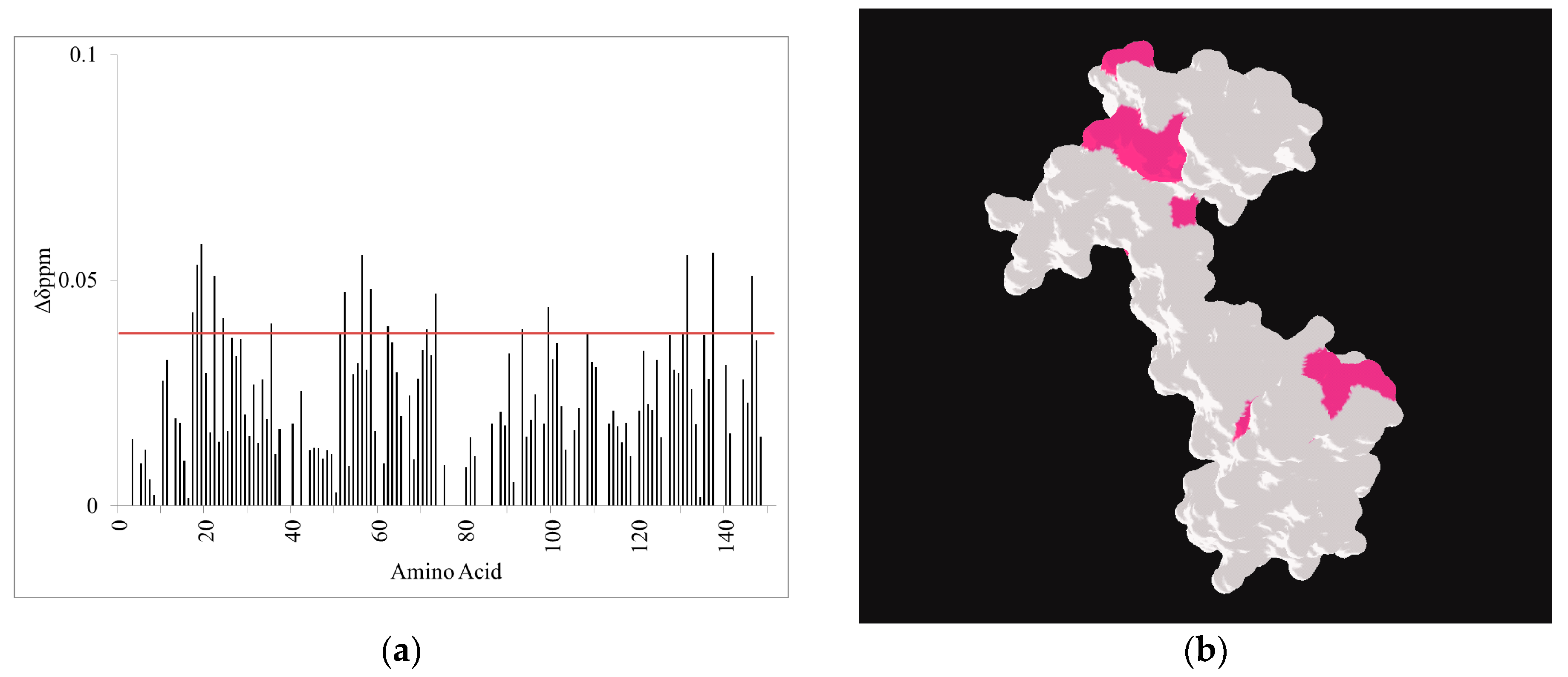
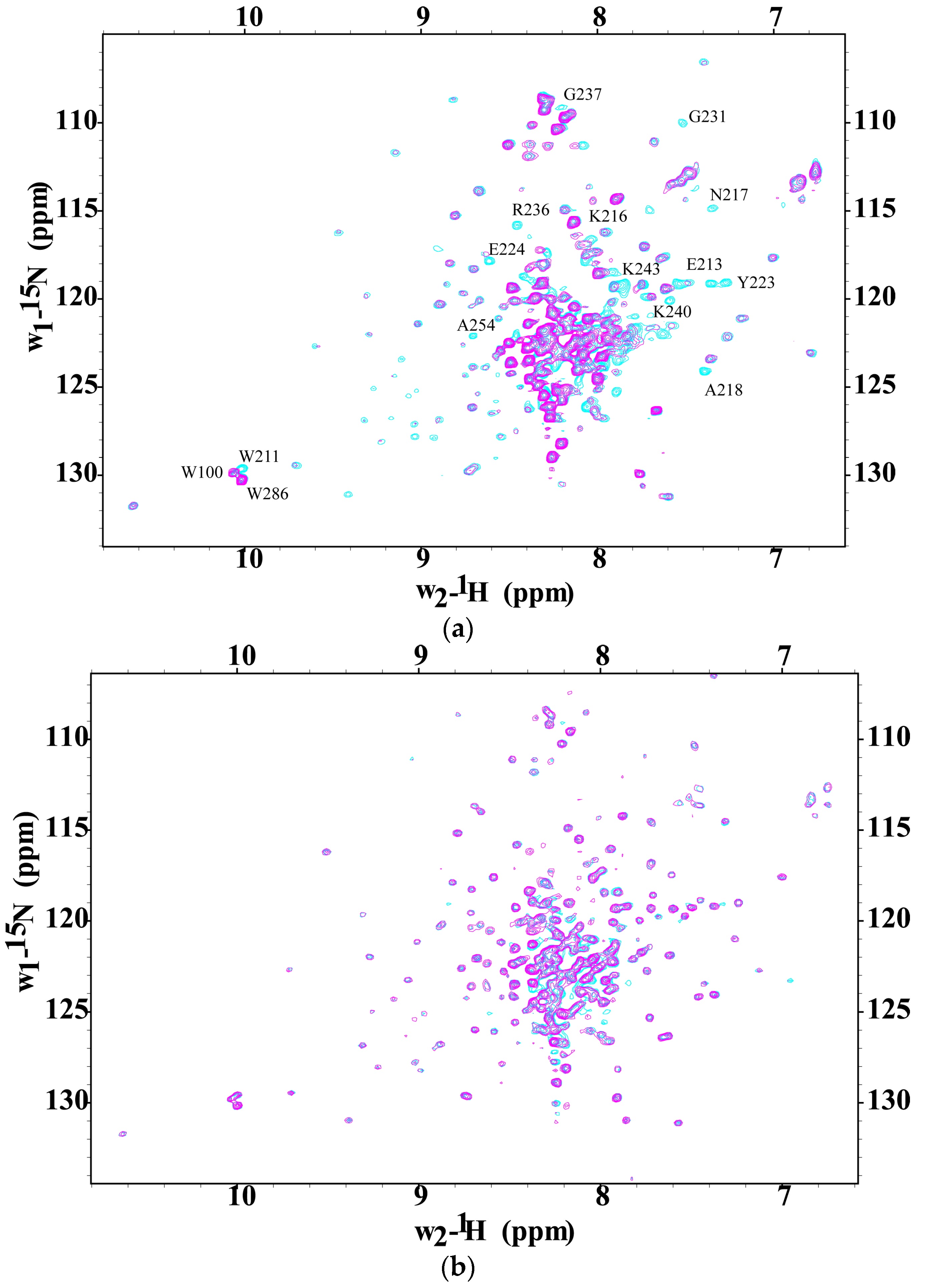
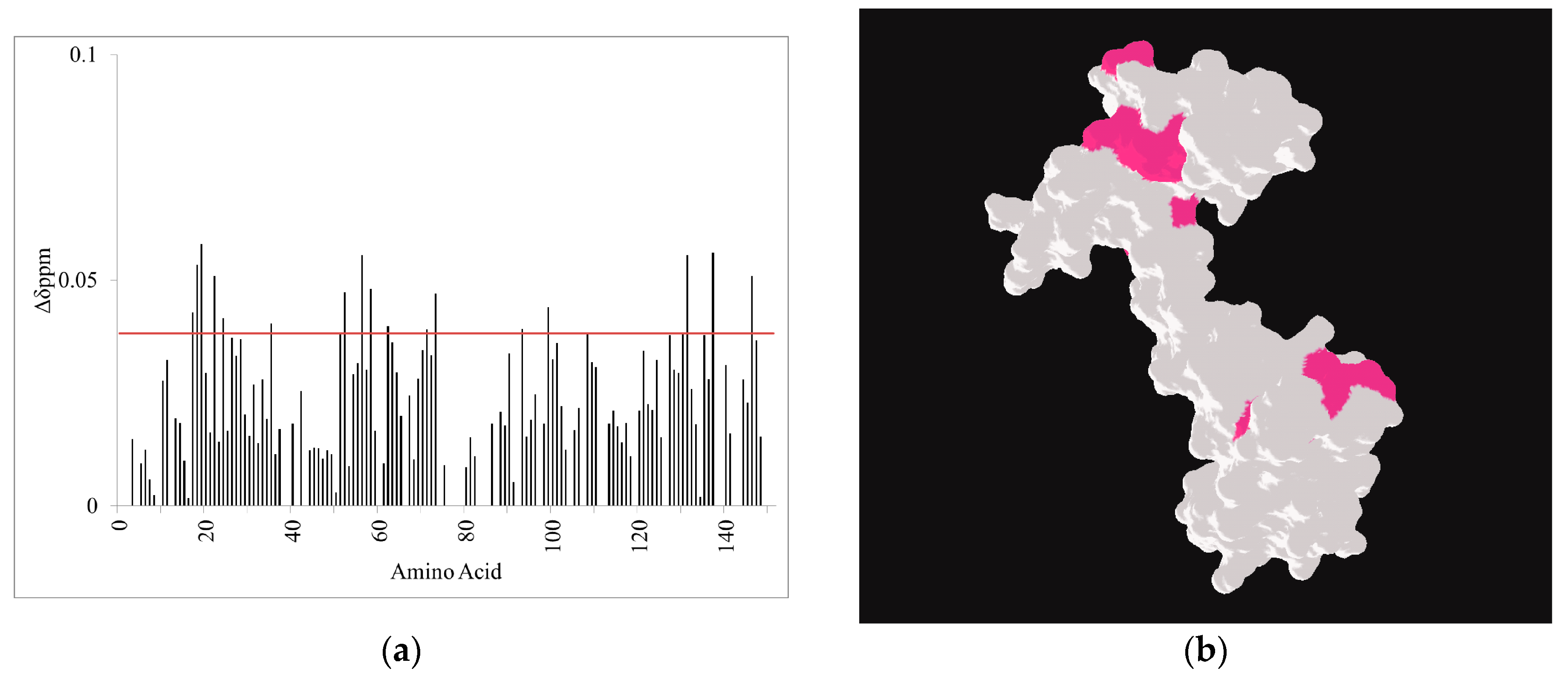
2.3. CaM-Induced Conformational Modulation of ssC1C2 Is Ca2+-Dependent and Localized Primarily to the M-Motif
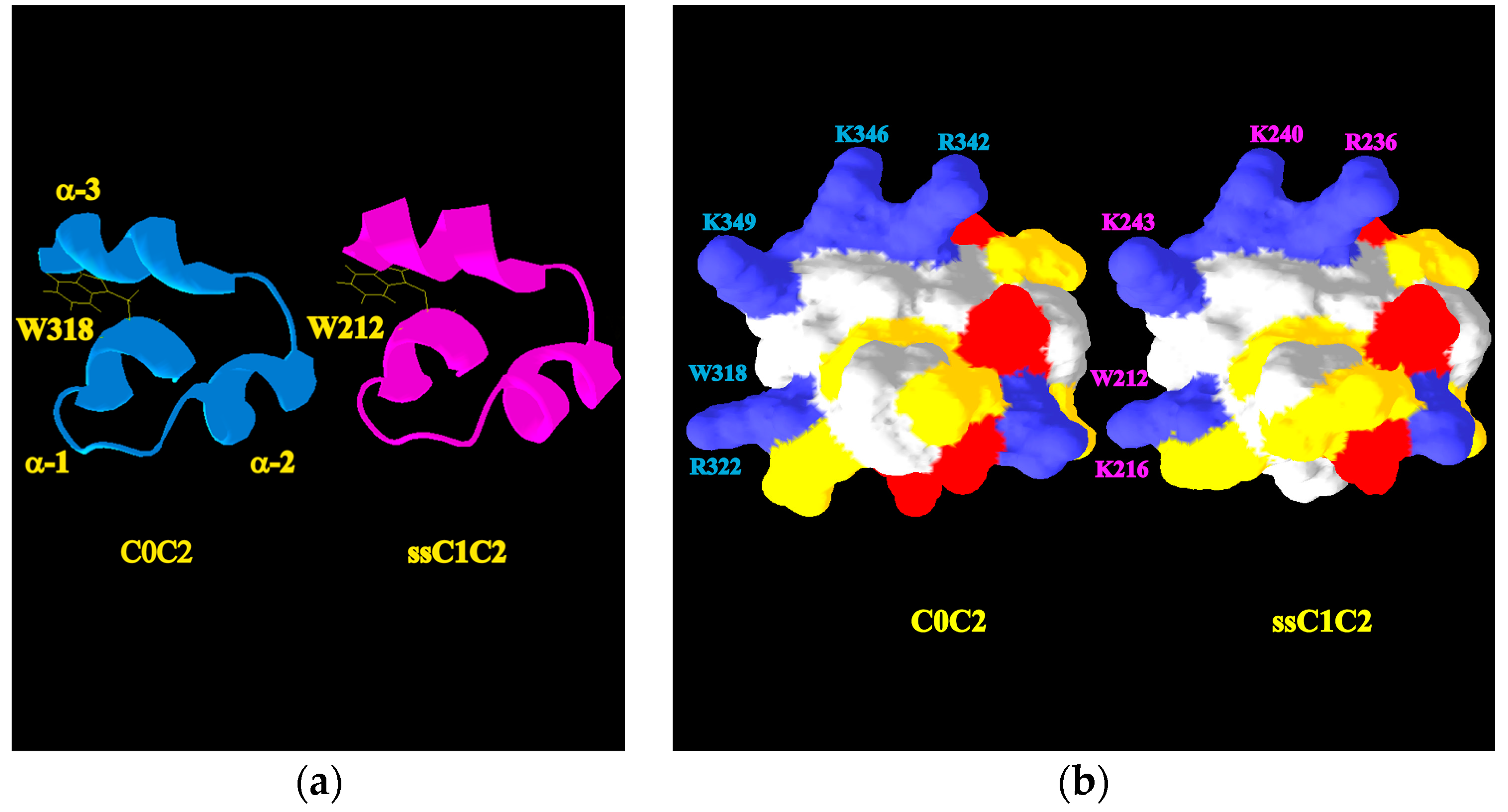
2.4. A Molecular Model Sheds Light on How the M-Motif of MyBP-C Engages Protein Targets
3. Discussion
4. Materials and Methods
4.1. Recombinant Protein Expression and Purification
4.2. Differential Scanning Fluorimetry (DSF) Experiments
4.3. Nuclear Magnetic Resonance Spectroscopy (NMR)
4.4. Homology Modeling and Bioinformatics
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Luther, P.K.; Winkler, H.; Taylor, K.; Zoghbi, M.E.; Craig, R.; Padron, R.; Squire, J.M.; Liu, J. Direct visualization of myosin-binding protein C bridging myosin and actin filaments in intact muscle. Proc. Natl. Acad. Sci. USA 2011, 108, 11423–11428. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, J.F.; Kensler, R.W.; Harris, S.P. The myosin-binding protein C motif binds to F-actin in a phosphorylation-sensitive manner. J. Biol. Chem. 2009, 284, 12318–12327. [Google Scholar] [CrossRef] [PubMed]
- Kensler, R.W.; Shaffer, J.F.; Harris, S.P. Binding of the N-terminal fragment C0–C2 of cardiac MyBP-C to cardiac F-actin. J. Struct. Biol. 2011, 174, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Kwan, A.H.; Trewhella, J.; Jeffries, C.M. The C0C1 fragment of human cardiac myosin binding protein C has common binding determinants for both actin and myosin. J. Mol. Biol. 2011, 413, 908–913. [Google Scholar] [CrossRef] [PubMed]
- Rybakova, I.N.; Greaser, M.L.; Moss, R.L. Myosin Binding Protein C Interaction with Actin: Characterization and Mapping of the Binding Site. J. Biol. Chem. 2011, 286, 2008–2016. [Google Scholar] [CrossRef] [PubMed]
- Bhuiyan, M.S.; Gulick, J.; Osinska, H.; Gupta, M.; Robbins, J. Determination of the critical residues responsible for cardiac myosin binding protein {C’s} interactions. J. Mol. Cell. Cardiol. 2012, 53, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Whitten, A.E.; Jeffries, C.M.; Harris, S.P.; Trewhella, J. Cardiac myosin-binding protein C decorates F-actin: Implications for cardiac function. Proc. Natl. Acad. Sci. USA 2008, 105, 18360–18365. [Google Scholar] [CrossRef] [PubMed]
- Sadayappan, S.; Gulick, J.; Osinska, H.; Barefield, D.; Cuello, F.; Avkiran, M.; Lasko, V.M.; Lorenz, J.N.; Maillet, M.; Martin, J.L.; et al. A critical function for Ser-282 in cardiac myosin binding protein-C phosphorylation and cardiac function. Circ. Res. 2011, 109, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Okagaki, T.; Weber, F.E.; Fischman, D.A.; Vaughan, K.T.; Mikawa, T.; Reinach, F.C. The Major Myosin-Binding Domain of Skeletal Muscle MyBP-C (C-Protein) Resides in the COOH-Terminal, Immunoglobulin-C2 Motif. J. Cell Biol. 1993, 123, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Gruen, M.; Prinz, H.; Gautel, M. cAPK-phosphorylation controls the interaction of the regulatory domain of cardiac myosin binding protein C with myosin-S2 in an on-off fashion. FEBS Lett. 1999, 453, 254–259. [Google Scholar] [CrossRef]
- Ratti, J.; Rostkova, E.; Gautel, M.; Pfuhl, M. Structure and interactions of myosin-binding protein C domain C0: Cardiac-specific regulation of myosin at its neck? J. Biol. Chem. 2011, 286, 12650–12658. [Google Scholar] [CrossRef] [PubMed]
- Freiburg, A.; Gautel, M. A molecular map of the interactions between titin and myosin-binding protein C. Implications for sarcomeric assembly in familial hypertrophic cardiomyopathy. Eur. J. Biochem. 1996, 235, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, C.A.; Fischman, D.A.; Reinach, F.C. The interface between MyBP-C and myosin: Site-directed mutagenesis of the CX myosin-binding domain of MyBP-C. J. Muscle Res. Cell Motil. 1999, 20, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Flashman, E.; Watkins, H.; Redwood, C. Localization of the binding site of the C-terminal domain of cardiac myosin-binding protein-C on the myosin rod. Biochem. J. 2007, 401, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Kuster, D.W.D.; Govindan, S.; Springer, T.I.; Martin, J.L.; Finley, N.L.; Sadayappan, S. A hypertrophic cardiomyopathy-associated MYBPC3 mutation common in populations of South Asian descent causes contractile dysfunction. J. Biol. Chem. 2015, 290, 5855–5867. [Google Scholar] [CrossRef] [PubMed]
- Weber, F.E.; Vaughan, K.T.; Reinach, F.C.; Fischman, D.A. Complete sequence of human fast-type and slow-type muscle myosin-binding-protein C (MyBP-C). Differential expression, conserved domain structure and chromosome assignment. Eur. J. Biochem. 1993, 216, 661–669. [Google Scholar] [PubMed]
- Lin, B.; Govindan, S.; Lee, K.; Zhao, P.; Han, R.; Runte, K.E.; Craig, R.; Palmer, B.M.; Sadayappan, S. Cardiac Myosin Binding Protein-C Plays No Regulatory Role in Skeletal Muscle Structure and Function. PLoS ONE 2013, 8, e69671. [Google Scholar] [CrossRef] [PubMed]
- Alyonycheva, T.N.; Mikawa, T.; Reinach, F.C.; Fischman, D.A. Isoform-specific interaction of the myosin-binding proteins (MyBPs) with skeletal and cardiac myosin is a property of the C-terminal immunoglobulin domain. J. Biol. Chem. 1997, 272, 20866–20872. [Google Scholar] [CrossRef] [PubMed]
- Idowu, S.M.; Gautel, M.; Perkins, S.J.; Pfuhl, M. Structure, stability and dynamics of the central domain of cardiac myosin binding protein C (MyBP-C): Implications for multidomain assembly and causes for cardiomyopathy. J. Mol. Biol. 2003, 329, 745–761. [Google Scholar] [CrossRef]
- Cecconi, F.; Guardiani, C.; Livi, R. Analyzing pathogenic mutations of C5 domain from cardiac myosin binding protein C through MD simulations. Eur. Biophys. J. 2008, 37, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, J.F.; Razumova, M.V.; Tu, A.Y.; Regnier, M.; Harris, S.P. Myosin S2 is not required for effects of myosin binding protein-C on motility. FEBS Lett. 2007, 581, 1501–1504. [Google Scholar] [CrossRef] [PubMed]
- Mun, J.Y.; Kensler, R.W.; Harris, S.P.; Craig, R. The cMyBP-C HCM variant L348P enhances thin filament activation through an increased shift in tropomyosin position. J. Mol. Cell. Cardiol. 2016, 91, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Bezold, K.L.; Shaffer, J.F.; Khosa, J.K.; Hoye, E.R.; Harris, S.P. A gain-of-function mutation in the M-domain of cardiac myosin-binding protein-C increases binding to actin. J. Biol. Chem. 2013, 288, 21496–21505. [Google Scholar] [CrossRef] [PubMed]
- Orlova, A.; Galkin, V.E.; Jeffries, C.M.J.; Egelman, E.H.; Trewhella, J. The N-terminal domains of myosin binding protein C can bind polymorphically to F-actin. J. Mol. Biol. 2011, 412, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Kwan, A.H.; Jeffries, C.M.; Guss, J.M.; Trewhella, J. The motif of human cardiac myosin-binding protein C is required for its Ca2+-dependent interaction with calmodulin. J. Biol. Chem. 2012, 287, 31596–31607. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.A.; Kontrogianni-Konstantopoulos, A. Myosin binding protein-C slow is a novel substrate for protein kinase A (PKA) and C (PKC) in skeletal muscle. J. Proteome Res. 2011, 10, 4547–4555. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, M.A.; Rivard, A.F.; Bachinger, H.P.; Adelman, J.P. Structure of the gating domain of a Ca2+-activated K+ channel complexed with Ca2+/calmodulin. Nature 2001, 410, 1120–1124. [Google Scholar] [CrossRef] [PubMed]
- Springer, T.I.; Goebel, E.; Hariraju, D.; Finley, N.L. Mutation in the beta-hairpin of the Bordetella pertussis adenylate cyclase toxin modulates N-lobe conformation in calmodulin. Biochem. Biophys. Res. Commun. 2014, 453, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Heller, W.T.; Krueger, J.K.; Trewhella, J. Further insights into calmodulin-myosin light chain kinase interaction from solution scattering and shape restoration. Biochemistry 2003, 42, 10579–10588. [Google Scholar] [CrossRef] [PubMed]
- Howarth, J.W.; Ramisetti, S.; Nolan, K.; Sadayappan, S.; Rosevear, P.R. Structural insight into unique cardiac myosin-binding protein-C motif: A partially folded domain. J. Biol. Chem. 2012, 287, 8254–8262. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2014, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.A.; Ward, C.W.; Gurnett, C.; Kontrogianni-Konstantopoulos, A. Myosin Binding Protein-C Slow Phosphorylation is Altered in Duchenne Dystrophy and Arthrogryposis Myopathy in Fast-Twitch Skeletal Muscles. Sci. Rep. 2015, 5, 13235. [Google Scholar] [CrossRef] [PubMed]
- Lavinder, J.J.; Hari, S.B.; Sullivan, B.J.; Magliery, T.J. High-Throughput Thermal Scanning: A General, Rapid Dye-Binding Thermal Shift Screen for Protein Engineering. J. Am. Chem. Soc. 2009, 131, 3794–3795. [Google Scholar] [CrossRef] [PubMed]
- Cimmperman, P.; Baranauskienė, L.; Jachimovičiūtė, S.; Jachno, J.; Torresan, J.; Michailovienė, V.; Matulienė, J.; Sereikaitė, J.; Bumelis, V.; Matulis, D. A Quantitative Model of Thermal Stabilization and Destabilization of Proteins by Ligands. Biophys. J. 2008, 95, 3222–3231. [Google Scholar] [CrossRef] [PubMed]
- Jeffries, C.M.; Lu, Y.; Hynson, R.M.G.; Taylor, J.E.; Ballesteros, M.; Kwan, A.H.; Trewhella, J. Human cardiac myosin binding protein C: Structural flexibility within an extended modular architecture. J. Mol. Biol. 2011, 414, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Ababou, A.; Zhou, L.; Gautel, M.; Pfuhl, M. Sequence specific assignment of domain C1 of the N-terminal myosin-binding site of human cardiac myosin binding protein C (MyBP-C). J. Biomol. NMR 2004, 29, 431–432. [Google Scholar] [CrossRef] [PubMed]
- Ababou, A.; Gautel, M.; Pfuhl, M. Dissecting the N-terminal myosin binding site of human cardiac myosin-binding protein C: Structure and myosin binding of domain C2. J. Biol. Chem. 2007, 282, 9204–9215. [Google Scholar] [CrossRef] [PubMed]
- Michie, K.A.; Kwan, A.H.; Tung, C.S.; Guss, J.M.; Trewhella, J. A Highly Conserved Yet Flexible Linker Is Part of a Polymorphic Protein-Binding Domain in Myosin-Binding Protein C. Structure 2016, 24, 2000–2007. [Google Scholar] [CrossRef] [PubMed]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Cassarino, T.G.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef] [PubMed]
- Benkert, P.; Tosatto, S.C.E.; Schomburg, D. QMEAN: A comprehensive scoring function for model quality assessment. Proteins Struct. Funct. Bioinform. 2008, 71, 261–277. [Google Scholar] [CrossRef] [PubMed]
- De Beer, T.A.P.; Berka, K.; Thornton, J.M.; Laskowski, R.A. PDBsum additions. Nucleic Acids Res. 2014, 42, D292–D296. [Google Scholar] [CrossRef] [PubMed]
- Gasmi-Seabrook, G.M.; Howarth, J.W.; Finley, N.; Abusamhadneh, E.; Gaponenko, V.; Brito, R.M.; Solaro, R.J.; Rosevear, P.R. Solution structures of the C-terminal domain of cardiac troponin C free and bound to the N-terminal domain of cardiac troponin I. Biochemistry 1999, 38, 8313–8322. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, R.M.B.; Blumenschein, T.M.A.; Sykes, B.D. An interplay between protein disorder and structure confers the Ca2+ regulation of striated muscle. J. Mol. Biol. 2006, 361, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Gurnett, C.A.; Desruisseau, D.M.; McCall, K.; Choi, R.; Meyer, Z.I.; Talerico, M.; Miller, S.E.; Ju, J.S.; Pestronk, A.; Connolly, A.M.; et al. Myosin binding protein C1: A novel gene for autosomal dominant distal arthrogryposis type 1. Hum. Mol. Genet. 2010, 19, 1165–1173. [Google Scholar] [CrossRef] [PubMed]
- Michalek, A.J.; Howarth, J.W.; Gulick, J.; Previs, M.J.; Robbins, J.; Rosevear, P.R.; Warshaw, D.M. Phosphorylation modulates the mechanical stability of the cardiac myosin-binding protein C motif. Biophys. J. 2013, 104, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Tavi, P.; Westerblad, H. The role of in vivo Ca2+ signals acting on Ca2+-calmodulin-dependent proteins for skeletal muscle plasticity. J. Physiol. 2011, 589, 5021–5031. [Google Scholar] [CrossRef] [PubMed]
- Abbott, M.B.; Dong, W.J.; Dvoretsky, A.; DaGue, B.; Caprioli, R.M.; Cheung, H.C.; Rosevear, P.R. Modulation of cardiac troponin c-cardiac troponin I regulatory interactions by the amino-terminus of cardiac troponin I. Biochemistry 2001, 40, 5992–6001. [Google Scholar] [CrossRef] [PubMed]
- Finley, N.; Dvoretsky, A.; Rosevear, P.R. Magnesium—Calcium Exchange in Cardiac Troponin C Bound to Cardiac Troponin I. J. Mol. Cell. Cardiol. 2000, 1446, 1439–1446. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.J.; Chandra, M.; Xing, J.; She, M.; Solaro, R.J.; Cheung, H.C. Phosphorylation-induced distance change in a cardiac muscle troponin I mutant. Biochemistry 1997, 36, 6754–6761. [Google Scholar] [CrossRef] [PubMed]
- Sakthivel, S.; Finley, N.L.; Rosevear, P.R.; Lorenz, J.N.; Gulick, J.; Kim, S.; VanBuren, P.; Martin, L.A.; Robbins, J. In Vivo and in Vitro Analysis of Cardiac Troponin I Phosphorylation. J. Biol. Chem. 2005, 280, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Baryshnikova, O.K.; Li, M.X.; Sykes, B.D. Modulation of cardiac troponin C function by the cardiac-specific N-terminus of troponin I: Influence of PKA phosphorylation and involvement in cardiomyopathies. J. Mol. Biol. 2008, 375, 735–751. [Google Scholar] [CrossRef] [PubMed]
- Finley, N.; Abbott, M.B.; Abusamhadneh, E.; Gaponenko, V.; Dong, W.; Gasmi-Seabrook, G.; Howarth, J.W.; Rance, M.; Solaro, R.J.; Cheung, H.C.; et al. NMR analysis of cardiac troponin C-troponin I complexes: Effects of phosphorylation. FEBS Lett. 1999, 453, 107–112. [Google Scholar] [CrossRef]
- Sadayappan, S.; Osinska, H.; Klevitsky, R.; Lorenz, J.N.; Sargent, M.; Molkentin, J.D.; Seidman, C.E.; Seidman, J.G.; Robbins, J. Cardiac myosin binding protein C phosphorylation is cardioprotective. Proc. Natl. Acad. Sci. USA 2006, 103, 16918–16923. [Google Scholar] [CrossRef] [PubMed]
- Colson, B.A.; Rybakova, I.N.; Prochniewicz, E.; Moss, R.L.; Thomas, D.D. Cardiac myosin binding protein-C restricts intrafilament torsional dynamics of actin in a phosphorylation-dependent manner. Proc. Natl. Acad. Sci. USA 2012, 109, 20437–20442. [Google Scholar] [CrossRef] [PubMed]
- Weith, A.; Sadayappan, S.; Gulick, J.; Previs, M.J.; VanBuren, P.; Robbins, J.; Warshaw, D.M. Unique single molecule binding of cardiac myosin binding protein-C to actin and phosphorylation-dependent inhibition of actomyosin motility requires 17 amino acids of the motif domain. J. Mol. Cell. Cardiol. 2012, 52, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Kooij, V.; Boontje, N.; Zaremba, R.; Jaquet, K.; dos Remedios, C.; Stienen, G.J.M.; van der Velden, J. Protein kinase C alpha and epsilon phosphorylation of troponin and myosin binding protein C reduce Ca2+ sensitivity in human myocardium. Basic Res. Cardiol. 2010, 105, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Tong, C.W.; Gaffin, R.D.; Zawieja, D.C.; Muthuchamy, M. Roles of phosphorylation of myosin binding protein-C and troponin I in mouse cardiac muscle twitch dynamics. J. Physiol. 2004, 558, 927–941. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Liu, Z.; Cao, J.; Ma, Q.; Gao, X.; Wang, Q.; Jin, C.; Zhou, Y.; Wen, L.; Ren, J. GPS 2.1: Enhanced prediction of kinase-specific phosphorylation sites with an algorithm of motif length selection. Protein Eng. Des. Sel. 2011, 24, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Offer, G.; Moos, C.; Starr, R. A new protein of the thick filaments of vertebrate skeletal myofibrils. J. Mol. Biol. 1973, 74, 653–676. [Google Scholar] [CrossRef]
- Squire, J.M.; Luther, P.K.; Knupp, C. Structural evidence for the interaction of C-protein (MyBP-C) with actin and sequence identification of a possible actin-binding domain. J. Mol. Biol. 2003, 331, 713–724. [Google Scholar] [CrossRef]
- Harris, S.P.; Lyons, R.G.; Bezold, K.L. In the thick of it: HCM-causing mutations in myosin binding proteins of the thick filament. Circ. Res. 2011, 108, 751–764. [Google Scholar] [CrossRef] [PubMed]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Goddard, T.D.; Kneller, D.G. Sparky—NMR Assignment and Integration Software. Available online: https://www.cgl.ucsf.edu/home/sparky/ (accessed on 16 December 2017).
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Springer, T.I.; Johns, C.W.; Cable, J.; Lin, B.L.; Sadayappan, S.; Finley, N.L. Calcium-Dependent Interaction Occurs between Slow Skeletal Myosin Binding Protein C and Calmodulin. Magnetochemistry 2018, 4, 1. https://doi.org/10.3390/magnetochemistry4010001
Springer TI, Johns CW, Cable J, Lin BL, Sadayappan S, Finley NL. Calcium-Dependent Interaction Occurs between Slow Skeletal Myosin Binding Protein C and Calmodulin. Magnetochemistry. 2018; 4(1):1. https://doi.org/10.3390/magnetochemistry4010001
Chicago/Turabian StyleSpringer, Tzvia I., Christian W. Johns, Jana Cable, Brian Leei Lin, Sakthivel Sadayappan, and Natosha L. Finley. 2018. "Calcium-Dependent Interaction Occurs between Slow Skeletal Myosin Binding Protein C and Calmodulin" Magnetochemistry 4, no. 1: 1. https://doi.org/10.3390/magnetochemistry4010001