Proteomic and Transcriptomic Techniques to Decipher the Molecular Evolution of Venoms


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
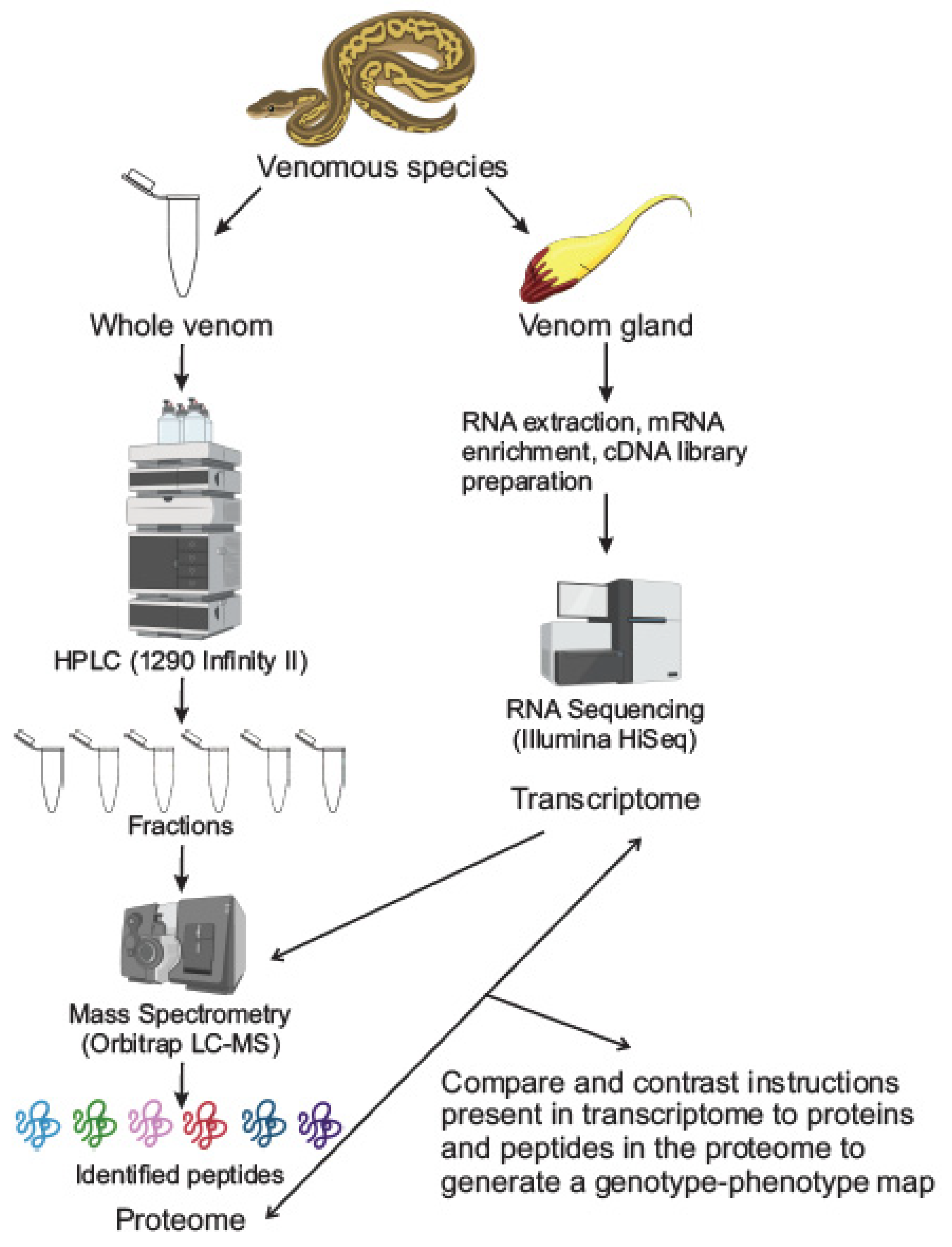
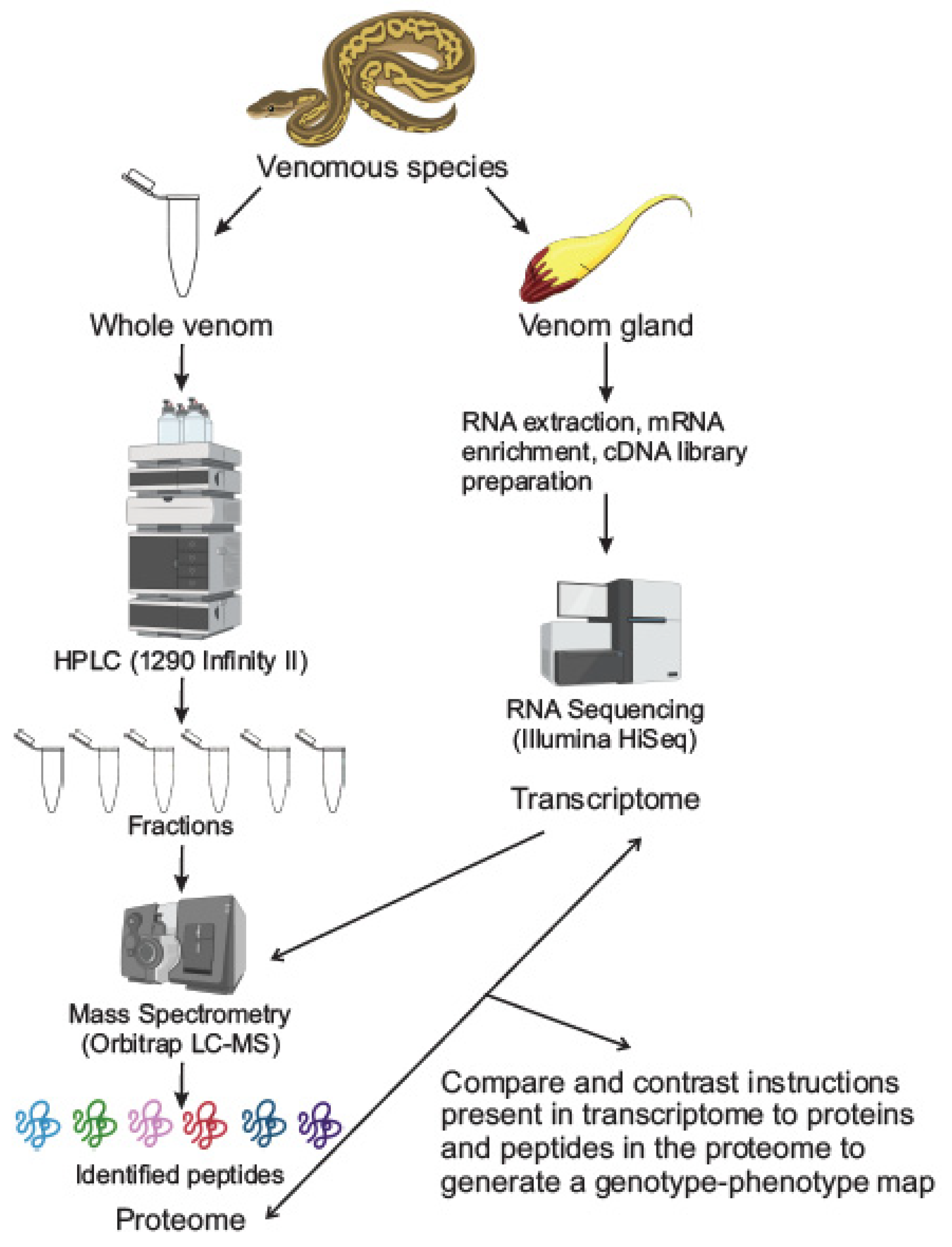
2. Techniques in Venomics
2.1. Proteomics: Separation and Mass Spectrometry Techniques
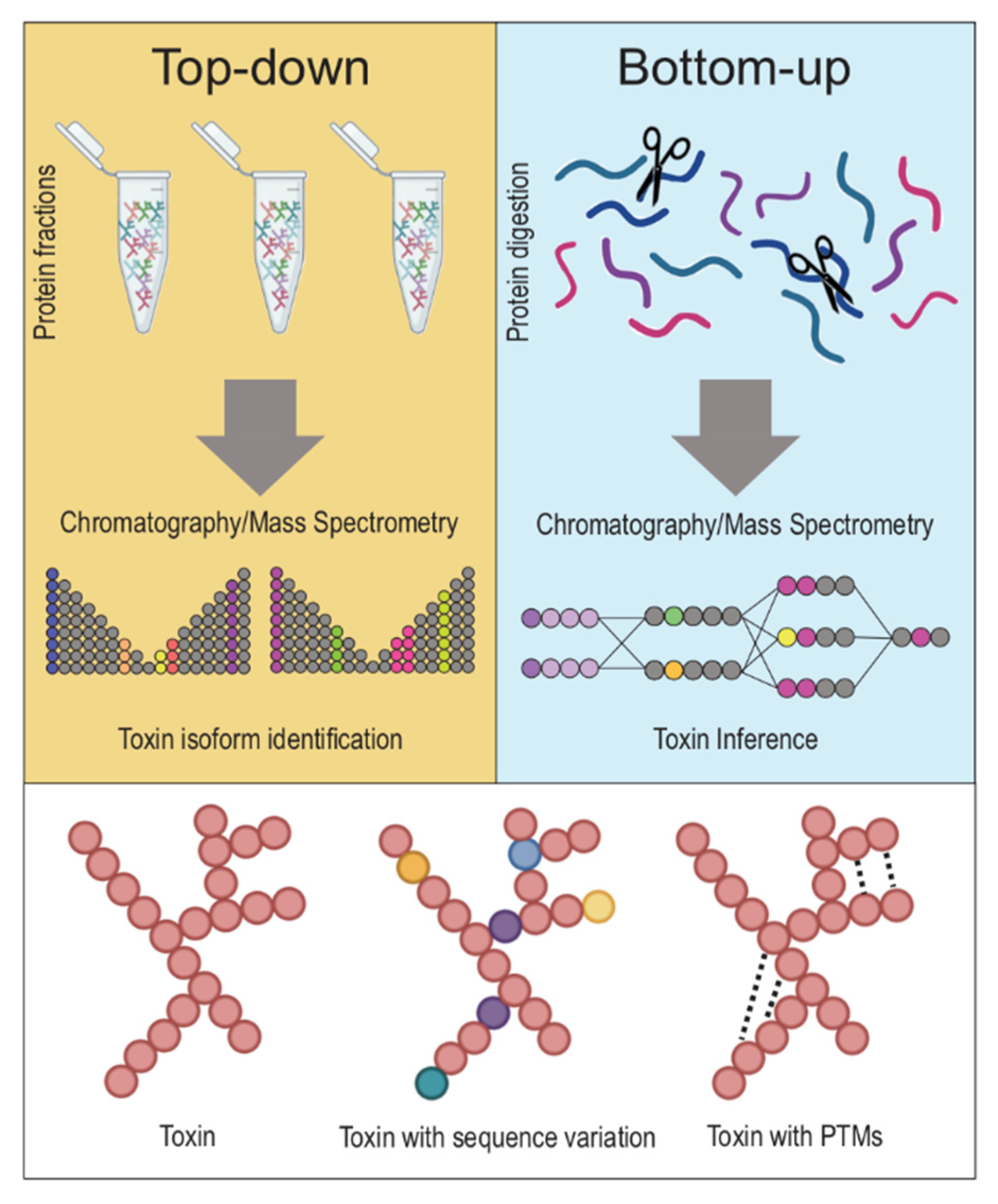
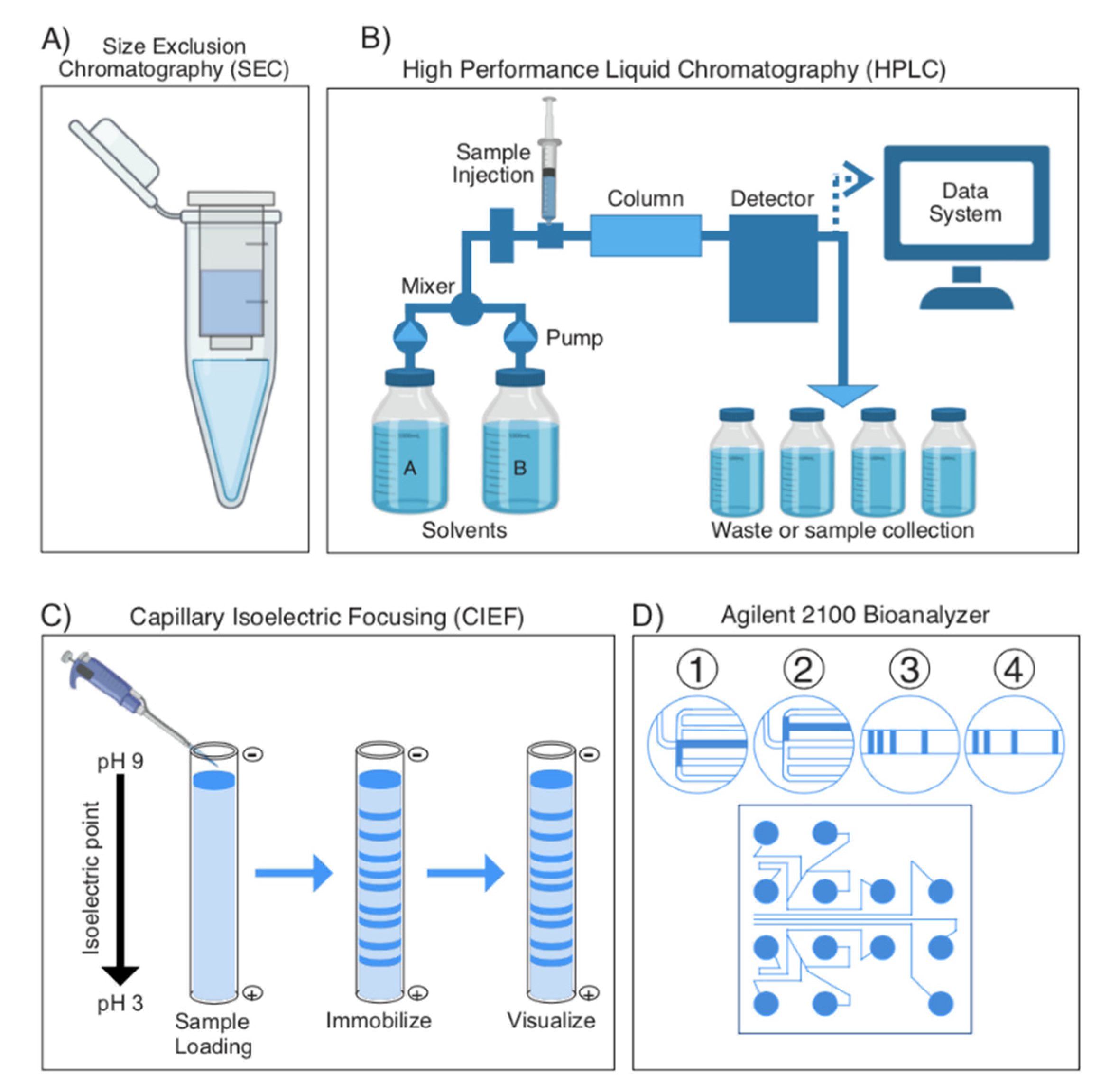
2.1.1. General Overview of Separating Proteins, Bottom up vs. Top down
2.1.2. Separation Techniques
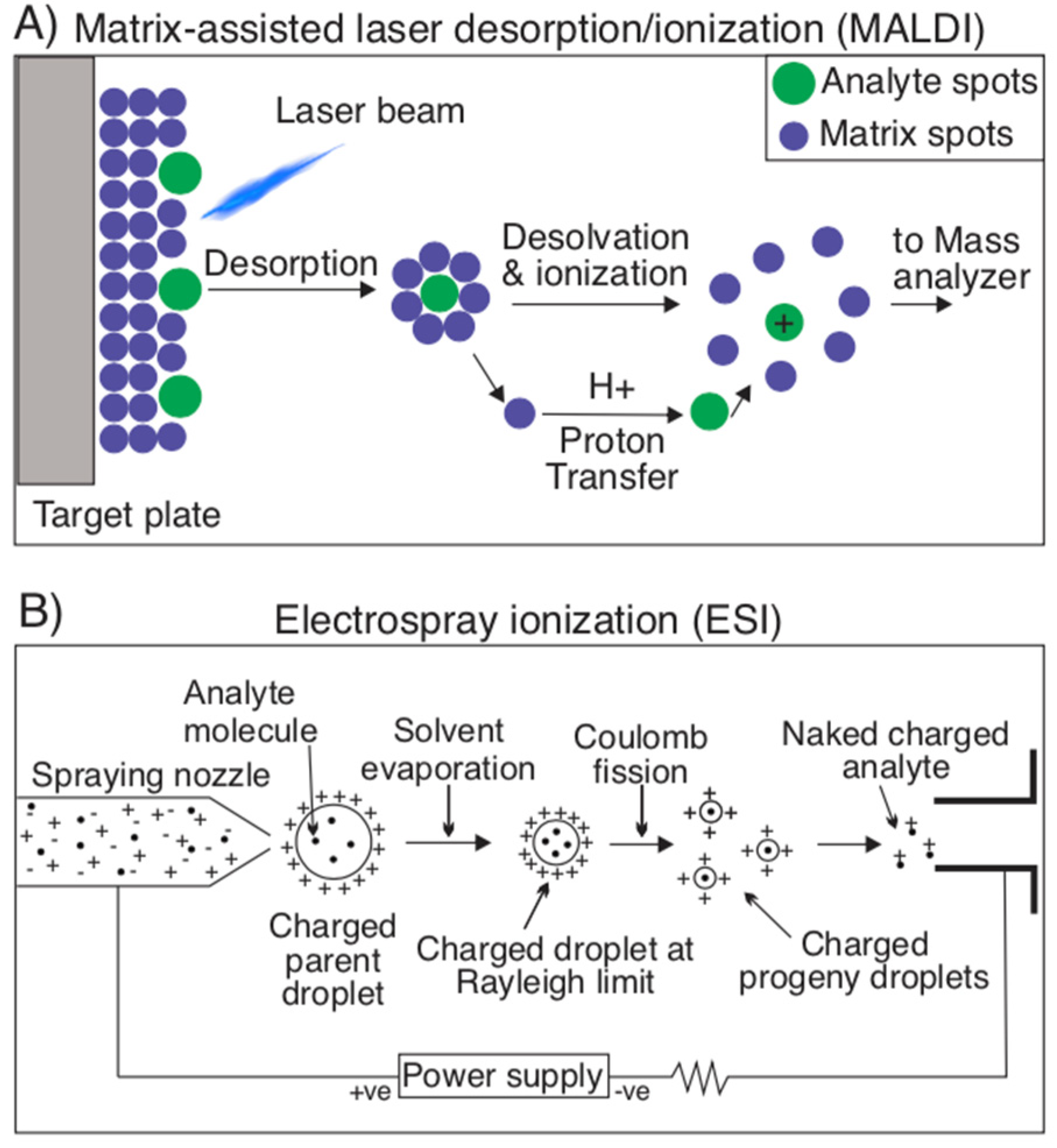
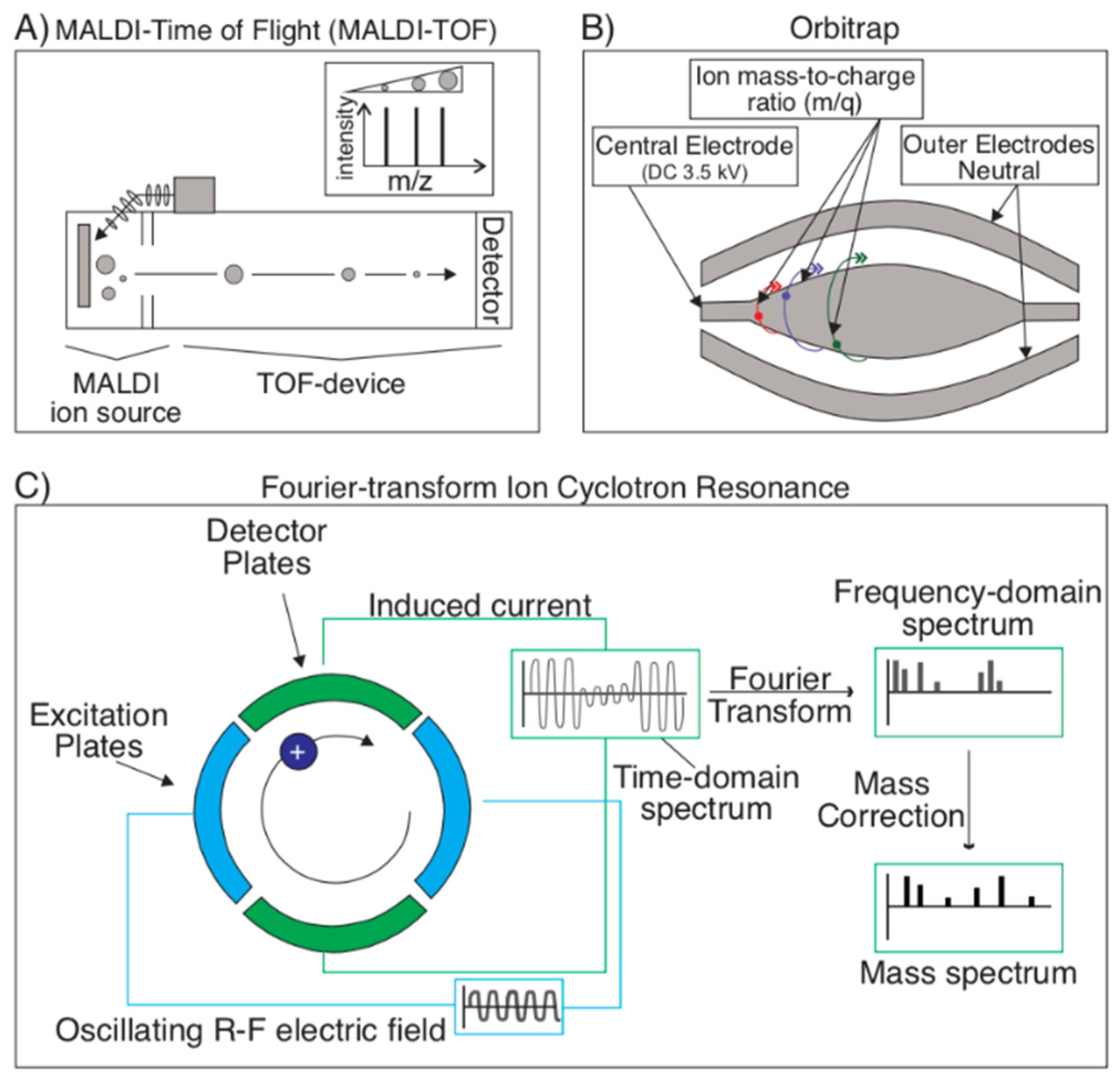
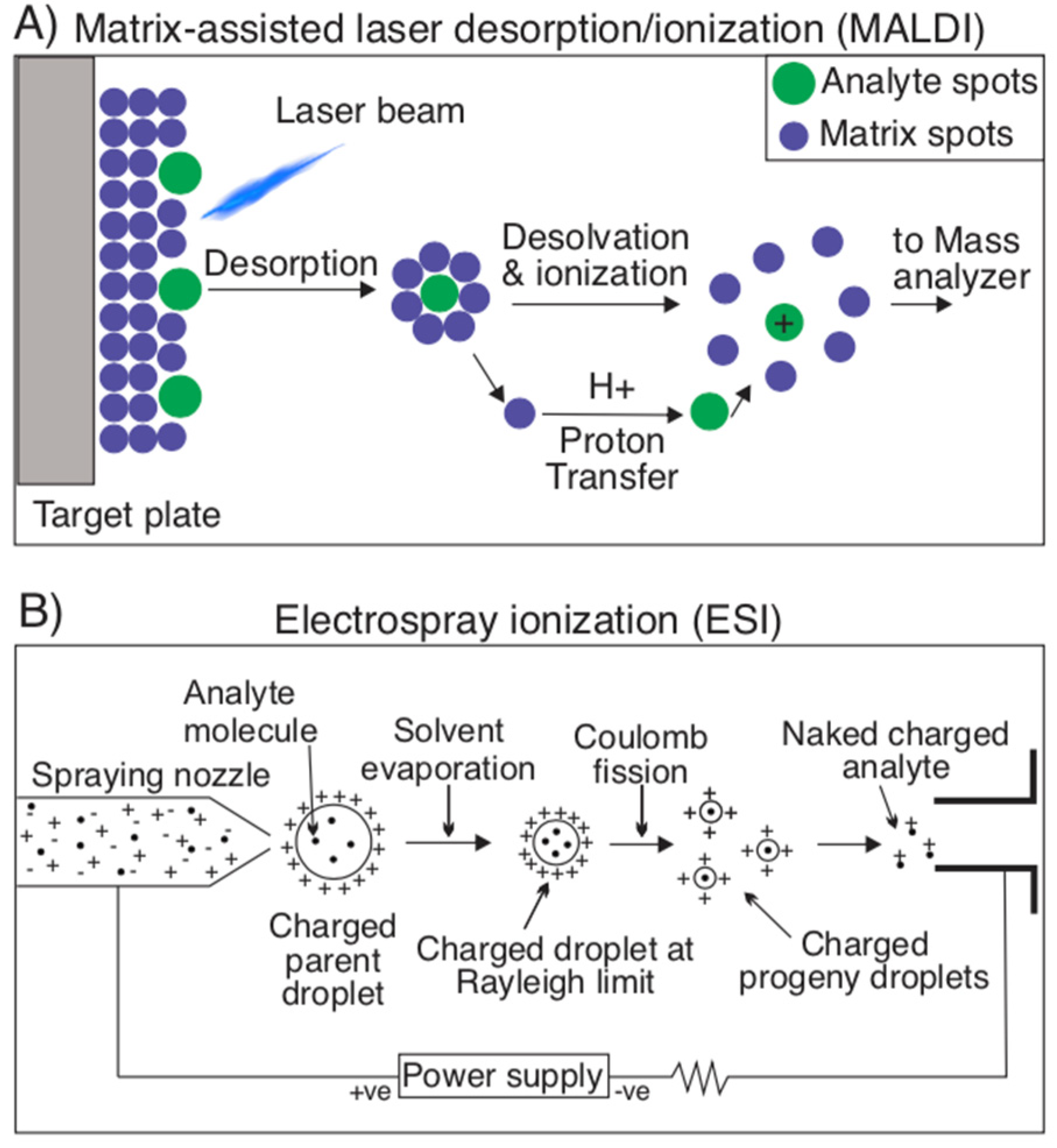
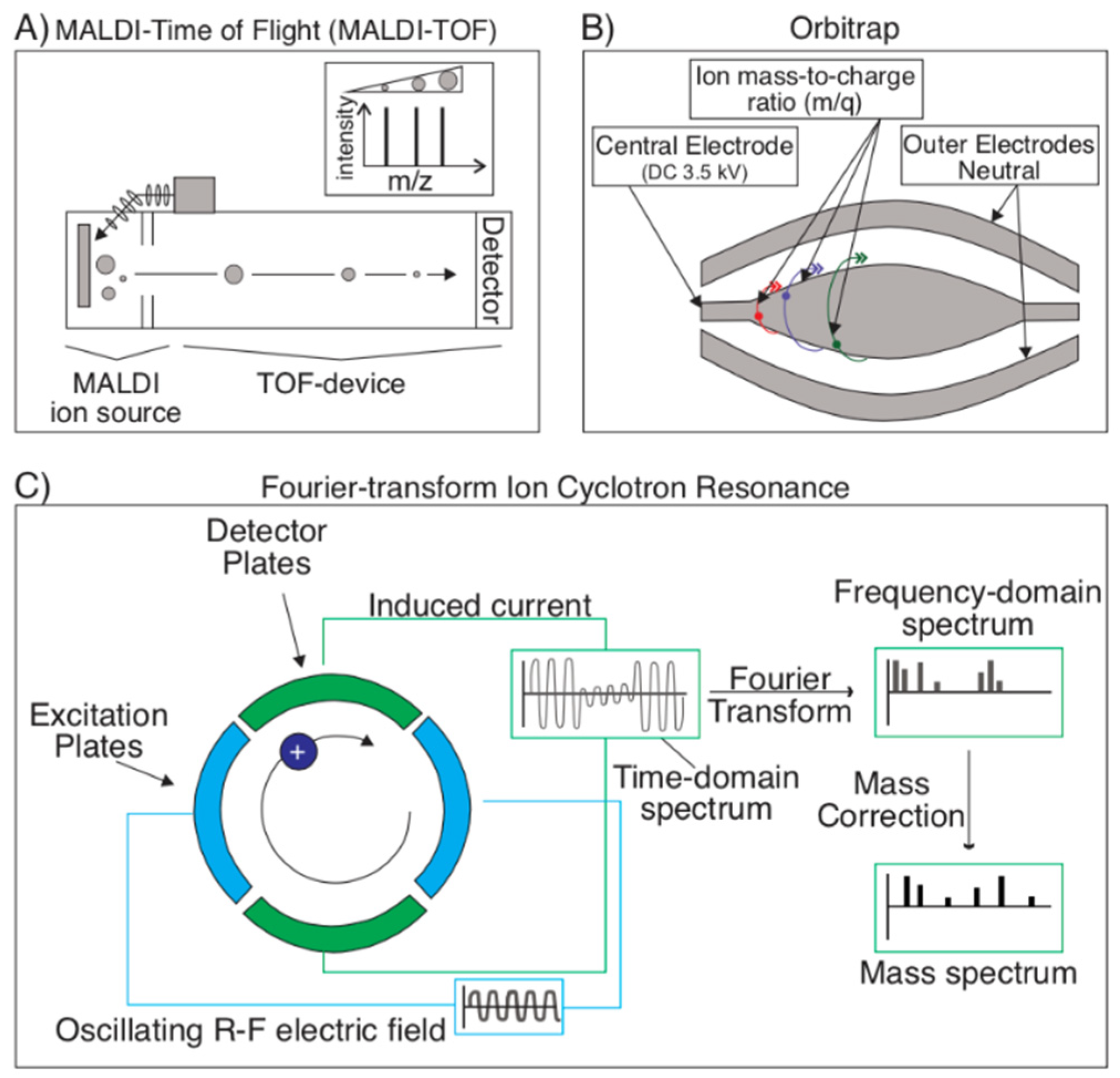
2.1.3. Mass Spectrometry Techniques
2.2. Transcriptomics
2.2.1. Transcriptomics: Sequencing Techniques
2.2.2. Current Gold Standards in Transcriptomics
2.3. Integrated Proteomic-Transcriptomic Techniques
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Von Reumont, B.M. Studying smaller and neglected organisms in modern evolutionary venomics implementing RNASEq (transcriptomics)—A critical guide. Toxins 2018, 10, 292. [Google Scholar] [CrossRef] [Green Version]
- Sunagar, K.; Morgenstern, D.; Reitzel, A.M.; Moran, Y. Ecological venomics: How genomics, transcriptomics and proteomics can shed new light on the ecology and evolution of venom. J. Proteom. 2016, 135, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.; Daly, N.L. Venomics: A Mini-Review. High Throughput 2018, 7, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.R.; Bubner, E.R.; Jovcevski, B.; Mittal, P.; Pukala, T.L. Interrogating the higher order structures of snake venom proteins using an integrated mass spectrometric approach. J. Proteom. 2020, 216, 103680. [Google Scholar] [CrossRef] [PubMed]
- Calvete, J.J. Venomics: Integrative venom proteomics and beyond. Biochem. J. 2017, 474, 611–634. [Google Scholar] [CrossRef] [PubMed]
- Daltry, J.C.; Wüster, W.; Thorpe, R.S. Diet and snake venom evolution. Nat. Cell Biol. 1996, 379, 537–540. [Google Scholar] [CrossRef]
- Holding, M.L.; Drabeck, D.H.; Jansa, S.A.; Gibbs, H.L. Venom resistance as a model for understanding the molecular basis of complex coevolutionary adaptations. Integr. Comp. Biol. 2016, 56, 1032–1043. [Google Scholar] [CrossRef] [PubMed]
- da Silva, N.J., Jr.; Aird, S.D. Prey specificity, comparative lethality and compositional differences of coral snake venoms. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2001, 128, 425–456. [Google Scholar] [CrossRef]
- Terlau, H.; Olivera, B.M. Conus venoms: A rich source of novel ion channel-targeted peptides. Physiol. Rev. 2004, 84, 41–68. [Google Scholar] [CrossRef] [Green Version]
- Terlau, H.; Shon, K.J.; Grilley, M.; Stocker, M.; Stühmer, W.; Olivera, B.M. Strategy for rapid immobilization of prey by a fish-hunting marine sn. Nat. Commun. 1996, 381, 148–151. [Google Scholar] [CrossRef]
- Olivera, B.M.; Gray, W.R.; Zeikus, R.; McIntosh, J.M.; Varga, J.; Rivier, J.; De Santos, V.; Cruz, L.J. Peptide neurotoxins from fish-hunting cone snails. Science 1985, 230, 1338–1343. [Google Scholar] [CrossRef] [PubMed]
- Melani, R.D.; Nogueira, F.C.S.; Domont, G.B. It is time for top-down venomics. J. Venom. Anim. Toxins Incl. Trop. Dis. 2017, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lomonte, B.; Calvete, J.J. Strategies in ‘snake venomics’ aiming at an integrative view of compositional, functional, and immunological characteristics of venoms. J. Venom. Anim. Toxins Incl. Trop. Dis. 2017, 23, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Nesvizhskii, A.I.; Aebersold, R. Interpretation of shotgun proteomic data: The protein inference problem. Mol. Cell. Proteom. 2005, 4, 1419–1440. [Google Scholar] [CrossRef] [Green Version]
- Ainsworth, S.; Petras, D.; Engmark, M.; Süssmuth, R.D.; Whiteley, G.; Albulescu, L.-O.; Kazandjian, T.D.; Wagstaff, S.C.; Rowley, P.; Wüster, W.; et al. The medical threat of mamba envenoming in sub-Saharan Africa revealed by genus-wide analysis of venom composition, toxicity and antivenomics profiling of available antivenoms. J. Proteom. 2018, 172, 173–189. [Google Scholar] [CrossRef]
- Petras, D.; Heiss, P.; Süssmuth, R.D.; Calvete, J.J. Venom proteomics of indonesian king cobra, ophiophagus hannah: Integrating top-down and bottom-up approaches. J. Proteome Res. 2015, 14, 2539–2556. [Google Scholar] [CrossRef] [PubMed]
- Petras, D.; Heiss, P.; Harrison, R.A.; Süssmuth, R.; Calvete, J.J. Top-down venomics of the East African green mamba, Dendroaspis angusticeps, and the black mamba, Dendroaspis polylepis, highlight the complexity of their toxin arsenals. J. Proteom. 2016, 146, 148–164. [Google Scholar] [CrossRef]
- Verano-Braga, T.; Dutra, A.A.; León, I.R.; Melo-Braga, M.N.; Roepstorff, P.; Pimenta, A.M.; Kjeldsen, F. Moving pieces in a venomic puzzle: Unveiling post-translationally modified toxins from Tityus serrulatus. J. Proteome Res. 2013, 12, 3460–3470. [Google Scholar] [CrossRef]
- Hempel, B.-F.; Damm, M.; Mrinalini, M.; Göçmen, B.; Karış, M.; Nalbantsoy, A.; Kini, R.M.; Suessmuth, R.D. Extended snake venomics by top-down in-source decay: Investigating the newly discovered anatolian meadow viper subspecies, Vipera anatolica senliki. J. Proteome Res. 2020, 19, 1731–1749. [Google Scholar] [CrossRef]
- Calvete, J.J.; Juárez, P.; Sanz, L. Snake venomics. Strategy and applications. J. Mass Spectrom. 2007, 42, 1405–1414. [Google Scholar] [CrossRef]
- Pla, D.; Petras, D.; Saviola, A.J.; Modahl, C.M.; Sanz, L.; Pérez, A.; Juárez, E.; Frietze, S.; Dorrestein, P.C.; Mackessy, S.P.; et al. Transcriptomics-guided bottom-up and top-down venomics of neonate and adult specimens of the arboreal rear-fanged Brown Treesnake, Boiga irregularis, from Guam. J. Proteom. 2018, 174, 71–84. [Google Scholar] [CrossRef]
- Fukuyama, Y.; Iwamoto, S.; Tanaka, K. Rapid sequencing and disulfide mapping of peptides containing disulfide bonds by using 1,5-diaminonaphthalene as a reductive matrix. J. Mass Spectrom. 2005, 41, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.; Ma, B.; Lajoie, G. De Novo Sequencing Methods in Proteomics; Springer Nature: Berlin, Germany, 2010. [Google Scholar]
- Barlow, A.; Pook, C.E.; Harrison, R.A.; Wüster, W. Coevolution of diet and prey-specific venom activity supports the role of selection in snake venom evolution. Proc. R. Soc. B Biol. Sci. 2009, 276, 2443–2449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, R.J.; Arbuckle, K. Tempo and mode of the evolution of venom and poison in tetrapods. Toxins 2016, 8, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, E.-L.; Arbuckle, K. Coevolution of snake venom toxic activities and diet: Evidence that ecological generalism favours toxicological diversity. Toxins 2019, 11, 711. [Google Scholar] [CrossRef] [Green Version]
- Modahl, C.M.; Mrinalini Frietze, S.; Mackessy, S.P. Adaptive evolution of distinct prey-specific toxin genes in rear-fanged snake venom. Proc. R. Soc. B 2018, 285, 20181003. [Google Scholar] [CrossRef] [PubMed]
- Amorim, F.G.; Costa, T.R.; Baiwir, D.; De Pauw, E.; Quinton, L.; Sampaio, S.V. Proteopeptidomic, functional and immunoreactivity characterization of Bothrops moojeni snake venom: Influence of snake gender on venom composition. Toxins 2018, 10, 177. [Google Scholar] [CrossRef] [Green Version]
- Lyons, K.; Dugon, M.M.; Healy, K. Diet breadth mediates the prey specificity of venom potency in snakes. Toxins 2020, 12, 74. [Google Scholar] [CrossRef] [Green Version]
- Healy, K.; Carbone, C.; Jackson, A.L. Snake venom potency and yield are associated with prey-evolution, predator metabolism and habitat structure. Ecol. Lett. 2019, 22, 527–537. [Google Scholar] [CrossRef]
- Sousa, L.F.; Portes, J.A., Jr.; Nicolau, C.A.; Bernardoni, J.L.; Nishiyama, M.Y., Jr.; Amazonas, D.R.; Freitas-De-Sousa, L.A.; Mourão, R.H.; Chalkidis, H.M.; Valente, R.H.; et al. Functional proteomic analyses of Bothrops atrox venom reveals phenotypes associated with habitat variation in the Amazon. J. Proteom. 2017, 159, 32–46. [Google Scholar] [CrossRef]
- Conlon, J.M. Purification of naturally occurring peptides by reversed-phase HPLC. Nat. Protoc. 2007, 2, 191–197. [Google Scholar] [CrossRef]
- Biardi, J.E.; Ho, C.; Marcinczyk, J.; Nambiar, K. Isolation and identification of a snake venom metalloproteinase inhibitor from California ground squirrel (Spermophilus beecheyi) blood sera. Toxicon 2011, 58, 486–493. [Google Scholar] [CrossRef]
- Margres, M.J.; McGivern, J.J.; Wray, K.P.; Seavy, M.; Calvin, K.; Rokyta, D.R. Linking the transcriptome and proteome to characterize the venom of the eastern diamondback rattlesnake (Crotalus adamanteus). J. Proteom. 2014, 96, 145–158. [Google Scholar] [CrossRef]
- Prashanth, J.R.; Hasaballah, N.; Vetter, I. Pharmacological screening technologies for venom peptide discovery. Neuropharmacology 2017, 127, 4–19. [Google Scholar] [CrossRef] [PubMed]
- Juárez, P.; Sanz, L.; Calvete, J.J. Snake venomics: Characterization of protein families in Sistrurus barbouri venom by cysteine mapping, N-terminal sequencing, and tandem mass spectrometry analysis. Proteomics 2004, 4, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Nicolau, C.A.; Carvalho, P.C.; Junqueira-De-Azevedo, I.L.; Teixeira-Ferreira, A.; Junqueira, M.; Perales, J.; Neves-Ferreira, A.G.C.; Valente, R.H. An in-depth snake venom proteopeptidome characterization: Benchmarking Bothrops jararaca. J. Proteom. 2017, 151, 214–231. [Google Scholar] [CrossRef] [PubMed]
- Asakawa, M.; Matsumoto, T.; Umezaki, K.; Kaneko, K.; Yu, X.; Gomez-Delan, G.; Tomano, S.; Noguchi, T.; Ohtsuka, S.; Yu, X. Toxicity and toxin composition of the greater blue-ringed octopus Hapalochlaena lunulata from Ishigaki Island, Okinawa Prefecture, Japan. Toxins 2019, 11, 245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besenius, P.; Cormack, P.A.; Ludlow, R.F.; Otto, S.; Sherrington, D.C. Affinity chromatography in dynamic combinatorial libraries: One-pot amplification and isolation of a strongly binding receptor. Org. Biomol. Chem. 2010, 8, 2414–2418. [Google Scholar] [CrossRef]
- Vestal, M.L. High-performance liquid chromatography-mass spectrometry. Science 1984, 226, 275–281. [Google Scholar] [CrossRef]
- Chen, H.; Horváth, C. High-speed high-performance liquid chromatography of peptides and proteins. J. Chromatogr. A 1995, 705, 3–20. [Google Scholar] [CrossRef]
- Wang, C.; Lee, C.S. Capillary electrophoresis–mass spectrometry for proteomic and metabolic analysis. Proteom. Metab. Approaches Biomark. Discov. 2013, 163–173, 163–173. [Google Scholar] [CrossRef]
- Smoluch, M.; Mielczarek, P.; Drabik, A.; Silberring, J. Online and offline sample fractionation. Proteom. Profiling Anal. Chem. 2016, 63–99, 63–99. [Google Scholar] [CrossRef]
- Trenerry, V.C.; Rochfort, S.J. Natural products research and metabolomics. Compr. Nat. Prod. II 2010, 2, 595–628. [Google Scholar] [CrossRef]
- Nagy, K.; Vékey, K. Separation methods. Med Appl. Mass Spectrom. 2008, 61–92, 61–92. [Google Scholar] [CrossRef]
- Moldoveanu, S.C.; David, V. Short overviews of the main analytical techniques containing a separation step. Sel. HPLC Method Chem. Anal. 2017, 55–85, 55–85. [Google Scholar] [CrossRef]
- Striegel, A. Size-exclusion chromatography. Liq. Chromatogr. 2013, 193–223, 193–223. [Google Scholar] [CrossRef]
- Tasoulis, T.; Silva, A.; Veerati, P.M.; Baker, M.; Hodgson, W.C.; Dunstan, N.; Isbiter, G.K. Coastal taipan Oxyuranus scutellatus. Toxins 2020, 12, 485. [Google Scholar] [CrossRef]
- Kunalan, S.; Othman, I.; Hassan, S.S.; Hodgson, W.C. Proteomic characterization of two medically important malaysian snake venoms, Calloselasma rhodostoma (Malayan Pit Viper) and Ophiophagus hannah (King Cobra). Toxins 2018, 10, 434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aslam, B.; Basit, M.; Nisar, M.A.; Khurshid, M.; Rasool, M.H. Proteomics: Technologies and their applications. J. Chromatogr. Sci. 2017, 55, 182–196. [Google Scholar] [CrossRef] [Green Version]
- Bocian, A.; Urbanik, M.; Hus, K.K.; Łyskowski, A.; Petrilla, V.; Andrejčáková, Z.; Petrillová, M.; Legáth, J. Proteomic analyses of agkistrodon contortrix contortrix venom using 2D electrophoresis and MS techniques. Toxins 2016, 8, 372. [Google Scholar] [CrossRef]
- Klose, J.; Kobalz, U. Two-dimensional electrophoresis of proteins: An updated protocol and implications for a functional analysis of the genome. Electrophoresis 1995, 16, 1034–1059. [Google Scholar] [CrossRef] [PubMed]
- Martins-De-Souza, D. 2DE Gels: A story of love and hate in proteomics. Proteomics 2018, 18, e1700472. [Google Scholar] [CrossRef] [Green Version]
- Viala, V.L.; Hildebrand, D.; Trusch, M.; Arni, R.K.; Pimenta, D.C.; Schlüter, H.; Betzel, C.; Spencer, P.J. Pseudechis guttatus venom proteome: Insights into evolution and toxin clustering. J. Proteom. 2014, 110, 32–44. [Google Scholar] [CrossRef]
- Barkan, N.P.; Bayazit, M.B.; Demiralp, D.O. Proteomic characterization of the venom of five bombus (Thoracobombus) species. Toxins 2017, 9, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittig, I.; Karas, M.; Schägger, H. High resolution clear native electrophoresis for in-gel functional assays and fluorescence studies of membrane protein complexes. Mol. Cell. Proteomics 2007, 6, 1215–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittig, I.; Braun, H.-P.; Schägger, H. Blue native PAGE. Nat. Protoc. 2006, 1, 418–428. [Google Scholar] [CrossRef]
- Nowakowski, A.B.; Wobig, W.J.; Petering, D.H. Native SDS-PAGE: High resolution electrophoretic separation of proteins with retention of native properties including bound metal ions. Metallomics 2014, 6, 1068–1078. [Google Scholar] [CrossRef] [Green Version]
- Zancolli, G.; Sanz, L.; Calvete, J.J.; Wüster, W. Venom on-a-chip: A Fast and efficient method for comparative venomics. Toxins 2017, 9, 179. [Google Scholar] [CrossRef] [Green Version]
- Kuschel, M.; Neumann, T.; Barthmaier, P.; Kratzmeier, M. Use of lab-on-a-chip technology for protein sizing and quantitation. J. Biomol. Tech. 2002, 13, 172–178. [Google Scholar]
- Ohashi, R.; Otero, J.M.; Chwistek, A.; Hamel, J.-F.P. Determination of monoclonal antibody production in cell culture using novel microfluidic and traditional assays. Electrophoresis 2002, 23, 3623–3629. [Google Scholar] [CrossRef]
- Schmut, O.; Horwath-Winter, J.; Zenker, A.; Trummer, G. The effect of sample treatment on separation profiles of tear fluid proteins: Qualitative and semi-quantitative protein determination by an automated analysis system. Graefe’s Arch. Clin. Exp. Ophthalmol. 2002, 240, 900–905. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Kwak, S.; Karpowicz, S.J. Re-use of commercial microfluidics chips for DNA, RNA, and protein electrophoresis. Biotechniques 2014, 57, 267–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escoubas, P.; Quinton, L.; Nicholson, G.M. Venomics: Unravelling the complexity of animal venoms with mass spectrometry. J. Mass Spectrom. 2008, 43, 279–295. [Google Scholar] [CrossRef] [PubMed]
- Greco, V.; Piras, C.; Pieroni, L.; Ronci, M.; Putignani, L.; Roncada, P.; Urbani, A. Applications of MALDI-TOF mass spectrometry in clinical proteomics. Expert Rev. Proteom. 2018, 15, 683–696. [Google Scholar] [CrossRef]
- Andersen, J.S.; Svensson, B.; Roepstorff, P. Electrospray ionization and matrix assisted laser desorption/ionization mass spectrometry: Powerful analytical tools in recombinant protein chemistry. Nat. Biotechnol. 1996, 14, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Fenn, J.B. Electrospray ion source. Another variation on the free-jet theme. J. Phys. Chem. 1984, 88, 4451–4459. [Google Scholar] [CrossRef]
- Fenn, J.B.; Mann, M.; Meng, C.K.; Wong, S.F.; Whitehouse, C.M. Electrospray ionization for mass spectrometry of large biomolecules. Science 1989, 246, 64–71. [Google Scholar] [CrossRef]
- Nadler, W.M.; Waidelich, D.; Kerner, A.; Hanke, S.; Berg, R.; Trumpp, A.; Rösli, C. MALDI versus ESI: The Impact of the Ion Source on Peptide Identification. J. Proteome Res. 2017, 16, 1207–1215. [Google Scholar] [CrossRef]
- Roberts, L.D. Defining the Metabolic Effect of Peroxisome Proliferator-Activated Receptor δ Activation. Ph.D. Thesis, University of Cambridge, Cambridge, UK, 2016. [Google Scholar]
- Eichhorn, P.; Pérez, S.; Barceló, D. Chapter 5-Time-of-Flight Mass Spectrometry Versus Orbitrap-Based Mass Spectrometry for the Screening and Identification of Drugs and Metabolites: Is There a Winner? In TOF-MS within Food and Environmental Analysis; Fernandez-Alba, A.R., Ed.; Elsevier: Amsterdam, The Netherlands, 2012; Volume 58, pp. 217–272. [Google Scholar]
- Ghaste, M.; Mistrik, R.; Shulaev, V. Applications of fourier transform ion cyclotron resonance (FT-ICR) and orbitrap based high resolution mass spectrometry in metabolomics and lipidomics. Int. J. Mol. Sci. 2016, 17, 816. [Google Scholar] [CrossRef]
- Breitkopf, S.B.; Ricoult, S.J.; Yuan, M.; Xu, Y.; Peake, D.A.; Manning, B.D.; Asara, J.M. A relative quantitative positive/negative ion switching method for untargeted lipidomics via high resolution LC-MS/MS from any biological source. Metabolomics 2017, 13, 30. [Google Scholar] [CrossRef] [Green Version]
- Quinton, L.; Le Caër, J.-P.; Vinh, J.; Gilles, N.; Chamot-Rooke, J. Fourier transform mass spectrometry: A powerful tool for toxin analysis. Toxicon 2006, 47, 715–726. [Google Scholar] [CrossRef]
- Quinton, L.; Le Caer, J.-P.; Phan, G.; Ligny-Lemaire, C.; Jomaro, J.B.; Ducancel, F.; Chamot-Rooke, J. Characterization of toxins within crude venoms by combined use of fourier transform mass spectrometry and cloning. Anal. Chem. 2005, 77, 6630–6639. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.W.; Ma, L.; Nelson, K.; Sherman, N.E.; Serrano, S.M.T. Comparison of indirect and direct approaches using ion-trap and fourier transform ion cyclotron resonance mass spectrometry for exploring viperid venom proteomes. Toxicon 2006, 47, 700–714. [Google Scholar] [CrossRef] [PubMed]
- Luna-Ramírez, K.S.; Quintero-Hernández, V.; Juárez-González, V.R.; Possani, L.D. Whole transcriptome of the venom gland from urodacus yaschenkoi scorpion. PLoS ONE 2015, 10, e0127883. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef] [PubMed]
- Van Den Berge, K.; Hembach, K.M.; Soneson, C.; Tiberi, S.; Clement, L.; Love, M.I.; Patro, B.; Robinson, M.D. RNA Sequencing Data: Hitchhiker’s Guide to Expression Analysis introduction: Overview of the RNA sequencing assay. Annu. Rev. Biomed. Data Sci. 2019, 2, 139–173. [Google Scholar] [CrossRef] [Green Version]
- Shaina, H.; Abdin, Z.U.; Webb, B.A.; Arif, M.J.; Jamil, A. De novo sequencing and transcriptome analysis of venom glands of endoparasitoid Aenasius arizonensis (Girault) (=Aenasius bambawalei Hayat) (Hymenoptera, Encyrtidae). Toxicon 2016, 121, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Manzoor, A.; Ulabdin, Z.; Webb, B.A.; Arif, M.J.; Jamil, A. De novo sequencing and transcriptome analysis of female venom glands of ectoparasitoid Bracon hebetor (Say.) (Hymenoptera: Braconidae). Comp. Biochem. Physiol. Part D Genom. Proteom. 2016, 20, 101–110. [Google Scholar] [CrossRef]
- Tan, C.H.; Tan, K.Y.; Fung, S.Y.; Tan, N.H. Venom-gland transcriptome and venom proteome of the Malaysian king cobra (Ophiophagus hannah). BMC Genom. 2015, 16, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Rokyta, D.R.; Ward, M.J. Venom-gland transcriptomics and venom proteomics of the black-back scorpion (Hadrurus spadix) reveal detectability challenges and an unexplored realm of animal toxin diversity. Toxicon 2017, 128, 23–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rokyta, D.R.; Lemmon, A.R.; Margres, M.J.; Aronow, K. The venom-gland transcriptome of the eastern diamondback rattlesnake (Crotalus adamanteus). BMC Genom. 2012, 13, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiezel, G.A.; Shibao, P.Y.T.; Cologna, C.T.; Filho, R.M.; Ueira-Vieira, C.; De Pauw, E.; Quinton, L.; Arantes, E.C. In-depth venome of the brazilian rattlesnake crotalus durissus terrificus: An integrative approach combining its venom gland transcriptome and venom proteome. J. Proteome Res. 2018, 17, 3941–3958. [Google Scholar] [CrossRef] [PubMed]
- Von Reumont, B.M.; Undheim, E.A.B.; Jauss, R.-T.; Jenner, R.A. Venomics of remipede crustaceans reveals novel peptide diversity and illuminates the venom’s biological role. Toxins 2017, 9, 234. [Google Scholar] [CrossRef] [Green Version]
- Romero-Gutierrez, T.; Peguero-Sanchez, E.; Cevallos, M.A.; Batista, C.V.; Ortiz, E.; Possani, L.D. A deeper examination of thorellius atrox scorpion venom components with omic techonologies. Toxins 2017, 9, 399. [Google Scholar] [CrossRef] [Green Version]
- Ward, M.J.; Ellsworth, S.A.; Rokyta, D.R. Venom-gland transcriptomics and venom proteomics of the Hentz striped scorpion (Centruroides hentzi; Buthidae) reveal high toxin diversity in a harmless member of a lethal family. Toxicon 2018, 142, 14–29. [Google Scholar] [CrossRef]
- Amorim, F.G.; Morandi-Filho, R.; Fujimura, P.T.; Ueira-Vieira, C.; Sampaio, S.V. New findings from the first transcriptome of the Bothrops moojeni snake venom gland. Toxicon 2017, 140, 105–117. [Google Scholar] [CrossRef]
- Brinkman, D.L.; Jia, X.; Potriquet, J.; Kumar, D.; Dash, D.; Kvaskoff, D.; Mulvenna, J. Transcriptome and venom proteome of the box jellyfish Chironex fleckeri. BMC Genom. 2015, 16, 407. [Google Scholar] [CrossRef] [Green Version]
- Yee, K.T.; Tongsima, S.; Vasieva, O.; Ngamphiw, C.; Wilantho, A.; Wilkinson, M.; Somparn, P.; Pisitkun, T.; Rojnuckarin, P. Analysis of snake venom metalloproteinases from Myanmar Russell’s viper transcriptome. Toxicon 2018, 146, 31–41. [Google Scholar] [CrossRef]
- Christensen, K.A.; Davidson, W.S. Autopolyploidy genome duplication preserves other ancient genome duplications in Atlantic salmon (Salmo salar). PLoS ONE 2017, 12, e0173053. [Google Scholar] [CrossRef]
- Kazemi-Lomedasht, F.; Khalaj, V.; Bagheri, K.P.; Behdani, M.; Shahbazzadeh, D. The first report on transcriptome analysis of the venom gland of Iranian scorpion, Hemiscorpius lepturus. Toxicon 2017, 125, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Mansoor, S.E.; Lü, W.; Oosterheert, W.; Shekhar, M.; Tajkhorshid, E.; Gouaux, E. X-ray structures define human P2X3 receptor gating cycle and antagonist action. Nat. Cell Biol. 2016, 538, 66–71. [Google Scholar] [CrossRef] [Green Version]
- SEQC/MAQC-III Consortium. A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the sequencing quality control consortium. Nat. Biotechnol. 2014, 32, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Frankish, A.; Diekhans, M.; Ferreira, A.-M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oikonomopoulos, S.; Wang, Y.C.; Djambazian, H.; Badescu, D.; Ragoussis, J. Benchmarking of the oxford nanopore minion sequencing for quantitative and qualitative assessment of cDNA populations. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Cartolano, M.; Huettel, B.; Hartwig, B.; Reinhardt, R.; Schneeberger, K. cDNA library enrichment of full length transcripts for SMRT long read sequencing. PLoS ONE 2016, 11, e0157779. [Google Scholar] [CrossRef]
- Sharon, D.; Tilgner, H.; Grubert, F.; Snyder, M. A single-molecule long-read survey of the human transcriptome. Nat. Biotechnol. 2013, 31, 1009–1014. [Google Scholar] [CrossRef]
- Silva, F.; Huang, Y.; Yang, V.; Mu, X.; Shi, Q.; Antunes, A. Transcriptomic characterization of the south American freshwater stingray Potamotrygon motoro venom apparatus. Toxins 2018, 10, 544. [Google Scholar] [CrossRef] [Green Version]
- Xie, B.; Huang, Y.; Baumann, K.; Fry, B.G.; Shi, Q. From marine venoms to drugs: Efficiently supported by a combination of transcriptomics and proteomics. Mar. Drugs 2017, 15, 103. [Google Scholar] [CrossRef]
- Robinson, S.D.; Undheim, E.A.B.; Ueberheide, B.; King, G.F. Venom peptides as therapeutics: Advances, challenges and the future of venom-peptide discovery. Expert Rev. Proteom. 2017, 14, 931–939. [Google Scholar] [CrossRef]
- Casewell, N.R.; Wüster, W.; Vonk, F.J.; Harrison, R.A.; Fry, B.G. Complex cocktails: The evolutionary novelty of venoms. Trends Ecol. Evol. 2013, 28, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Casewell, N.R.; Wagstaff, S.C.; Harrison, R.A.; Renjifo, C.; Wüster, W. Domain loss facilitates accelerated evolution and neofunctionalization of duplicate snake venom metalloproteinase toxin genes. Mol. Biol. Evol. 2011, 28, 2637–2649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nei, M.; Gu, X.; Sitnikova, T. Evolution by the birth-and-death process in multigene families of the vertebrate immune system. Proc. Natl. Acad. Sci. USA 1997, 94, 7799–7806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nei, M.; Rooney, A.P. Concerted and birth-and-death evolution of multigene families. Annu. Rev. Genet. 2005, 39, 121–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponce, D.; Brinkman, D.L.; Potriquet, J.; Mulvenna, J. Tentacle transcriptome and venom proteome of the Pacific Sea Nettle, Chrysaora fuscescens (Cnidaria: Scyphozoa). Toxins 2016, 8, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durban, J.; Sanz, L.; Trevisan-Silva, D.; Neri-Castro, E.; Alagon, A.; Calvete, J.J. Integrated venomics and venom gland transcriptome analysis of juvenile and adult mexican rattlesnakes Crotalus simus, C. tzabcan, and C. culminatus revealed miRNA-modulated ontogenetic shifts. J. Proteome Res. 2017, 16, 3370–3390. [Google Scholar]
- Aili, S.R.; Touchard, A.; Hayward, R.J.; Robinson, S.D.; Pineda, S.S.; Lalagüe, H.; Vetter, I.; Undheim, E.A.B.; Kini, R.M.; Escoubas, P.; et al. An Integrated proteomic and transcriptomic analysis reveals the venom complexity of the bullet ant Paraponera clavata. Toxins 2020, 12, 324. [Google Scholar] [CrossRef]
- de Oliveira, U.C.; Nishiyama, M.Y., Jr.; Dos Santos, M.B.V.; Santos-da-Silva, A.D.P.; Chalkidis, H.D.M.; Souza-Imberg, A.; Candido, D.M.; Yamanouye, N.; Dorce, V.A.C.; Junqueira-de-Azevedo, I.D.L.M. Proteomic endorsed transcriptomic profiles of venom glands from Tityus obscurus and T. serrulatus scorpions. PLoS ONE 2018, 13, e0193739. [Google Scholar]
- Violette, A.; Biass, D.; Dutertre, S.; Koua, D.; Piquemal, D.; Pierrat, F.; Stöcklin, R.; Favreau, P. Large-scale discovery of conopeptides and conoproteins in the injectable venom of a fish-hunting cone snail using a combined proteomic and transcriptomic approach. J. Proteom. 2012, 75, 5215–5225. [Google Scholar] [CrossRef]
- Arbuckle, K.; Rodriguez de la Vega, R.C.; Casewell, N.R. Coevolution takes the sting out of it: Evolutionary biology and mechanisms of toxin resistance in animals. Toxicon 2017, 140, 118–131. [Google Scholar] [CrossRef]
- Holding, M.L.; Biardi, J.E.; Gibbs, H.L. Coevolution of venom function and venom resistance in a rattlesnake predator and its squirrel prey. Proc. R. Soc. B Biol. Sci. 2016, 283, 20152841. [Google Scholar] [CrossRef]
- Feldman, C.R.; Brodie, E.D.; Pfrender, M.E. Genetic architecture of a feeding adaptation: Garter snake (Thamnophis) resistance to tetrodotoxin bearing prey. Proc. R. Soc. B Biol. Sci. 2010, 277, 3317–3325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biardi, J.E.; Coss, R.G. Rock squirrel (Spermophilus variegatus) blood sera affects proteolytic and hemolytic activities of rattlesnake venoms. Toxicon 2011, 57, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Margres, M.J.; Aronow, K.; Loyacano, J.; Rokyta, D.R. The venom-gland transcriptome of the eastern coral snake (Micrurus fulvius) reveals high venom complexity in the intragenomic evolution of venoms. BMC Genom. 2013, 14, 531. [Google Scholar] [CrossRef] [Green Version]
- Dawkins, R.; Krebs, J.R. Arms races between and within species. Proc. R. Soc. Lond. Ser. B Biol. Sci. 1979, 205, 489–511. [Google Scholar] [CrossRef]
- Brodie, E.D., III; Brodie, E.D., Jr. Predator-prey arms races: Asymmetrical selection on predators and prey may be reduced when prey are dangerous. Bioscience 1999, 49, 557–568. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mouchbahani-Constance, S.; Sharif-Naeini, R. Proteomic and Transcriptomic Techniques to Decipher the Molecular Evolution of Venoms. Toxins 2021, 13, 154. https://doi.org/10.3390/toxins13020154
Mouchbahani-Constance S, Sharif-Naeini R. Proteomic and Transcriptomic Techniques to Decipher the Molecular Evolution of Venoms. Toxins. 2021; 13(2):154. https://doi.org/10.3390/toxins13020154
Chicago/Turabian StyleMouchbahani-Constance, Stephanie, and Reza Sharif-Naeini. 2021. "Proteomic and Transcriptomic Techniques to Decipher the Molecular Evolution of Venoms" Toxins 13, no. 2: 154. https://doi.org/10.3390/toxins13020154
APA StyleMouchbahani-Constance, S., & Sharif-Naeini, R. (2021). Proteomic and Transcriptomic Techniques to Decipher the Molecular Evolution of Venoms. Toxins, 13(2), 154. https://doi.org/10.3390/toxins13020154