Aspects of Newborn Screening in Isovaleric Acidemia


1
Experimental Pediatrics and Metabolism, Department of General Pediatrics, Neonatology and Pediatric Cardiology, Heinrich Heine University Düsseldorf, Moorenstrasse 5, 40225 Düsseldorf, Germany
2
Department of Obstetrics and Gynecology, Hospital Altötting-Burghausen, Teaching Hospital of the Ludwig-Maximilians-Universität München, Vinzenz-von-Paul-Strasse 10, 84503 Altötting, Germany
*
Author to whom correspondence should be addressed.
Int. J. Neonatal Screen. 2018, 4(1), 7; https://doi.org/10.3390/ijns4010007
Submission received: 17 December 2017
/
Revised: 19 January 2018
/
Accepted: 22 January 2018
/
Published: 29 January 2018
Abstract
:Isovaleric acidemia (IVA), an inborn error of leucine catabolism, is caused by mutations in the isovaleryl-CoA dehydrogenase (IVD) gene, resulting in the accumulation of derivatives of isovaleryl-CoA including isovaleryl (C5)-carnitine, the marker metabolite used for newborn screening (NBS). The inclusion of IVA in NBS programs in many countries has broadened knowledge of the variability of the condition, whereas prior to NBS, two distinct clinical phenotypes were known, an “acute neonatal” and a “chronic intermittent” form. An additional biochemically mild and potentially asymptomatic form of IVA and its association with a common missense mutation, c.932C>T (p.A282V), was discovered in subjects identified through NBS. Deficiency of short/branched chain specific acyl-CoA dehydrogenase (2-methylbutyryl-CoA dehydrogenase), a defect of isoleucine degradation whose clinical significance remains unclear, also results in elevated C5-carnitine, and may therefore be detected by NBS for IVA. Treatment strategies for the long-term management of symptomatic IVA comprise the prevention of catabolism, dietary restriction of natural protein or leucine intake, and supplementation with l-carnitine and/or l-glycine. Recommendations on how to counsel and manage individuals with the mild phenotype detected by NBS are required.
1. Introduction
Newborn screening (NBS) for organic acidurias such as isovaleric acidemia (IVA) has been a matter of debate [1,2,3]. Yet, IVA (OMIM #243500) has been included in many NBS programs, most recently in the United Kingdom [4] (Table 1).
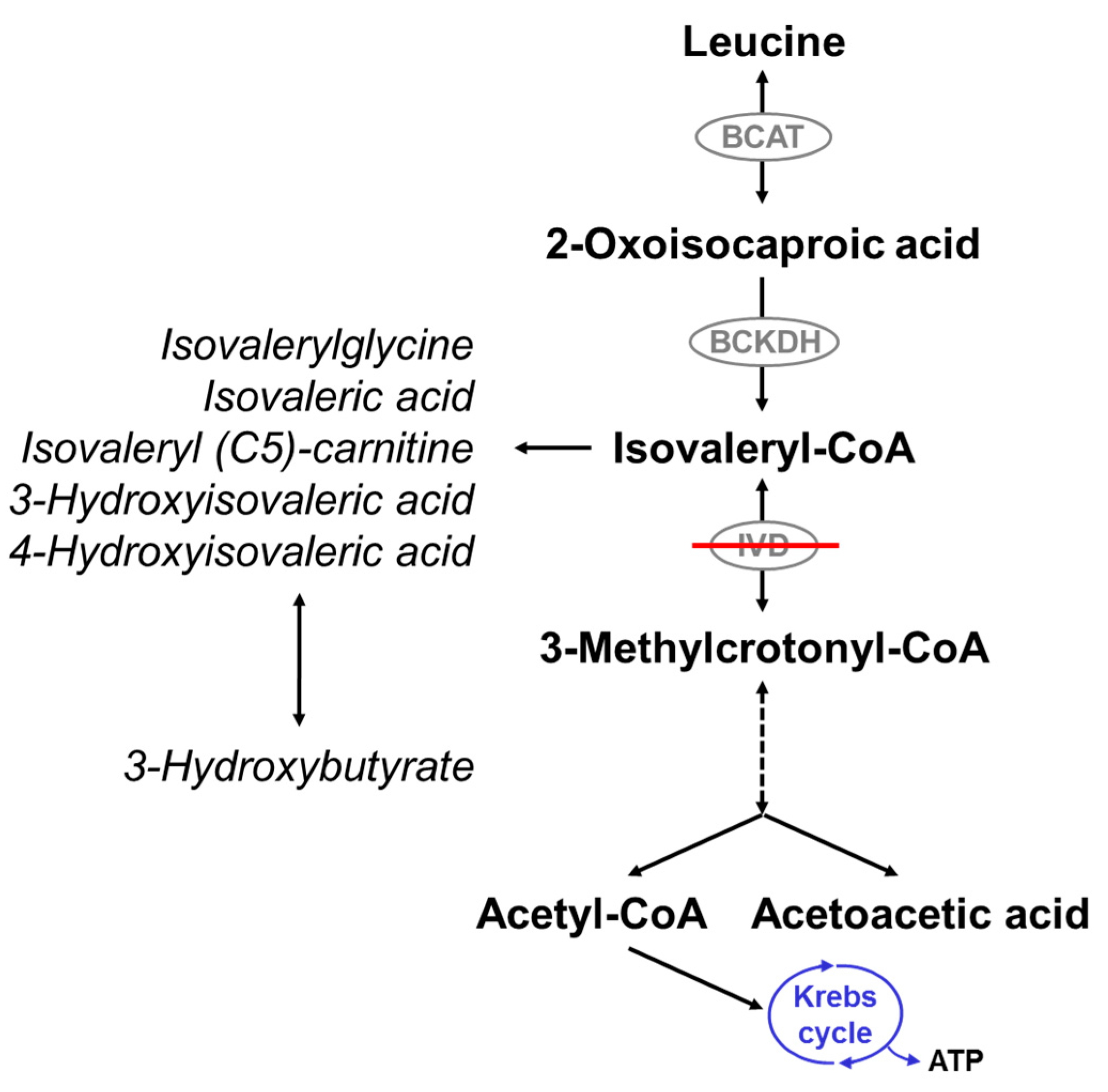
IVA is due to a defect of isovalery-CoA dehydrogenase (IVD; Mendelian Inheritance in Man [MIM] #607036; enzyme commission [EC] 1.3.8.4), an acyl-CoA dehydrogenase (ACAD) catalyzing the third step in the intramitochondrial breakdown of leucine (Figure 1). It is caused by mutations in the isovaleryl-CoA dehydrogenase (IVD) gene and is inherited as an autosomal recessive trait. More than 60 disease-causing mutations in the IVD gene have been described. The majority are point mutations, but splice site mutations, nonsense mutations, missense mutations, deletions, and insertions have also been described [18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33].
Deficiency of IVD results in an accumulation of derivatives of isovaleryl-coenzyme A (CoA), the metabolite before the block, such as isovaleric acid, 3-hydroxyisovaleric acid, isovaleryl (C5)-carnitine, and isovalerylglycine (IVG) (Figure 1). The pathogenesis of the disease is still not fully understood. Mechanisms thought to be involved include the induction of oxidative stress through accumulating metabolites as seen in the rat brain cortex [34], the reduction of Na+, K+-ATPase activity by free isovaleric acid as shown in synaptic membranes from the cerebral cortex in young rats [35], and abnormal cellular growth signaling through activation of the mammalian target of rapamycin complex 1 (mTORC1), as suggested from studies with human IVD deficient cells [36].
The phenotypical spectrum of IVA is wide and has been further broadened by NBS. Available data suggests that early diagnosis by NBS may improve the clinical outcome of IVA, as supported by reports of less frequent relapsing episodes of metabolic decompensation [37] and short-term improvement of neurodevelopmental symptoms [37,38], even though studies on the long-term outcome of screened patients are still lacking.
Two clinical phenotypes have been observed in unscreened patients. They may become symptomatic within the first days or weeks of life, presenting with poor feeding or vomiting and severe metabolic acidosis accompanied by neurological signs including lethargy, potentially progressing to coma or death [39]. Alternatively, patients may present later in childhood with acute acidotic episodes often triggered by catabolic stress such as intercurrent illness [32,40]. In 1966, IVA was first reported by Tanaka and coworkers [41], who described two siblings of preschool age with recurrent episodes of vomiting and lethargy and an unusual odor of “sweaty feet”, in whom a massive urinary excretion of isovalerylglycine and other metabolites of isovaleryl-CoA were detected using gas chromatography (GC) and mass spectrometry (MS) [42].
Besides, a third distinct phenotype of IVA has been identified by NBS [30]. Individuals show a less pronounced accumulation of isovaleric acid and its derivatives than clinically detected patients and present with a potentially asymptomatic phenotype. So far, no severe metabolic crises have been reported in these subjects. A certain missense mutation, c.932C>T (p.A282V), in either a homozygous or compound heterozygous state, is associated with this “mild” form of IVA [30]. However, the long-term outcome of screened individuals with different types of IVA still needs to be defined.
2. IVA Newborn Screening: Diagnosis, Birth Prevalence and Differential Diagnosis
Introduction of tandem mass spectrometry (MS/MS) for NBS allowed the detection of elevated levels of C5-carnitine in dried blood spots [43]. In urine, the elevation of IVG confirms the metabolic diagnosis of IVA [44]. The first countries that introduced IVA to their NBS programs were Australia, where IVA was included in the New South Wales NBS program in 1998 [45], and Germany, where it was first included in the Bavarian NBS program in 1999 [8]. Since then, it has been implemented in national NBS programs in about 30 countries worldwide (Table 1), and most recently (2015), in England and Wales. In addition, in the absence of government-run screening programs, NBS for IVA is offered on a private basis in some countries, e.g., South Africa and Lebanon.
Using a data set of 1.6 million newborns from Germany, the birth prevalence of IVA was calculated to be 1 in 67,000 [8]. Prevalences from other countries were reported to be lower, such as 1 in 660,000 in Taiwan [46] or 1 in 105,000 in Portugal [47]. In Australia, the prevalence of IVA has been shown to be 1 in 775,600 in the unscreened population and 1 in 230,750 in a screened cohort [48]. Similarly, an analysis of available evidence by Dionisi-Vici et al. showed a more than four times higher incidence of IVA in the screened population as compared to clinical diagnosis [37], suggesting that the phenotypic spectrum of IVA detected by NBS is different and may include individuals that would not have presented clinically.
Because C5-carnitine represents several isomers, such as isovalerylcarnitine, 2-methylbutyrylcarnitine, or pivaloylcarnitine, elevated levels detected in NBS may account for several differential diagnoses of IVA, including short/branched chain specific acyl-CoA dehydrogenase deficiency (SBCADD) (also called 2-methylbutyryl-CoA dehydrogenase deficiency [2-MBCD] or 2-methylbutyrylglycinuria). SBCADD, an autosomal recessive condition caused by an error in the degradation pathway of l-isoleucine [49], is detected by IVA NBS programs because it shows elevated 2-methylbutyryl (C5)-carnitine, which has the same mass to charge on MS/MS as isovalerylcarnitine [49]. The first patient with SBCADD was reported in 1999 [50]. The ACADSB gene structure was described in 2000 [51], and several mutations in this gene have been reported [49,52,53]. In individuals with this disease, urine analysis reveals marked elevations of 2-methylbutyrylglycine [51,54]. Symptoms reported in the literature range from developmental delay, seizures, and autism to neonatal crises [49,55], and protein restriction and supplementation with l-carnitine have been suggested for treatment [49,55,56]. However, most patients seem to be asymptomatic despite metabolic abnormalities. While the frequency of SBCADD was found to be higher and its variability greater following the introduction of MS/MS into NBS programs, the last report focusing on this condition was published in 2013 [49]. There is little information on the long-term clinical outcome of individuals with SBCADD, but overall, this condition is assumed to be benign.
With NBS becoming an important part of pediatric preventive strategies worldwide, several diagnostic pitfalls have come to attention. Pivaloylcarnitine, a derivative of antibiotics containing pivalic acid, can be mistaken for isovalerylcarnitine in NBS blood samples, and treatment of mothers with these antibiotics before delivery has been blamed for a number of false positive NBS results [57,58,59,60,61]. Sivelestat, a neutrophil elastase inhibitor used to treat acute respiratory distress syndrome, also contains pivalic acid and can lead to false positive NBS results for IVA [62]. In order to differentiate SBCADD from IVA and to exclude interference from antibiotics, urine acylglycine analysis and/or quantitative organic acid analysis are performed. Furthermore, several strategies for second-tier testing in dried blood spots have been developed, including stable isotope dilution MS/MS analysis to determine isovalerylglycine [63,64] and ultra-performance liquid chromatography (UPLC)-MS/MS analysis of C5-carnitines [59,65].
3. Emerging Spectrum of the Disease
3.1. Clinical Presentation
Today, an increasing number of patients are diagnosed by NBS using MS/MS before the onset of symptoms. A large study analyzing 1.6 million newborns from Germany found that nearly half of the cases detected by NBS (11/24) were defined as “metabolically mild or intermediate” [8]. It may be assumed that individuals with the mild phenotype identified by NBS may remain asymptomatic throughout their lives [30]. This hypothesis is supported by family studies identifying asymptomatic individuals with biochemical evidence of IVA and genotypes identical to their younger siblings identified by NBS [30] and by the reported increase in the prevalence of organic acidurias in screened cohorts as compared to clinical ascertainment [32,37,48,66,67].
Potentially life-threatening episodes of metabolic acidosis associated with lethargy or impaired consciousness—often but not always following situations of catabolic stress—are common in IVA. The first life-threatening catabolic episode in patients with the acute form usually occurs by the end of the first week of life (“acute neonatal form”) [38,39,66]. Depending on the time of NBS sampling and turnaround time, patients with this form may even present clinically before the NBS result is reported [32]. Early symptoms are nonspecific. Newborns are feeding poorly and present with emesis often associated with dehydration, lethargy, and sometimes seizures [39,66,68]. Metabolic acidosis with an elevated anion gap representing the accumulation of organic acids, secondary hyperammonemia, and hyperglycemia or hypoglycemia are reported as laboratory findings [39,66,67]. An unpleasant smell of “sweaty feet” is characteristic for IVA and can be noticed in acutely sick infants [39], but may not be recognizable in otherwise well patients. If left untreated, the clinical condition can worsen to coma and ultimately death [39]. The incidence of acute life-threatening catabolic episodes is highest in early infancy and decreases with age. Still, patients may become sick later in childhood with intermittent bouts of illness with vomiting and metabolic acidosis [39,66,67], often precipitated by infections or other physiologic stressors [39]. Interestingly, in a study of 21 children with symptomatic IVA, no such event was observed after nine years of age [39], although acute metabolic decompensations have been reported in adult life [69,70].
Apart from life-threatening metabolic crises, unscreened patients may first present later in life with neurological symptoms and cognitive impairment (“chronic intermittent presentation”) [39,66]. Symptoms are often nonspecific and include feeding difficulties, vomiting, and failure to thrive and/or developmental delay and cognitive impairment. Neurological manifestations of IVA relate to EEG abnormalities or seizures and motor dysfunction [71,72]. Similar to findings in other organic acidurias [73], there have been single reports of patients with pancreatitis [39,74,75,76]. Liver fibrosis [28] and, most recently, optic nerve atrophy [71] have also been associated with IVA. Pregnancies of affected women have been reported as uneventful [77,78,79].
3.2. Management/Treatment
Long-term treatment strategies aim to: (1) reduce the production of toxic metabolites by the restriction of protein or leucine intake; and to (2) enhance the conjugation of potentially toxic free isovaleric acid to its non-toxic conjugates isovalerylcarnitine and isovalerylglycine, which are excreted by the kidneys via supplementation of l-carnitine and/or l-glycine [44,80,81].
Many patients with a clinically symptomatic type of IVA follow a protein-restricted diet to reduce their intake of leucine and limit the production of toxic metabolites. In order to cover age-appropriate amounts needed for normal growth and to avoid malnutrition, leucine-free amino acid supplements enriched with micronutrients may be needed. However, wide differences in actual dietary practices in IVA have been documented [82]. l-carnitine is usually given at a dosage of 100 mg/kg × day in three doses. Because it remains unknown if subjects with the mild IVA phenotype detected by NBS might experience metabolic crises or long-term neurological manifestations, these individuals may be advised to take l-carnitine, although it is unclear whether it prevents metabolic crises. A low dosage of 30 to 50 mg/kg × day has been proposed for these individuals [44]. l-glycine may be omitted from long-term treatment, especially in individuals with the mild phenotype. If given, the dosage is usually between 100 and 300 mg/kg × day in three doses [56].
During intercurrent illness, the production of isovaleryl-CoA might be increased due to a higher rate of breakdown of the endogenous protein. Therefore, the prevention of catabolic episodes is crucial. Anabolizing measures including oral glucose polymer solutions or high-dose glucose and potentially lipid infusions may be necessary to secure an adequate energy supply [44,83]. A short-term decrease of protein intake should also be part of the acute treatment protocol [44,83,84]. In order to prevent the accumulation of toxic metabolites, increased doses of l-carnitine (up to 400 mg/kg × day) [83,84] and l-glycine (by 50% to 100%) [85] have been recommended. As for other classic organic acidurias, treatment with N-carbaglutamate has been suggested for the treatment of acute neonatal hyperammonemia in IVA [86]. As it cannot be entirely excluded that periods of illness might trigger unfavorable effects in patients with the mild type of IVA, individuals with this condition should also be counseled to follow an emergency protocol and to increase the intake of l-carnitine and energy during febrile illnesses.
3.3. Outcome
Mortality is highest in patients with an early clinical onset: an analysis of 155 published patients with symptomatic IVA showed a mortality rate of about one-third during the initial metabolic crisis in patients diagnosed within the first five weeks of life, whereas patients diagnosed thereafter had a low mortality rate of only 3% [39]. Patients with an early presentation who died during the initial catabolic episode had a significantly earlier onset of symptoms than patients who survived this initial catabolic episode.
Early initiation of treatment in IVA, i.e., starting therapy during the first weeks of life, was shown to decrease the frequency of severe ketoacidotic crises and was associated with an overall good clinical outcome [85]. Patients diagnosed by NBS often appear asymptomatic [87,88], and early diagnosis of IVA has been reported to correlate with a good neurocognitive outcome: an extensive review of published patients found that in patients diagnosed in the first five weeks of life, 85% had an unremarkable neurocognitive outcome as opposed to only 45% of patients who were diagnosed after the fifth week of life [39]. Similarly, in a South African population, all patients diagnosed within the neonatal period, but only 43% of patients diagnosed thereafter, had a normal neurocognitive outcome [67].
However, not all studies available to date appear to support the relevance of early diagnosis: an analysis of 52 patients from the European Registry and Network for Intoxication Type Metabolic Diseases (E-IMD) showed a statistical trend for normal development in patients diagnosed by NBS as compared with patients who were diagnosed after the onset of symptoms. This trend disappeared after the omission of patients with the “mild” phenotype from the analysis [38]. Cognitive function in a series of 16 Spanish patients was shown to be within the normal range in both patients diagnosed clinically and patients detected by NBS [32]. Overall, neurological sequelae and organ manifestations in IVA have been shown to be less common as compared with other classic organic acidurias [39,71,89].
Whether patients with a biochemically mild type of IVA may develop any clinical symptoms under certain circumstances remains open, since long-term data on the outcome of individuals diagnosed by NBS are still lacking.
4. Conclusions
IVA Newborn Screening—Outlook and Challenges
The possibility of pre-symptomatic diagnosis through NBS and the apparent benefit that has been demonstrated for patients diagnosed and treated early [39] make IVA an ideal candidate for NBS programs. An additional “mild” form of IVA with only slight biochemical abnormalities and a potentially asymptomatic phenotype has been discovered by NBS. Still, there is little information on the long-term outcome of patients with this mild type of the disease, and it is not known whether these patients are actually at risk for severe catabolic episodes. With IVA being included as a target disorder of NBS programs in a growing number of countries worldwide, more of these individuals will be identified. Overall, longitudinal studies of screened individuals with IVA are needed to allow for a better understanding of the long-term outcome and clinical spectrum including the “mild” phenotype and to provide the basis for management recommendations and counseling. Results may also allow considering the adjustment of NBS cut-off levels in order to not detect individuals with benign variants.
Acknowledgments
We would like to thank Violeta Anastasovska, Laila Bastaki, François Boemer, Jim Bonham, Leifur Franzson, Viktor Kožich, and Maximilian Zeyda for sharing their experience with their national newborn screening programs.
Author Contributions
All authors of this work have contributed to the planning, writing, and revising this paper. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hoffmann, G.F.; Lindner, M.; Loeber, J.G. 50 years of newborn screening. J. Inherit. Metab. Dis. 2014, 37, 163–164. [Google Scholar] [CrossRef] [PubMed]
- Wilcken, B. Medicine. Newborn screening: Gaps in the evidence. Science 2013, 342, 197–198. [Google Scholar] [CrossRef] [PubMed]
- Wilcken, B.; Wiley, V. Fifty years of newborn screening. J. Paediatr. Child Health 2015, 51, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, N. Newborns in England are screened for four extra genetic conditions. BMJ 2015, 350, h10. [Google Scholar] [CrossRef] [PubMed]
- Therrell, B.L.; Padilla, C.D.; Loeber, J.G.; Kneisser, I.; Saadallah, A.; Borrajo, G.J.; Adams, J. Current status of newborn screening worldwide: 2015. Semin. Perinatol. 2015, 39, 171–187. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Knoll, D.; de Hora, M.; Kyle, C.; Glamuzina, E.; Webster, D. The risk of fatty acid oxidation disorders and organic acidemias in children with normal newborn screening. JIMD Rep. 2017, 35, 53–58. [Google Scholar] [PubMed]
- Sygdomme Som Indgår I Screeningen. Available online: https://www.ssi.dk/Diagnostik/Center%20for%20Neonatal%20Screening/Sygdomme%20som%20indgaer%20i%20screeningen.aspx (accessed on 22 February 2017).
- Ensenauer, R.; Fingerhut, R.; Maier, E.M.; Polanetz, R.; Olgemoller, B.; Roschinger, W.; Muntau, A.C. Newborn screening for isovaleric acidemia using tandem mass spectrometry: Data from 1.6 million newborns. Clin. Chem. 2011, 57, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Federal Ministry of Health and Social Security. Announcement of a Decision of the Federal Joint Committee on an Amendment to the Guidelines of the Federal Committee of Physicians and Health Insurance Funds on the Early Detection of Diseases in Children up to the Age of 6 (Children’s Guidelines) for the Introduction of Extended Neonatal Screening. 2005. Available online: https://www.g-ba.de/downloads/39-261-170/2004-12-21-Kinder-TMS.pdf (accessed on 29 January 2018). (In German).
- Javaher, P.; Nyoungui, E.; Kaariainen, H.; Kristoffersson, U.; Nippert, I.; Sequeiros, J.; Schmidtke, J. Genetic screening in Europe. Public Health Genom. 2010, 13, 524–537. [Google Scholar] [CrossRef] [PubMed]
- Newborn Screening in Canada Status Report. Available online: https://www.raredisorders.ca/content/uploads/Canada-NBS-status-updated-Sept.-3-2015.pdf (accessed on 22 February 2017).
- Patchchat 6: The Case for Newborn Screening in South Africa: A Personal Perspective. Available online: https://www.ampath.co.za/pathchat/newborn-screening/ (accessed on 22 February 2017).
- Abdel-Hamid, M.; Tisocki, K.; Sharaf, L.; Ramadan, D. Development, validation and application of tandem mass spectrometry for screening of inborn metabolic disorders in Kuwaiti infants. Med. Princ. Pract. 2007, 16, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Khneisser, I.; Adib, S.; Assaad, S.; Megarbane, A.; Karam, P. Cost-benefit analysis: Newborn screening for inborn errors of metabolism in Lebanon. J. Med. Screen. 2015, 22, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Krotoski, D.; Namaste, S.; Raouf, R.K.; El Nekhely, I.; Hindi-Alexander, M.; Engelson, G.; Hanson, J.W.; Howell, R.R.; Committee, M.N.S. Conference report: Second conference of the Middle East and North Africa newborn screening initiative: Partnerships for sustainable newborn screening infrastructure and research opportunities. Genet. Med. 2009, 11, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Lindner, M.; Abdoh, G.; Fang-Hoffmann, J.; Shabeck, N.; Al-Sayrafi, M.; Al-Janahi, M.; Ho, S.; Abdelrahman, M.O.; Ben-Omran, T.; Bener, A.; et al. Implementation of extended neonatal screening and a metabolic unit in the state of Qatar: Developing and optimizing strategies in cooperation with the neonatal screening center in Heidelberg. J. Inherit. Metab. Dis. 2007, 30, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Al Hosani, H.; Salah, M.; Osman, H.M.; Farag, H.M.; El-Assiouty, L.; Saade, D.; Hertecant, J. Expanding the comprehensive national neonatal screening programme in the United Arab Emirates from 1995 to 2011. East. Mediterr. Health J. 2014, 20, 17–23. [Google Scholar] [PubMed]
- Mohsen, A.W.; Anderson, B.D.; Volchenboum, S.L.; Battaile, K.P.; Tiffany, K.; Roberts, D.; Kim, J.J.; Vockley, J. Characterization of molecular defects in isovaleryl-CoA dehydrogenase in patients with isovaleric acidemia. Biochemistry 1998, 37, 10325–10335. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Ikeda, Y.; Matsubara, Y.; Hyman, D.B. Molecular basis of isovaleric acidemia and medium-chain acyl-CoA dehydrogenase deficiency. Enzyme 1987, 38, 91–107. [Google Scholar] [CrossRef] [PubMed]
- Vockley, J.; Parimoo, B.; Nagao, M.; Tanaka, K. Identification of the molecular defects responsible for the various genotypes of isovaleric acidemia. Prog. Clin. Biol. Res. 1992, 375, 533–540. [Google Scholar] [PubMed]
- Vockley, J.; Parimoo, B.; Tanaka, K. Molecular characterization of four different classes of mutations in the isovaleryl-CoA dehydrogenase gene responsible for isovaleric acidemia. Am. J. Hum. Genet. 1991, 49, 147–157. [Google Scholar] [PubMed]
- Vockley, J.; Rogan, P.K.; Anderson, B.D.; Willard, J.; Seelan, R.S.; Smith, D.I.; Liu, W. Exon skipping in IVD RNA processing in isovaleric acidemia caused by point mutations in the coding region of the IVD gene. Am. J. Hum. Genet. 2000, 66, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.D.; Wang, C.H.; Lee, C.C.; Lai, C.C.; Tsai, Y.; Tsai, F.J. Genetic mutation profile of isovaleric acidemia patients in Taiwan. Mol. Genet. Metab. 2007, 90, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.W.; Lee, D.H.; Vockley, J.; Kim, N.D.; Lee, Y.K.; Ki, C.S. Different spectrum of mutations of isovaleryl-CoA dehydrogenase (IVD) gene in Korean patients with isovaleric acidemia. Mol. Genet. Metab. 2007, 92, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Vatanavicharn, N.; Liammongkolkul, S.; Sakamoto, O.; Sathienkijkanchai, A.; Wasant, P. Phenotypic and mutation spectrums of Thai patients with isovaleric acidemia. Pediatr. Int. 2011, 53, 990–994. [Google Scholar] [CrossRef] [PubMed]
- Hertecant, J.L.; Ben-Rebeh, I.; Marah, M.A.; Abbas, T.; Ayadi, L.; Ben Salem, S.; Al-Jasmi, F.A.; Al-Gazali, L.; Al-Yahyaee, S.A.; Ali, B.R. Clinical and molecular analysis of isovaleric acidemia patients in the United Arab Emirates reveals remarkable phenotypes and four novel mutations in the IVD gene. Eur. J. Med. Genet. 2012, 55, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Bei, F.; Sun, J.H.; Yu, Y.G.; Jia, J.; Zheng, Z.J.; Fu, Q.H.; Cai, W. Two novel isovaleryl-CoA dehydrogenase gene mutations in a Chinese infant. Gene 2013, 524, 396–400. [Google Scholar] [CrossRef] [PubMed]
- Ozgul, R.K.; Karaca, M.; Kilic, M.; Kucuk, O.; Yucel-Yilmaz, D.; Unal, O.; Hismi, B.; Aliefendioglu, D.; Sivri, S.; Tokatli, A.; et al. Phenotypic and genotypic spectrum of Turkish patients with isovaleric acidemia. Eur. J. Med. Genet. 2014, 57, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Kaya, N.; Colak, D.; Al-Bakheet, A.; Al-Younes, B.; Tulbah, S.; Daghestani, M.; Al-Mutairi, F.; Al-Amoudi, M.; Al-Odaib, A.; Al-Aqeel, A.I. Identification of a novel IVD mutation in a consanguineous family with isovaleric acidemia. Gene 2013, 513, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Ensenauer, R.; Vockley, J.; Willard, J.M.; Huey, J.C.; Sass, J.O.; Edland, S.D.; Burton, B.K.; Berry, S.A.; Santer, R.; Grunert, S.; et al. A common mutation is associated with a mild, potentially asymptomatic phenotype in patients with isovaleric acidemia diagnosed by newborn screening. Am. J. Hum. Genet. 2004, 75, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.M.; Lee, B.H.; Kim, G.H.; Kim, Y.M.; Choi, J.H.; Yoo, H.W. Chronic intermittent form of isovaleric aciduria in a 2-year-old boy. Korean J. Pediatr. 2013, 56, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Couce, M.L.; Aldamiz-Echevarria, L.; Bueno, M.A.; Barros, P.; Belanger-Quintana, A.; Blasco, J.; Garcia-Silva, M.T.; Marquez-Armenteros, A.M.; Vitoria, I.; Vives, I.; et al. Genotype and phenotype characterization in a Spanish cohort with isovaleric acidemia. J. Hum. Genet. 2017, 62, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, O.; Arai-Ichinoi, N.; Mitsubuchi, H.; Chinen, Y.; Haruna, H.; Maruyama, H.; Sugawara, H.; Kure, S. Phenotypic variability and newly identified mutations of the IVD gene in Japanese patients with isovaleric acidemia. Tohoku J. Exp. Med. 2015, 236, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Solano, A.F.; Leipnitz, G.; De Bortoli, G.M.; Seminotti, B.; Amaral, A.U.; Fernandes, C.G.; Latini, A.S.; Dutra-Filho, C.S.; Wajner, M. Induction of oxidative stress by the metabolites accumulating in isovaleric acidemia in brain cortex of young rats. Free Radic. Res. 2008, 42, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.A.; Balestro, F.; Grando, V.; Wajner, M. Isovaleric acid reduces Na+, K+-ATPase activity in synaptic membranes from cerebral cortex of young rats. Cell. Mol. Neurobiol. 2007, 27, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Schriever, S.C.; Deutsch, M.J.; Adamski, J.; Roscher, A.A.; Ensenauer, R. Cellular signaling of amino acids towards mTORC1 activation in impaired human leucine catabolism. J. Nutr. Biochem. 2013, 24, 824–831. [Google Scholar] [CrossRef] [PubMed]
- Dionisi-Vici, C.; Deodato, F.; Roschinger, W.; Rhead, W.; Wilcken, B. ‘Classical’ organic acidurias, propionic aciduria, methylmalonic aciduria and isovaleric aciduria: Long-term outcome and effects of expanded newborn screening using tandem mass spectrometry. J. Inherit. Metab. Dis. 2006, 29, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Heringer, J.; Valayannopoulos, V.; Lund, A.M.; Wijburg, F.A.; Freisinger, P.; Baric, I.; Baumgartner, M.R.; Burgard, P.; Burlina, A.B.; Chapman, K.A.; et al. Impact of age at onset and newborn screening on outcome in organic acidurias. J. Inherit. Metab. Dis. 2016, 39, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Grunert, S.C.; Wendel, U.; Lindner, M.; Leichsenring, M.; Schwab, K.O.; Vockley, J.; Lehnert, W.; Ensenauer, R. Clinical and neurocognitive outcome in symptomatic isovaleric acidemia. Orphanet J. Rare Dis. 2012, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K. Isovaleric acidemia: Personal history, clinical survey and study of the molecular basis. Prog. Clin. Biol. Res. 1990, 321, 273–290. [Google Scholar] [PubMed]
- Tanaka, K.; Budd, M.A.; Efron, M.L.; Isselbacher, K.J. Isovaleric acidemia: A new genetic defect of leucine metabolism. Proc. Natl. Acad. Sci. USA 1966, 56, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Isselbacher, K.J. The isolation and identification of N-isovalerylglycine from urine of patients with isovaleric acidemia. J. Biol. Chem. 1967, 242, 2966–2972. [Google Scholar] [PubMed]
- Fingerhut, R.; Olgemoller, B. Newborn screening for inborn errors of metabolism and endocrinopathies: An update. Anal. Bioanal. Chem. 2009, 393, 1481–1497. [Google Scholar] [CrossRef] [PubMed]
- Vockley, J.; Ensenauer, R. Isovaleric acidemia: New aspects of genetic and phenotypic heterogeneity. Am. J. Med. Genet. C Semin. Med. Genet. 2006, 142c, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Estrella, J.; Wilcken, B.; Carpenter, K.; Bhattacharya, K.; Tchan, M.; Wiley, V. Expanded newborn screening in New South Wales: Missed cases. J. Inherit. Metab. Dis. 2014, 37, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Niu, D.M.; Chien, Y.H.; Chiang, C.C.; Ho, H.C.; Hwu, W.L.; Kao, S.M.; Chiang, S.H.; Kao, C.H.; Liu, T.T.; Chiang, H.; et al. Nationwide survey of extended newborn screening by tandem mass spectrometry in Taiwan. J. Inherit. Metab. Dis. 2010, 33, S295–S305. [Google Scholar] [CrossRef] [PubMed]
- Vilarinho, L.; Rocha, H.; Sousa, C.; Marcao, A.; Fonseca, H.; Bogas, M.; Osorio, R.V. Four years of expanded newborn screening in Portugal with tandem mass spectrometry. J. Inherit. Metab. Dis. 2010, 33 (Suppl. 3), S133–S138. [Google Scholar] [CrossRef] [PubMed]
- Wilcken, B.; Haas, M.; Joy, P.; Wiley, V.; Bowling, F.; Carpenter, K.; Christodoulou, J.; Cowley, D.; Ellaway, C.; Fletcher, J.; et al. Expanded newborn screening: Outcome in screened and unscreened patients at age 6 years. Pediatrics 2009, 124, e241–e248. [Google Scholar] [CrossRef] [PubMed]
- Van Calcar, S.C.; Baker, M.W.; Williams, P.; Jones, S.A.; Xiong, B.; Thao, M.C.; Lee, S.; Yang, M.K.; Rice, G.M.; Rhead, W.; et al. Prevalence and mutation analysis of short/branched chain acyl-CoA dehydrogenase deficiency (SBCADD) detected on newborn screening in Wisconsin. Mol. Genet. Metab. 2013, 110, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Gibson, K.M.; Sacks, M.; Kiss, D.; Pohowalla, P.; Linck, L.; Steiner, R.D.; Burlingame, T. 2-methylbutyrylglycinuria in a neonate with CNS dysfunction: Evidence for isolated 2-methylbutyryl-CoA dehydrogenase deficiency, an inborn error of L-leucine metabolism. J. Inherit. Metab. Dis. 1999, 22 (Suppl. 1), 16. [Google Scholar]
- Andresen, B.S.; Christensen, E.; Corydon, T.J.; Bross, P.; Pilgaard, B.; Wanders, R.J.; Ruiter, J.P.; Simonsen, H.; Winter, V.; Knudsen, I.; et al. Isolated 2-methylbutyrylglycinuria caused by short/branched-chain acyl-CoA dehydrogenase deficiency: Identification of a new enzyme defect, resolution of its molecular basis, and evidence for distinct acyl-CoA dehydrogenases in isoleucine and valine metabolism. Am. J. Hum. Genet. 2000, 67, 1095–1103. [Google Scholar] [PubMed]
- Alfardan, J.; Mohsen, A.W.; Copeland, S.; Ellison, J.; Keppen-Davis, L.; Rohrbach, M.; Powell, B.R.; Gillis, J.; Matern, D.; Kant, J.; et al. Characterization of new ACADSB gene sequence mutations and clinical implications in patients with 2-methylbutyrylglycinuria identified by newborn screening. Mol. Genet. Metab. 2010, 100, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Madsen, P.P.; Kibaek, M.; Roca, X.; Sachidanandam, R.; Krainer, A.R.; Christensen, E.; Steiner, R.D.; Gibson, K.M.; Corydon, T.J.; Knudsen, I.; et al. Short/branched-chain acyl-CoA dehydrogenase deficiency due to an IVS3+3A>G mutation that causes exon skipping. Hum. Genet. 2006, 118, 680–690. [Google Scholar] [CrossRef] [PubMed]
- Gibson, K.M.; Burlingame, T.G.; Hogema, B.; Jakobs, C.; Schutgens, R.B.; Millington, D.; Roe, C.R.; Roe, D.S.; Sweetman, L.; Steiner, R.D.; et al. 2-methylbutyryl-coenzyme A dehydrogenase deficiency: A new inborn error of L-isoleucine metabolism. Pediatr. Res. 2000, 47, 830–833. [Google Scholar] [CrossRef] [PubMed]
- Kanavin, O.J.; Woldseth, B.; Jellum, E.; Tvedt, B.; Andresen, B.S.; Stromme, P. 2-methylbutyryl-CoA dehydrogenase deficiency associated with autism and mental retardation: A case report. J. Med. Case Rep. 2007, 1, 98. [Google Scholar] [CrossRef] [PubMed]
- Knerr, I.; Weinhold, N.; Vockley, J.; Gibson, K.M. Advances and challenges in the treatment of branched-chain amino/keto acid metabolic defects. J. Inherit. Metab. Dis. 2012, 35, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Abdenur, J.E.; Chamoles, N.A.; Guinle, A.E.; Schenone, A.B.; Fuertes, A.N. Diagnosis of isovaleric acidaemia by tandem mass spectrometry: False positive result due to pivaloylcarnitine in a newborn screening programme. J. Inherit. Metab. Dis. 1998, 21, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Boemer, F.; Schoos, R.; de Halleux, V.; Kalenga, M.; Debray, F.G. Surprising causes of C5-carnitine false positive results in newborn screening. Mol. Genet. Metab. 2014, 111, 52–54. [Google Scholar] [CrossRef] [PubMed]
- Cloppenborg, T.; Janzen, N.; Wagner, H.; Steuerwald, U.; Peter, M.; Das, A. Application of a second-tier newborn screening assay for C5 isoforms. JIMD Rep. 2014, 13, 23–26. [Google Scholar] [PubMed]
- Minkler, P.E.; Stoll, M.S.K.; Ingalls, S.T.; Hoppel, C.L. Selective and accurate C5 acylcarnitine quantitation by UHPLC-MS/MS: Distinguishing true isovaleric acidemia from pivalate derived interference. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2017, 1061–1062, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Fukuda, S.; Yamada, K.; Hasegawa, Y.; Takahashi, T.; Purevsuren, J.; Yamaguchi, S. Clinical features of carnitine deficiency secondary to pivalate-conjugated antibiotic therapy. J. Pediatr. 2016, 173, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Kobayashi, H.; Bo, R.; Takahashi, T.; Hasegawa, Y.; Nakamura, M.; Ishige, N.; Yamaguchi, S. Elevation of pivaloylcarnitine by sivelestat sodium in two children. Mol. Genet. Metab. 2015, 116, 192–194. [Google Scholar] [CrossRef] [PubMed]
- Shigematsu, Y.; Hata, I.; Tajima, G. Useful second-tier tests in expanded newborn screening of isovaleric acidemia and methylmalonic aciduria. J. Inherit. Metab. Dis. 2010, 33, S283–S288. [Google Scholar] [CrossRef] [PubMed]
- Shigematsu, Y.; Hata, I.; Tanaka, Y. Stable-isotope dilution measurement of isovalerylglycine by tandem mass spectrometry in newborn screening for isovaleric acidemia. Clin. Chim. Acta 2007, 386, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Janzen, N.; Steuerwald, U.; Sander, S.; Terhardt, M.; Peter, M.; Sander, J. UPLC-MS/MS analysis of C5-acylcarnitines in dried blood spots. Clin. Chim. Acta 2013, 421, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Kolker, S.; Cazorla, A.G.; Valayannopoulos, V.; Lund, A.M.; Burlina, A.B.; Sykut-Cegielska, J.; Wijburg, F.A.; Teles, E.L.; Zeman, J.; Dionisi-Vici, C.; et al. The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 1: The initial presentation. J. Inherit. Metab. Dis. 2015, 38, 1041–1045. [Google Scholar] [CrossRef] [PubMed]
- Dercksen, M.; Duran, M.; Ijlst, L.; Mienie, L.J.; Reinecke, C.J.; Ruiter, J.P.; Waterham, H.R.; Wanders, R.J. Clinical variability of isovaleric acidemia in a genetically homogeneous population. J. Inherit. Metab. Dis. 2012, 35, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Tokatli, A.; Coskun, T.; Ozalp, I. Isovaleric acidemia. Clinical presentation of 6 cases. Turk. J. Pediatr. 1998, 40, 111–119. [Google Scholar] [PubMed]
- Feinstein, J.A.; O’Brien, K. Acute metabolic decompensation in an adult patient with isovaleric acidemia. South. Med. J. 2003, 96, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Kimmoun, A.; Abboud, G.; Strazeck, J.; Merten, M.; Gueant, J.L.; Feillet, F. Acute decompensation of isovaleric acidemia induced by Graves’ disease. Intensive Care Med. 2008, 34, 2315–2316. [Google Scholar] [CrossRef] [PubMed]
- Kolker, S.; Valayannopoulos, V.; Burlina, A.B.; Sykut-Cegielska, J.; Wijburg, F.A.; Teles, E.L.; Zeman, J.; Dionisi-Vici, C.; Baric, I.; Karall, D.; et al. The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 2: The evolving clinical phenotype. J. Inherit. Metab. Dis. 2015, 38, 1059–1074. [Google Scholar] [CrossRef] [PubMed]
- Sezer, T.; Balci, O. Infantile spasms during acute metabolic decompensation in an infant with isovaleric acidemia. J. Clin. Neurol. 2016, 12, 376–377. [Google Scholar] [CrossRef] [PubMed]
- Marquard, J.; El Scheich, T.; Klee, D.; Schmitt, M.; Meissner, T.; Mayatepek, E.; Oh, J. Chronic pancreatitis in branched-chain organic acidurias—A case of methylmalonic aciduria and an overview of the literature. Eur. J. Pediatr. 2011, 170, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Kahler, S.G.; Sherwood, W.G.; Woolf, D.; Lawless, S.T.; Zaritsky, A.; Bonham, J.; Taylor, C.J.; Clarke, J.T.; Durie, P.; Leonard, J.V. Pancreatitis in patients with organic acidemias. J. Pediatr. 1994, 124, 239–243. [Google Scholar] [CrossRef]
- Mantadakis, E.; Chrysafis, I.; Tsouvala, E.; Evangeliou, A.; Chatzimichael, A. Acute pancreatitis with rapid clinical improvement in a child with isovaleric acidemia. Case Rep. Pediatr. 2013, 2013, 721871. [Google Scholar] [CrossRef] [PubMed]
- Sag, E.; Cebi, A.H.; Kaya, G.; Karaguzel, G.; Cakir, M. A rare cause of recurrent acute pancreatitis in a child: Isovaleric acidemia with novel mutation. Pediatr. Gastroenterol. Hepatol. Nutr. 2017, 20, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Castelnovi, C.; Moseley, K.; Yano, S. Maternal isovaleric acidemia: Observation of distinctive changes in plasma amino acids and carnitine profiles during pregnancy. Clin. Chim. Acta 2010, 411, 2101–2103. [Google Scholar] [CrossRef] [PubMed]
- Habets, D.D.; Schaper, N.C.; Rogozinski, H.; van Spronsen, F.J.; van Rijn, M.; Bierau, J.; Bakker, J.A. Biochemical monitoring and management during pregnancy in patients with isovaleric acidaemia is helpful to prevent metabolic decompensation. JIMD Rep. 2012, 3, 83–89. [Google Scholar] [PubMed]
- Shih, V.E.; Aubry, R.H.; DeGrande, G.; Gursky, S.F.; Tanaka, K. Maternal isovaleric acidemia. J. Pediatr. 1984, 105, 77–78. [Google Scholar] [CrossRef]
- Roe, C.R.; Millington, D.S.; Maltby, D.A.; Kahler, S.G.; Bohan, T.P. L-carnitine therapy in isovaleric acidemia. J. Clin. Investig. 1984, 74, 2290–2295. [Google Scholar] [CrossRef] [PubMed]
- Yudkoff, M.; Cohn, R.M.; Puschak, R.; Rothman, R.; Segal, S. Glycine therapy in isovaleric acidemia. J. Pediatr. 1978, 92, 813–817. [Google Scholar] [CrossRef]
- Pinto, A.; Daly, A.; Evans, S.; Almeida, M.F.; Assoun, M.; Belanger-Quintana, A.; Bernabei, S.; Bollhalder, S.; Cassiman, D.; Champion, H.; et al. Dietary practices in isovaleric acidemia: A European survey. Mol. Genet. Metab. Rep. 2017, 12, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Ogier de Baulny, H. Management and emergency treatments of neonates with a suspicion of inborn errors of metabolism. Semin. Neonatol. 2002, 7, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Prietsch, V.; Lindner, M.; Zschocke, J.; Nyhan, W.L.; Hoffmann, G.F. Emergency management of inherited metabolic diseases. J. Inherit. Metab. Dis. 2002, 25, 531–546. [Google Scholar] [CrossRef] [PubMed]
- Berry, G.T.; Yudkoff, M.; Segal, S. Isovaleric acidemia: Medical and neurodevelopmental effects of long-term therapy. J. Pediatr. 1988, 113, 58–64. [Google Scholar] [CrossRef]
- Kasapkara, C.S.; Ezgu, F.S.; Okur, I.; Tumer, L.; Biberoglu, G.; Hasanoglu, A. N-carbamylglutamate treatment for acute neonatal hyperammonemia in isovaleric acidemia. Eur. J. Pediatr. 2011, 170, 799–801. [Google Scholar] [CrossRef] [PubMed]
- Schulze-Bergkamen, A.; Okun, J.G.; Spiekerkotter, U.; Lindner, M.; Haas, D.; Kohlmuller, D.; Mayatepek, E.; Schulze-Bergkamen, H.; Greenberg, C.R.; Zschocke, J.; et al. Quantitative acylcarnitine profiling in peripheral blood mononuclear cells using in vitro loading with palmitic and 2-oxoadipic acids: Biochemical confirmation of fatty acid oxidation and organic acid disorders. Pediatr. Res. 2005, 58, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Lindner, M.; Gramer, G.; Haege, G.; Fang-Hoffmann, J.; Schwab, K.O.; Tacke, U.; Trefz, F.K.; Mengel, E.; Wendel, U.; Leichsenring, M.; et al. Efficacy and outcome of expanded newborn screening for metabolic diseases—Report of 10 years from South-West Germany. Orphanet J. Rare Dis. 2011, 6, 44. [Google Scholar] [CrossRef] [PubMed]
- Nizon, M.; Ottolenghi, C.; Valayannopoulos, V.; Arnoux, J.B.; Barbier, V.; Habarou, F.; Desguerre, I.; Boddaert, N.; Bonnefont, J.P.; Acquaviva, C.; et al. Long-term neurological outcome of a cohort of 80 patients with classical organic acidurias. Orphanet J. Rare Dis. 2013, 8, 148. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Leucine catabolism pathway. Isovaleryl-CoA dehydrogenase (IVD) catalyzes the degradation of isovaleryl-CoA to 3-methylcrotonyl-CoA. Deficiency of IVD leads to the accumulation of derivatives of isovaleryl-CoA and reduced production of acetyl-CoA and acetoacetate. BCAT: branched-chain amino acid aminotransferase; BCKDH: branched-chain alpha-ketoacid dehydrogenase.
Figure 1.
Leucine catabolism pathway. Isovaleryl-CoA dehydrogenase (IVD) catalyzes the degradation of isovaleryl-CoA to 3-methylcrotonyl-CoA. Deficiency of IVD leads to the accumulation of derivatives of isovaleryl-CoA and reduced production of acetyl-CoA and acetoacetate. BCAT: branched-chain amino acid aminotransferase; BCKDH: branched-chain alpha-ketoacid dehydrogenase.


{kind=link}
Table 1.
Countries with published experience in NBS for IVA.
| Region | Country | Local Specifics | IVA Targeted by NBS Since (as Available) | Reference/Source |
|---|---|---|---|---|
| Asia Pacific | Australia | [5] | ||
| China | No full population screening | [5] | ||
| India | No full population screening; program not government funded | [5] | ||
| Japan | [5] | |||
| Malaysia | No full population screening | [5] | ||
| New Zealand | 2006 | [6] | ||
| Philippines | No full population screening | [5] | ||
| Singapore | [5] | |||
| South Korea | [5] | |||
| Thailand | No full population screening | [5] | ||
| Taiwan | [5] | |||
| Europe | Austria | 2002 a | [5] | |
| Belgium | 2009 (Pilot 2007) b | [5] | ||
| Czech Republic | 2010 c | [5] | ||
| Denmark | 2012 | [5,7] | ||
| Estonia | No full population screening | [5] | ||
| Germany | Bavaria 1999 Nationwide 2005 | [8,9] | ||
| Greece | [10] | |||
| Hungary | [5] | |||
| Iceland | 2008 d | [5] | ||
| Italy | No full population screening | [5] | ||
| Liechtenstein | [5] | |||
| Macedonia | No full population screening | 2013 e | Personal communication e | |
| Netherlands | [5] | |||
| Norway | [5] | |||
| Poland | [5] | |||
| Portugal | [5] | |||
| Russia | [5] | |||
| San Marino | No full population screening | [5] | ||
| Spain | [5] | |||
| Sweden | [5] | |||
| Switzerland | [5] | |||
| United Kingdom | Not in Scotland and Northern Ireland | 2015 (Pilot 2012) f | [5] | |
| North America | United States | IVA included in all states but District of Columbia and Massachusetts | [5] | |
| Canada | IVA included in all provinces/territories but Newfoundland & Labrador; IVA screened by urine in Quebec | [11] | ||
| South America | Argentina | Offered exclusively in the private sector | [5] | |
| Brazil | Offered exclusively in the private sector | [5] | ||
| Chile | Offered as selective screening | [5] | ||
| Colombia | No full population screening; offered in the private sector | [5] | ||
| Costa Rica | [5] | |||
| Dominican Republic | Offered exclusively in the private sector | [5] | ||
| Mexico | No full population screening | [5] | ||
| Uruguay | No full population screening | [5] | ||
| Venezuela | Offered exclusively in the private sector | [5] | ||
| Africa | South Africa | Offered exclusively in the private sector | [12] | |
| Middle East | Kuwait | Pilot 2004–2006 | [13] Personal communication g | |
| Lebanon | Offered exclusively in the private sector | 2006 | [14,15] | |
| Saudi Arabia | [15] | |||
| Qatar | 2004 | [15,16] | ||
| United Arab Emirates | 2011 | [17] |
IVA: isovaleric acidemia; NBS: newborn screening; a Personal communication by Maximilian Zeyda; b Personal communication by François Boemer; c Personal communication by Viktor Kožich; d Personal communication by Leifur Franzson; e Personal communication by Violeta Anastasovska; f Personal communication by Jim Bonham; g Personal communication by Laila Bastaki.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Schlune, A.; Riederer, A.; Mayatepek, E.; Ensenauer, R. Aspects of Newborn Screening in Isovaleric Acidemia. Int. J. Neonatal Screen. 2018, 4, 7. https://doi.org/10.3390/ijns4010007
AMA Style
Schlune A, Riederer A, Mayatepek E, Ensenauer R. Aspects of Newborn Screening in Isovaleric Acidemia. International Journal of Neonatal Screening. 2018; 4(1):7. https://doi.org/10.3390/ijns4010007
Chicago/Turabian StyleSchlune, Andrea, Anselma Riederer, Ertan Mayatepek, and Regina Ensenauer. 2018. "Aspects of Newborn Screening in Isovaleric Acidemia" International Journal of Neonatal Screening 4, no. 1: 7. https://doi.org/10.3390/ijns4010007
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.