
Age-Related Alterations at Neuromuscular Junction: Role of Oxidative Stress and Epigenetic Modifications




Abstract
1. Introduction
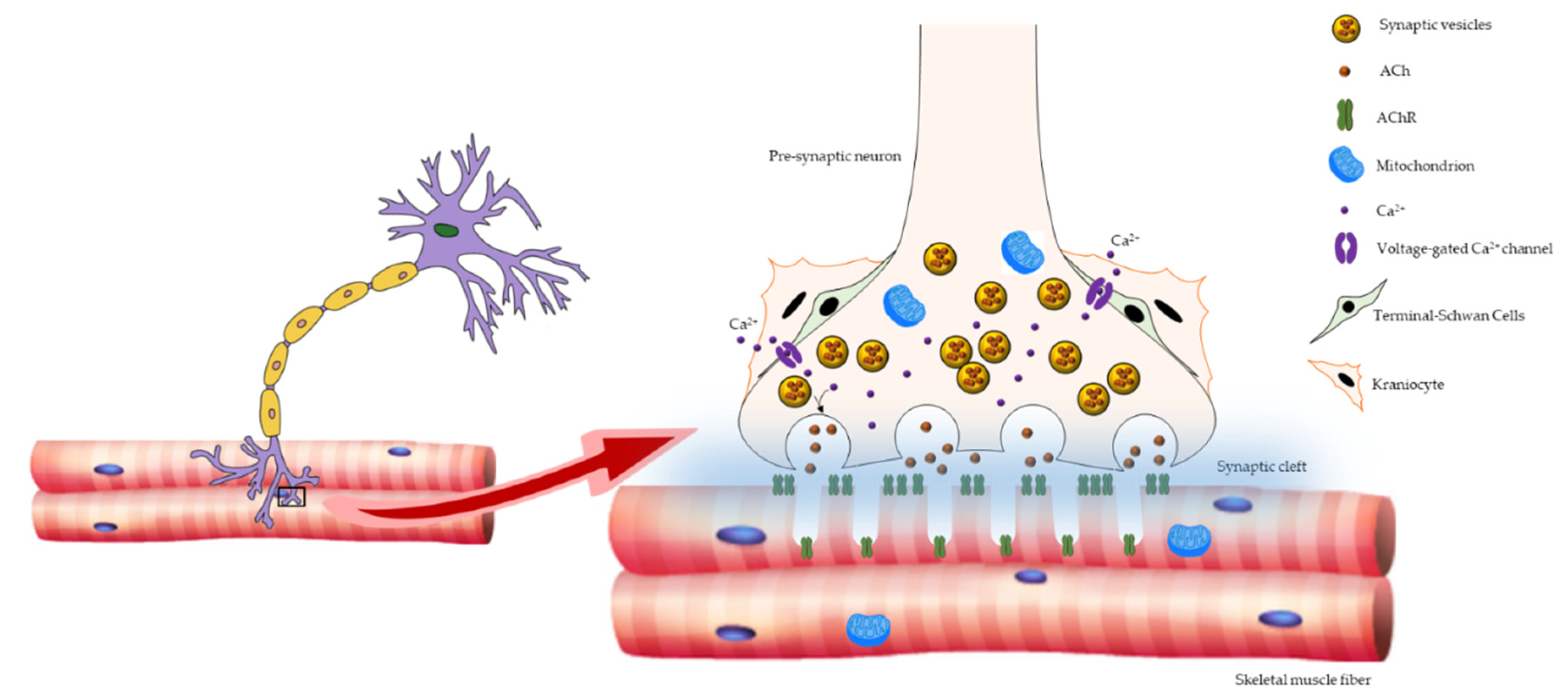
2. Age-Related Changes in NMJ
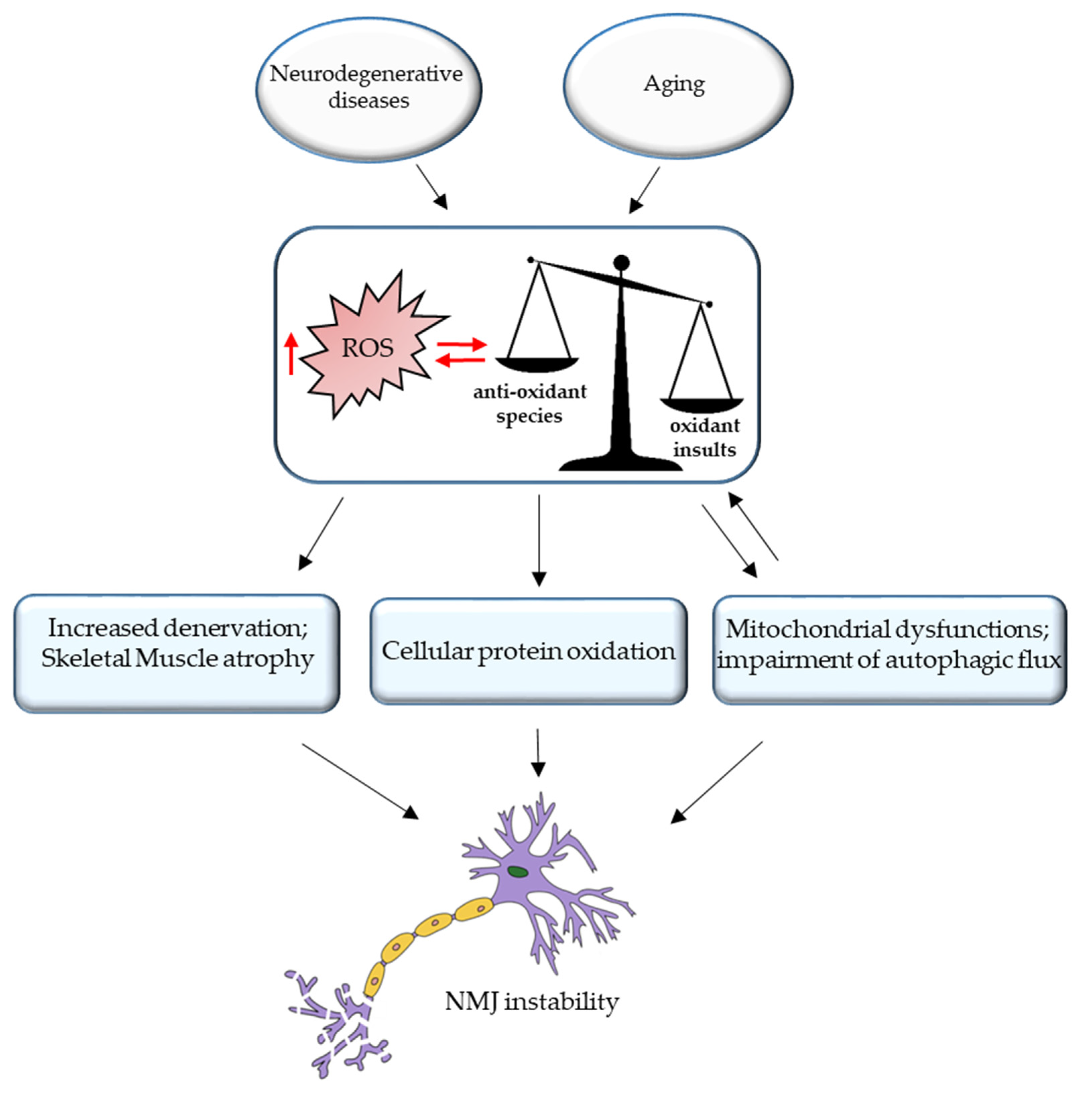
3. ROS and NMJ Degeneration
4. Epigenetic Regulation of NMJ Dysfunction
4.1. DNA Methylation
4.2. Histone Acetylation
4.3. miRNAs
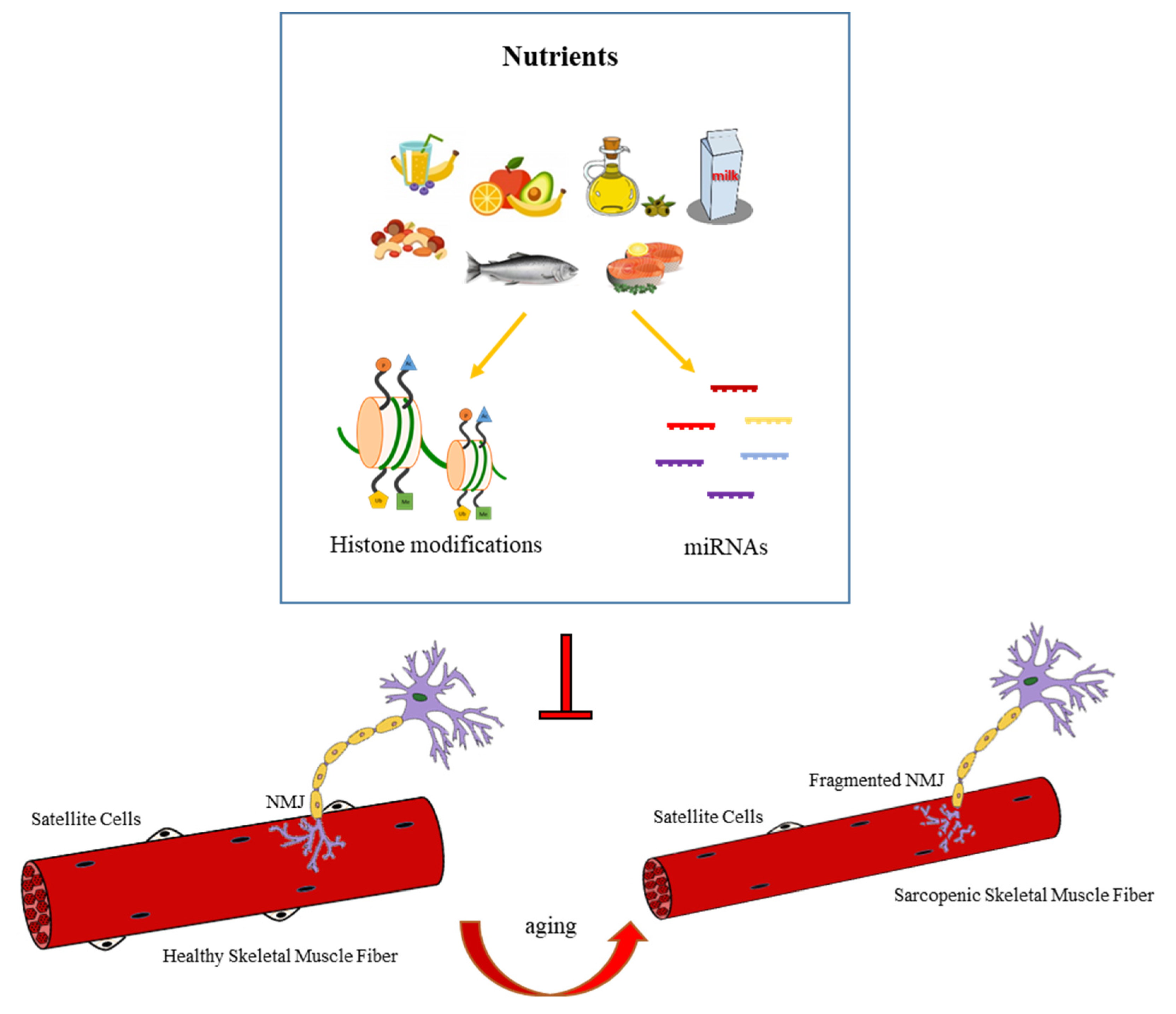
4.4. Nutrition-Dependent Epigenetic Regulation of NMJ
4.5. Exercise-Dependent Regulation of NMJs
5. Conclusions and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Chhetri, J.K.; de Souto Barreto, P.; Fougère, B.; Rolland, Y.; Vellas, B.; Cesari, M. Chronic inflammation and sarcopenia: A regenerative cell therapy perspective. Exp. Gerontol. 2018, 103, 115–123. [Google Scholar] [CrossRef]
- Dickinson, J.M.; Volpi, E.; Rasmussen, B.B. Exercise and Nutrition to Target Protein Synthesis Impairments in Aging Skeletal Muscle. Exerc. Sport Sci. Rev. 2013, 41, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Marzetti, E.; Calvani, R.; Cesari, M.; Buford, T.W.; Lorenzi, M.; Behnke, B.J.; Leeuwenburgh, C. Mitochondrial dysfunction and sarcopenia of aging: From signaling pathways to clinical trials. Int. J. Biochem. Cell Biol. 2013, 45, 2288–2301. [Google Scholar] [CrossRef] [PubMed]
- Sousa-Victor, P.; Muñoz-Cánoves, P. Regenerative decline of stem cells in sarcopenia. Mol. Aspects Med. 2016, 50, 109–117. [Google Scholar] [CrossRef]
- Larsson, L.; Degens, H.; Li, M.; Salviati, L.; Lee, Y.I.; Thompson, W.; Kirkland, J.L.; Sandri, M. Sarcopenia: Aging-related loss of muscle mass and function. Physiol. Rev. 2019, 99, 427–511. [Google Scholar] [CrossRef] [PubMed]
- Hepple, R.T.; Rice, C.L. Innervation and neuromuscular control in ageing skeletal muscle. J. Physiol. 2016, 594, 1965–1978. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; De Vito, G.; Delahunt, E.; Ditroilo, M. Age-related Changes in Motor Function (I)Mechanical and Neuromuscular Factors. Int. J. Sports Med. 2020, 41, 709–719. [Google Scholar] [CrossRef]
- Rudolf, R.; Khan, M.M.; Witzemann, V. Motor Endplate—Anatomical, Functional, and Molecular Concepts in the Historical Perspective. Cells 2019, 8, 387. [Google Scholar] [CrossRef]
- Mège, R.M.; Goudou, D.; Giaume, C.; Nicolet, M.; Rieger, F. Is intercellular communication via Gap junctions required for myoblast fusion?? Cell Commun. Adhes. 1994, 2, 329–343. [Google Scholar] [CrossRef]
- Schiaffino, S.; Reggiani, C. Molecular diversity of myofibrillar proteins: Gene regulation and functional significance. Physiol. Rev. 1996, 76, 371–423. [Google Scholar] [CrossRef]
- Hughes, S.M. Muscle development: Electrical control of gene expression. Curr. Biol. 1998, 8, R892–R894. [Google Scholar] [CrossRef]
- Olson, E.N.; Williams, R.S. Calcineurin signaling and muscle remodeling. Cell 2000, 101, 689–692. [Google Scholar] [CrossRef]
- Buller, A.J.; Eccles, J.C.; Eccles, R.M. Differentiation of fast and slow muscles in the cat hind limb. J. Physiol. 1960, 150, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Pette, D.; Vrbová, G. Invited review: Neural control of phenotypic expression in mammalian muscle fibers. Muscle Nerve 1985, 8, 676–689. [Google Scholar] [CrossRef] [PubMed]
- Klitgaard, H.; Zhou, M.; Schiaffino, S.; Betto, R.; Salviati, G.; Saltin, B. Ageing alters the myosin heavy chain composition of single fibres from human skeletal muscle. Acta Physiol. Scand. 1990, 140, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-J. Age-related functional changes and susceptibility to eccentric contraction-induced damage in skeletal muscle cell. Integr. Med. Res. 2016, 5, 171–175. [Google Scholar] [CrossRef]
- Bao, Z.; Cui, C.; Chow, S.K.H.; Qin, L.; Wong, R.M.Y.; Cheung, W.H. AChRs Degeneration at NMJ in Aging-Associated Sarcopenia–A Systematic Review. Front. Aging Neurosci. 2020, 12, 597811. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, D.W. Effects of advancing age on peripheral nerve regeneration. J. Comp. Neurol. 1992, 323, 219–237. [Google Scholar] [CrossRef] [PubMed]
- Delmonico, M.J.; Harris, T.B.; Visser, M.; Park, S.W.; Conroy, M.B.; Velasquez-Mieyer, P.; Boudreau, R.; Manini, T.M.; Nevitt, M.; Newman, A.B.; et al. Longitudinal study of muscle strength, quality, and adipose tissue infiltration. Am. J. Clin. Nutr. 2009, 90, 1579–1585. [Google Scholar]
- Lexell, J.; Taylor, C.C.; Sjöström, M. What is the cause of the ageing atrophy?. Total number, size and proportion of different fiber types studied in whole vastus lateralis muscle from 15- to 83-year-old men. J. Neurol. Sci. 1988, 84, 275–294. [Google Scholar] [CrossRef]
- Wokke, J.H.J.; Jennekens, F.G.I.; van den Oord, C.J.M.; Veldman, H.; Smit, L.M.E.; Leppink, G.J. Morphological changes in the human end plate with age. J. Neurol. Sci. 1990, 95, 291–310. [Google Scholar] [CrossRef]
- Rowan, S.L.; Rygiel, K.; Purves-Smith, F.M.; Solbak, N.M.; Turnbull, D.M.; Hepple, R.T. Denervation causes fiber atrophy and myosin heavy chain co-expression in senescent skeletal muscle. PLoS ONE 2012, 7, 29082. [Google Scholar] [CrossRef] [PubMed]
- Valdez, G.; Tapia, J.C.; Kang, H.; Clemenson, G.D.; Gage, F.H.; Lichtman, J.W.; Sanes, J.R. Attenuation of age-related changes in mouse neuromuscular synapses by caloric restriction and exercise. Proc. Natl. Acad. Sci. USA 2010, 107, 14863–14868. [Google Scholar] [CrossRef] [PubMed]
- Cerrato, F.; Sparago, A.; Ariani, F.; Brugnoletti, F.; Calzari, L.; Coppedè, F.; De Luca, A.; Gervasini, C.; Giardina, E.; Gurrieri, F.; et al. DNA methylation in the diagnosis of monogenic diseases. Genes 2020, 11, 355. [Google Scholar] [CrossRef] [PubMed]
- Bennett, S.A.; Tanaz, R.; Cobos, S.N.; Torrente, M.P. Epigenetics in amyotrophic lateral sclerosis: A role for histone post-translational modifications in neurodegenerative disease. Transl. Res. 2019, 204, 19–30. [Google Scholar] [CrossRef]
- Pratt, J.; De Vito, G.; Narici, M.; Boreham, C. Neuromuscular Junction Aging: A Role for Biomarkers and Exercise. J. Gerontol. A Biol. Sci. Med. Sci. 2021, 76, 576–585. [Google Scholar] [CrossRef]
- Lepore, E.; Casola, I.; Dobrowolny, G.; Musarò, A. Neuromuscular Junction as an Entity of Nerve-Muscle Communication. Cells 2019, 8, 906. [Google Scholar] [CrossRef]
- Ruegg, M.A. Organization of synaptic myonuclei by Syne proteins and their role during the formation of the nerve-muscle synapse. Proc. Natl. Acad. Sci. USA 2005, 102, 5643–5644. [Google Scholar] [CrossRef]
- Li, L.; Xiong, W.C.; Mei, L. Neuromuscular Junction Formation, Aging, and Disorders. Annu. Rev. Physiol. 2018, 80, 159–188. [Google Scholar] [CrossRef]
- Schaeffer, L.; De Kerchove D’Exaerde, A.; Changeux, J.P. Targeting transcription to the neuromuscular synapse. Neuron 2001, 31, 15–22. [Google Scholar] [CrossRef]
- Saha, R.N.; Pahan, K. HATs and HDACs in neurodegeneration: A tale of disconcerted acetylation homeostasis. Cell Death Differ. 2006, 13, 539–550. [Google Scholar] [CrossRef]
- Battey, E.; Stroud, M.J.; Ochala, J. Using nuclear envelope mutations to explore age-related skeletal muscle weakness. Clin. Sci. 2020, 134, 2177–2187. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xu, R.; Zhu, B.; Yang, X.; Ding, X.; Duan, S.; Xu, T.; Zhuang, Y.; Han, M. Syne-1 and Syne-2 play crucial roles in myonuclear anchorage and motor neuron innervation. Development 2007, 134, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Grady, R.M.; Starr, D.A.; Ackerman, G.L.; Sanes, J.R.; Han, M. Syne proteins anchor muscle nuclei at the neuromuscular junction. Proc. Natl. Acad. Sci. USA 2005, 102, 4359–4364. [Google Scholar] [CrossRef] [PubMed]
- Burke, B.; Stewart, C.L. The laminopathies: The functional architecture of the nucleus and its contribution to disease. Annu. Rev. Genomics Hum. Genet. 2006, 7, 369–405. [Google Scholar] [CrossRef]
- Burke, B.; Stewart, C.L. The nuclear lamins: Flexibility in function. Nat. Rev. Mol. Cell Biol. 2013, 14, 13–24. [Google Scholar] [CrossRef]
- Hutchison, C.J. Lamins: Building blocks or regulators of gene expression? Nat. Rev. Mol. Cell Biol. 2002, 3, 848–858. [Google Scholar] [CrossRef]
- Mounkes, L.; Kozlov, S.; Burke, B.; Stewart, C.L. The laminopathies: Nuclear structure meets disease. Curr. Opin. Genet. Dev. 2003, 13, 223–230. [Google Scholar] [CrossRef]
- Broers, J.L.V.; Ramaekers, F.C.S.; Bonne, G.; Ben Yaou, R.; Hutchison, C.J. Nuclear lamins: Laminopathies and their role in premature ageing. Physiol. Rev. 2006, 86, 967–1008. [Google Scholar] [CrossRef]
- De Sandre-Giovannoli, A.; Chaouch, M.; Kozlov, S.; Vallat, J.M.; Tazir, M.; Kassouri, N.; Szepetowski, P.; Hammadouche, T.; Vandenberghe, A.; Stewart, C.L.; et al. Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am. J. Hum. Genet. 2002, 70, 726–736. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T. Lamin A-dependent nuclear defects in human aging. Science 2006, 312, 1059–1063. [Google Scholar] [CrossRef] [PubMed]
- Gao, N.; Zhao, K.; Cao, Y.; Ren, X.; Jing, H.; Xing, G.; Xiong, W.C.; Mei, L. A role of lamin A/C in preventing neuromuscular junction decline in mice. J. Neurosci. 2020, 40, 7203–7215. [Google Scholar] [CrossRef] [PubMed]
- Coppedè, F. Epigenetics of neuromuscular disorders. Epigenomics 2020, 12, 2125–2139. [Google Scholar] [CrossRef] [PubMed]
- Azpurua, J.; Eaton, B.A. Neuronal epigenetics and the aging synapse. Front. Cell. Neurosci. 2015, 9. [Google Scholar] [CrossRef]
- Latcheva, N.K.; Viveiros, J.M.; Waddell, E.A.; Nguyen, P.T.T.; Liebl, F.L.W.; Marenda, D.R. Epigenetic crosstalk: Pharmacological inhibition of HDACs can rescue defective synaptic morphology and neurotransmission phenotypes associated with loss of the chromatin reader Kismet. Mol. Cell. Neurosci. 2018, 87, 77–85. [Google Scholar] [CrossRef]
- Osseni, A.; Ravel-Chapuis, A.; Thomas, J.-L.; Gache, V.; Schaeffer, L.; Jasmin, B.J. HDAC6 regulates microtubule stability and clustering of AChRs at neuromuscular junctions. J. Cell Biol. 2020, 219. [Google Scholar] [CrossRef]
- Lu, L.; Liu, Y.; Liu, Y.; Zhang, F.; Wang, H.; Zhang, Q.; Pan, F. Secreted miRNAs in the tripartite neuromuscular junction. ExRNA 2019, 1, 1–4. [Google Scholar] [CrossRef]
- Castro, R.; Taetzsch, T.; Vaughan, S.K.; Godbe, K.; Chappell, J.; Settlage, R.E.; Valdez, G. Specific labeling of synaptic schwann cells reveals unique cellular and molecular features. Elife 2020, 9, 1–19. [Google Scholar] [CrossRef]
- Feng, Z.; Ko, C.P. Schwann cells promote synaptogenesis at the neuromuscular junction via transforming growth factor-β1. J. Neurosci. 2008, 28, 9599–9609. [Google Scholar] [CrossRef]
- Sugiura, Y.; Lin, W. Neuron-glia interactions: The roles of Schwann cells in neuromuscular synapse formation and function. Biosci. Rep. 2011, 31, 295–302. [Google Scholar] [CrossRef]
- Court, F.A.; Gillingwater, T.H.; Melrose, S.; Sherman, D.L.; Greenshields, K.N.; Morton, A.J.; Harris, J.B.; Willison, H.J.; Ribchester, R.R. Identity, developmental restriction and reactivity of extralaminar cells capping mammalian neuromuscular junctions. J. Cell Sci. 2008, 121, 3901–3911. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Freire, M.; de Cabo, R.; Studenski, S.A.; Ferrucci, L. The neuromuscular junction: Aging at the crossroad between nerves and muscle. Front. Aging Neurosci. 2014, 6, 208. [Google Scholar] [CrossRef] [PubMed]
- Rudolf, R.; Khan, M.M.; Labeit, S.; Deschenes, M.R. Degeneration of neuromuscular junction in age and dystrophy. Front. Aging Neurosci. 2014, 6, 99. [Google Scholar] [CrossRef] [PubMed]
- Willadt, S.; Nash, M.; Slater, C. Age-related changes in the structure and function of mammalian neuromuscular junctions. Ann. N. Y. Acad. Sci. 2018, 1412, 41–53. [Google Scholar] [CrossRef]
- Deschenes, M.R.; Hurst, T.E.; Ramser, A.E.; Sherman, E.G. Presynaptic to postsynaptic relationships of the neuromuscular junction are held constant across age and muscle fiber type. Dev. Neurobiol. 2013, 73, 744–753. [Google Scholar] [CrossRef]
- Bowen, D.C.; Park, J.S.; Bodine, S.; Stark, J.L.; Valenzuela, D.M.; Stitt, T.N.; Yancopoulos, G.D.; Lindsay, R.M.; Glass, D.J.; Distefano, P.S. Localization and regulation of MuSK at the neuromuscular junction. Dev. Biol. 1998, 199, 309–319. [Google Scholar] [CrossRef]
- Itou, Y.; Nochi, R.; Kuribayashi, H.; Saito, Y.; Hisatsune, T. Cholinergic activation of hippocampal neural stem cells in aged dentate gyrus. Hippocampus 2011, 21, 446–459. [Google Scholar] [CrossRef]
- Taetzsch, T.; Valdez, G. NMJ maintenance and repair in aging. Curr. Opin. Physiol. 2018, 4, 57–64. [Google Scholar] [CrossRef]
- Jang, Y.C.; Van Remmen, H. Age-associated alterations of the neuromuscular junction. Exp. Gerontol. 2011, 46, 193–198. [Google Scholar] [CrossRef]
- Kang, H.; Tian, L.; Mikesh, M.; Lichtman, J.W.; Thompson, W.J. Terminal schwann cells participate in neuromuscular synapse remodeling during reinnervation following nerve injury. J. Neurosci. 2014, 34, 6323–6333. [Google Scholar] [CrossRef]
- Barik, A.; Lu, Y.; Sathyamurthy, A.; Bowman, A.; Shen, C.; Li, L.; Xiong, W.C.; Mei, L. LRP4 is critical for neuromuscular junction maintenance. J. Neurosci. 2014, 34, 13892–13905. [Google Scholar] [CrossRef]
- Ohno, K.; Ohkawara, B.; Ito, M. Agrin-LRP4-MuSK signaling as a therapeutic target for myasthenia gravis and other neuromuscular disorders. Expert Opin. Ther. Targets 2017, 21, 949–958. [Google Scholar] [CrossRef] [PubMed]
- DeChiara, T.M.; Bowen, D.C.; Valenzuela, D.M.; Simmons, M.V.; Poueymirou, W.T.; Thomas, S.; Kinetz, E.; Compton, D.L.; Rojas, E.; Park, J.S.; et al. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell 1996, 85, 501–512. [Google Scholar] [CrossRef]
- Bowen, D.C.; Sugiyama, J.; Ferns, M.; Hall, Z.W. Neural agrin activates a high-affinity receptor in C2 muscle cells that is unresponsive to muscle agrin. J. Neurosci. 1996, 16, 3791–3797. [Google Scholar] [CrossRef] [PubMed]
- Ferns, M.J.; Campanelli, J.T.; Hoch, W.; Scheller, R.H.; Hall, Z. The ability of agrin to cluster AChRs depends on alternative splicing and on cell surface proteoglycans. Neuron 1993, 11, 491–502. [Google Scholar] [CrossRef]
- Gesemann, M.; Denzer, A.J.; Ruegg, M.A. Acetylcholine receptor-aggregating activity of agrin isoforms and mapping of the active site. J. Cell Biol. 1995, 128, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Burgess, R.W.; Dominguez, B.; Pfaff, S.L.; Sanes, J.R.; Lee, K.F. Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature 2001, 410, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.F.; Cao, G.; Koirala, S.; Reddy, L.V.; Ko, C.P. Schwann cells express active agrin and enhance aggregation of acetylcholine receptors on muscle fibers. J. Neurosci. 2001, 21, 9572–9584. [Google Scholar] [CrossRef] [PubMed]
- Bütikofer, L.; Zurlinden, A.; Bolliger, M.F.; Kunz, B.; Sonderegger, P. Destabilization of the neuromuscular junction by proteolytic cleavage of agrin results in precocious sarcopenia. FASEB J. 2011, 25, 4378–4393. [Google Scholar] [CrossRef]
- Ibebunjo, C.; Chick, J.M.; Kendall, T.; Eash, J.K.; Li, C.; Zhang, Y.; Vickers, C.; Wu, Z.; Clarke, B.A.; Shi, J.; et al. Genomic and Proteomic Profiling Reveals Reduced Mitochondrial Function and Disruption of the Neuromuscular Junction Driving Rat Sarcopenia. Mol. Cell. Biol. 2013, 33, 194–212. [Google Scholar] [CrossRef]
- Dalkin, W.; Taetzsch, T.; Valdez, G. The fibular nerve injury method: A reliable assay to identify and test factors that repair neuromuscular junctions. J. Vis. Exp. 2016, 2016, e54186. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yi, J.; Fu, R.; Liu, E.; Siddique, T.; Rios, E.; Deng, H.X. Hyperactive intracellular calcium signaling associated with localized mitochondrial defects in skeletal muscle of an animal model of amyotrophic lateral sclerosis. J. Biol. Chem. 2010, 285, 705–712. [Google Scholar] [CrossRef]
- Muller, F.L.; Song, W.; Jang, Y.C.; Liu, Y.; Sabia, M.; Richardson, A.; Van Remmen, H. Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am. J. Physiol. Integr. Comp. Physiol. 2007, 293, 1159–1168. [Google Scholar] [CrossRef]
- Banker, B.Q.; Kelly, S.S.; Robbins, N. Neuromuscular transmission and correlative morphology in young and old mice. J. Physiol. 1983, 339, 355–377. [Google Scholar] [CrossRef] [PubMed]
- Dobrowolny, G.; Martini, M.; Scicchitano, B.M.; Romanello, V.; Boncompagni, S.; Nicoletti, C.; Pietrangelo, L.; De Panfilis, S.; Catizone, A.; Bouchè, M.; et al. Muscle Expression of SOD1 G93A Triggers the Dismantlement of Neuromuscular Junction via PKC-Theta. Antioxid. Redox Signal. 2018, 28, 1105–1119. [Google Scholar] [CrossRef]
- Rocha, M.C.; Pousinha, P.A.; Correia, A.M.; Sebastião, A.M.; Ribeiro, J.A. Early Changes of Neuromuscular Transmission in the SOD1(G93A) Mice Model of ALS Start Long before Motor Symptoms Onset. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Scialò, F.; Fernández-Ayala, D.J.; Sanz, A. Role of Mitochondrial Reverse Electron Transport in ROS Signaling: Potential Roles in Health and Disease. Front. Physiol. 2017, 8, 428. [Google Scholar] [CrossRef]
- Li, A.; Yi, J.; Li, X.; Zhou, J. Physiological Ca2+ Transients Versus Pathological Steady-State Ca2+ Elevation, Who Flips the ROS Coin in Skeletal Muscle Mitochondria. Front. Physiol. 2020, 11. [Google Scholar] [CrossRef]
- Hyatt, H.W.; Powers, S.K. Mitochondrial dysfunction is a common denominator linking skeletal muscle wasting due to disease, aging, and prolonged inactivity. Antioxidants 2021, 10, 588. [Google Scholar] [CrossRef]
- Simon, H.U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell. Longev. 2016, 2016, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Scicchitano, B.M.; Pelosi, L.; Sica, G.; Musarò, A. The physiopathologic role of oxidative stress in skeletal muscle. Mech. Ageing Dev. 2018, 170, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, L.; Gonzalez de Aguilar, J.L.; Echaniz-Laguna, A.; Eschbach, J.; Rene, F.; Oudart, H.; Halter, B.; Huze, C.; Schaeffer, L.; Bouillaud, F.; et al. Muscle mitochondrial uncoupling dismantles neuromuscular junction and triggers distal degeneration of motor neurons. PLoS ONE 2009, 4, e5390. [Google Scholar] [CrossRef] [PubMed]
- Fischer, L.R.; Li, Y.; Asress, S.A.; Jones, D.P.; Glass, J.D. Absence of SOD1 leads to oxidative stress in peripheral nerve and causes a progressive distal motor axonopathy. Exp. Neurol. 2012, 233, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Fischyer, L.R.; Culver, D.G.; Tennant, P.; Davis, A.A.; Wang, M.; Castellano-Sanchez, A.; Khan, J.; Polak, M.A.; Glass, J.D. Amyotrophic lateral sclerosis is a distal axonopathy: Evidence in mice and man. Exp. Neurol. 2004, 185, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef] [PubMed]
- Hayes, L.R.; Asress, S.A.; Li, Y.; Galkin, A.; Stepanova, A.; Kawamata, H.; Manfredi, G.; Glass, J.D. Distal denervation in the SOD1 knockout mouse correlates with loss of mitochondria at the motor nerve terminal. Exp. Neurol. 2019, 318, 251–257. [Google Scholar] [CrossRef]
- Pollari, E.; Goldsteins, G.; Bart, G.; Koistinaho, J.; Giniatullin, R. The role of oxidative stress in degeneration of the neuromuscular junction in amyotrophic lateral sclerosis. Front. Cell. Neurosci. 2014, 8, 131. [Google Scholar] [CrossRef]
- Martin, L.J.; Wong, M. Skeletal Muscle-Restricted Expression of Human SOD1 in Transgenic Mice Causes a Fatal ALS-Like Syndrome. Front. Neurol. 2020, 11. [Google Scholar] [CrossRef]
- Wong, M.; Martin, L.J. Skeletal muscle-restricted expression of human SOD1 causes motor neuron degeneration in transgenic mice. Hum. Mol. Genet. 2010, 19, 2284–2302. [Google Scholar] [CrossRef]
- Ji, L.L.; Yeo, D.; Kang, C.; Zhang, T. The role of mitochondria in redox signaling of muscle homeostasis. J. Sport Heal. Sci. 2020, 9, 386–393. [Google Scholar] [CrossRef]
- Giniatullin, A.; Petrov, A.; Giniatullin, R. The involvement of P2Y12 receptors, NADPH oxidase, and lipid rafts in the action of extracellular ATP on synaptic transmission at the frog neuromuscular junction. Neuroscience 2015, 285, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.J.; Vasilaki, A.; McArdle, A. Cellular mechanisms underlying oxidative stress in human exercise. Free Radic. Biol. Med. 2016, 98, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Cabrera, M.C.; Domenech, E.; Viña, J. Moderate exercise is an antioxidant: Upregulation of antioxidant genes by training. Free Radic. Biol. Med. 2008, 44, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Wright, V.P.; Reiser, P.J.; Clanton, T.L. Redox modulation of global phosphatase activity and protein phosphorylation in intact skeletal muscle. J. Physiol. 2009, 587, 5767–5781. [Google Scholar] [CrossRef] [PubMed]
- Bejma, J.; Ji, L.L. Rapid communication aging and acute exercise enhance free radical generation in rat skeletal muscle. J. Appl. Physiol. 1999, 87, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Damiano, S.; Muscariello, E.; La Rosa, G.; Di Maro, M.; Mondola, P.; Santillo, M. Dual role of reactive oxygen species in muscle function: Can antioxidant dietary supplements counteract age-related sarcopenia? Int. J. Mol. Sci. 2019, 20, 3815. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Ray, U.S.; Saha, M.; Singh, S.N.; Tomar, O.S. Antioxidant and redox status after maximal aerobic exercise at high altitude in acclimatized lowlanders and native highlanders. Eur. J. Appl. Physiol. 2009, 106, 807–814. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Singh, S.N.; Saha, M.; Kain, T.C.; Tyagi, A.K.; Ray, U.S. Antioxidant and oxidative stress responses of sojourners at high altitude in different climatic temperatures. Int. J. Biometeorol. 2010, 54, 85–92. [Google Scholar] [CrossRef]
- Carnio, S.; LoVerso, F.; Baraibar, M.A.; Longa, E.; Khan, M.M.; Maffei, M.; Reischl, M.; Canepari, M.; Loefler, S.; Kern, H.; et al. Autophagy Impairment in Muscle Induces Neuromuscular Junction Degeneration and Precocious Aging. Cell Rep. 2014, 8, 1509–1521. [Google Scholar] [CrossRef]
- You, J.S.; Singh, N.; Reyes-Ordonez, A.; Khanna, N.; Bao, Z.; Zhao, H.; Chen, J. ARHGEF3 Regulates Skeletal Muscle Regeneration and Strength through Autophagy. Cell Rep. 2021, 34. [Google Scholar] [CrossRef] [PubMed]
- Sharples, A.P.; Seaborne, R.A.; Stewart, C.E. Epigenetics of Skeletal Muscle Aging. In Epigenetics of Aging and Longevity; Elsevier BV: Amsterdam, The Netherlands, 2018; pp. 389–416. [Google Scholar]
- Gensous, N.; Bacalini, M.G.; Pirazzini, C.; Marasco, E.; Giuliani, C.; Ravaioli, F.; Mengozzi, G.; Bertarelli, C.; Palmas, M.G.; Franceschi, C.; et al. The epigenetic landscape of age-related diseases: The geroscience perspective. Biogerontology 2017, 18, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Calvanese, V.; Lara, E.; Kahn, A.; Fraga, M.F. The role of epigenetics in aging and age-related diseases. Ageing Res. Rev. 2009, 8, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Tyler, J.K. Epigenetics and aging. Sci. Adv. 2016, 2, e1600584. [Google Scholar] [CrossRef] [PubMed]
- Amir, R.E.; Van Den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl- CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef]
- Zhong, X.; Li, H.; Chang, Q. MeCP2 phosphorylation is required for modulating synaptic scaling through mGluR5. J. Neurosci. 2012, 32, 12841–12847. [Google Scholar] [CrossRef] [PubMed]
- Tajrishi, M.M.; Shin, J.; Hetman, M.; Kumar, A. DNA methyltransferase 3a and mitogen-activated protein kinase signaling regulate the expression of fibroblast growth factor-inducible 14 (Fn14) during denervation-induced skeletal muscle atrophy. J. Biol. Chem. 2014, 289, 19985–19999. [Google Scholar] [CrossRef]
- Mittal, A.; Bhatnagar, S.; Kumar, A.; Lach-Trifilieff, E.; Wauters, S.; Li, H.; Makonchuk, D.Y.; Glass, D.J.; Kumar, A. The TWEAK-Fn14 system is a critical regulator of denervation-induced skeletal muscle atrophy in mice. J. Cell Biol. 2010, 188, 833–849. [Google Scholar] [CrossRef]
- Bhatnagar, S.; Kumar, A. The TWEAK-Fn14 System: Breaking the Silence of Cytokine-Induced Skeletal Muscle Wasting. Curr. Mol. Med. 2011, 12, 3–13. [Google Scholar] [CrossRef]
- Purohit, J.S.; Chaturvedi, M.M. Chromatin and Aging. In Topics in Biomedical Gerontology; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2017; pp. 205–241. [Google Scholar]
- Consalvi, S.; Sandoná, M.; Saccone, V. Epigenetic reprogramming of muscle progenitors: Inspiration for clinical therapies. Stem Cells Int. 2016, 2016, 1–11. [Google Scholar] [CrossRef]
- Pigna, E.; Simonazzi, E.; Sanna, K.; Bernadzki, K.M.; Proszynski, T.; Heil, C.; Palacios, D.; Adamo, S.; Moresi, V. Histone deacetylase 4 protects from denervation and skeletal muscle atrophy in a murine model of amyotrophic lateral sclerosis. EBioMedicine 2019, 40, 717–732. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.H.; Valdez, G.; Moresi, V.; Qi, X.; McAnally, J.; Elliott, J.L.; Bassel-Duby, R.; Sanes, J.R.; Olson, E.N. MicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science 2009, 326, 1549–1554. [Google Scholar] [CrossRef] [PubMed]
- Rouaux, C.; Jokic, N.; Mbebi, C.; Boutillier, S.; Loeffler, J.P.; Boutillier, A.L. Critical loss of CBP/p300 histone acetylase activity by caspase-6 during neurodegeneration. EMBO J. 2003, 22, 6537–6549. [Google Scholar] [CrossRef] [PubMed]
- Rouaux, C.; Loeffler, J.-P.; Boutillier, A.-L. Targeting CREB-binding protein (CBP) loss of function as a therapeutic strategy in neurological disorders. Biochem. Pharmacol. 2004, 68, 1157–1164. [Google Scholar] [CrossRef]
- Langley, B.; Gensert, J.A.M.; Beal, M.F.; Ratan, R.R. Remodeling chromatin and stress resistance in the central nervous system: Histone deacetylase inhibitors as novel and broadly effective neuroprotective agents. Curr. Drug Targets CNS Neurol. Disord. 2005, 4, 41–50. [Google Scholar] [CrossRef]
- Yoo, Y.E.; Ko, C.P. Treatment with trichostatin A initiated after disease onset delays disease progression and increases survival in a mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2011, 231, 147–159. [Google Scholar] [CrossRef]
- Piepers, S.; Veldink, J.H.; De Jong, S.W.; Van Der Tweel, I.; Van Der Pol, W.L.; Uijtendaal, E.V.; Schelhaas, H.J.; Scheffer, H.; De Visser, M.; De Jong, J.M.B.V.; et al. Randomized sequential trial of valproic acid in amyotrophic lateral sclerosis. Ann. Neurol. 2009, 66, 227–234. [Google Scholar] [CrossRef]
- Rouaux, C.; Panteleeva, I.; René, F.; De Aguilar, J.L.G.; Echaniz-Laguna, A.; Dupuis, L.; Menger, Y.; Boutillier, A.L.; Loeffler, J.P. Sodium valproate exerts neuroprotective effects in vivo through CREB-binding protein-dependent mechanisms but does not improve survival in an amyotrophic lateral sclerosis mouse model. J. Neurosci. 2007, 27, 5535–5545. [Google Scholar] [CrossRef]
- Petri, S.; Kiaei, M.; Kipiani, K.; Chen, J.; Calingasan, N.Y.; Crow, J.P.; Beal, M.F. Additive neuroprotective effects of a histone deacetylase inhibitor and a catalytic antioxidant in a transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2006, 22, 40–49. [Google Scholar] [CrossRef]
- Ryu, H.; Smith, K.; Camelo, S.I.; Carreras, I.; Lee, J.; Iglesias, A.H.; Dangond, F.; Cormier, K.A.; Cudkowicz, M.E.; Brown, R.H.; et al. Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J. Neurochem. 2005, 93, 1087–1098. [Google Scholar] [CrossRef]
- Kim, H.J.; Im, W.; Kim, S.; Sung, H.K.; Sung, J.J.; Kim, M.; Lee, K.W. Calcium-influx increases SOD1 aggregates via nitric oxide in cultured motor neurons. Exp. Mol. Med. 2007, 39, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Ageta-Ishihara, N.; Nagatsu, S.; Takao, K.; Komine, O.; Endo, F.; Miyakawa, T.; Misawa, H.; Takahashi, R.; Kinoshita, M.; et al. SIRT1 overexpression ameliorates a mouse model of SOD1-linked amyotrophic lateral sclerosis via HSF1/HSP70i chaperone system. Mol. Brain 2014, 7, 62. [Google Scholar] [CrossRef]
- Herskovits, A.Z.; Hunter, T.A.; Maxwell, N.; Pereira, K.; Whittaker, C.A.; Valdez, G.; Guarente, L.P. SIRT1 deacetylase in aging-induced neuromuscular degeneration and amyotrophic lateral sclerosis. Aging Cell 2018, 17, e12839. [Google Scholar] [CrossRef] [PubMed]
- Snyder-Warwick, A.K.; Satoh, A.; Santosa, K.B.; Imai, S.-I.; Jablonka-Shariff, A. Hypothalamic Sirt1 protects terminal Schwann cells and neuromuscular junctions from age-related morphological changes. Aging Cell 2018, 17, e12776. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Ingolia, N.T.; Weissman, J.S.; Bartel, D.P. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 2010, 466, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Ørom, U.A.; Nielsen, F.C.; Lund, A.H. MicroRNA-10a Binds the 5′UTR of Ribosomal Protein mRNAs and Enhances Their Translation. Mol. Cell 2008, 30, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V.; Lee, R.C. Identification of microRNAs and other tiny noncoding RNAs by cDNA cloning. Methods Mol. Biol. 2004, 265, 131–158. [Google Scholar] [PubMed]
- Winter, J.; Jung, S.; Keller, S.; Gregory, R.I.; Diederichs, S. Many roads to maturity: MicroRNA biogenesis pathways and their regulation. Nat. Cell Biol. 2009, 11, 228–234. [Google Scholar] [CrossRef]
- Callis, T.E.; Deng, Z.; Chen, J.F.; Wang, D.Z. Muscling through the microRNA world. Exp. Biol. Med. 2008, 233, 131–138. [Google Scholar] [CrossRef]
- McCarthy, J.J. General Commentary: MicroRNA and skeletal muscle function: Novel potential roles in exercise, diseases, and aging. Front. Physiol. 2014. [Google Scholar] [CrossRef]
- van Rooij, E.; Quiat, D.; Johnson, B.A.; Sutherland, L.B.; Qi, X.; Richardson, J.A.; Kelm, R.J.; Olson, E.N. A Family of microRNAs Encoded by Myosin Genes Governs Myosin Expression and Muscle Performance. Dev. Cell 2009, 17, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.H.; Liu, N.; van Rooij, E.; Olson, E.N. MicroRNA control of muscle development and disease. Curr. Opin. Cell Biol. 2009, 21, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Zacharewicz, E.; Lamon, S.; Russell, A.P. MicroRNAs in skeletal muscle and their regulation with exercise, ageing, and disease. Front. Physiol. 2013, 4, 266. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Nie, Q.; Zhang, X. MicroRNAs Involved in Skeletal Muscle Differentiation. J. Genet. Genomics 2013, 40, 107–116. [Google Scholar] [CrossRef]
- McGregor, R.A.; Poppitt, S.D.; Cameron-Smith, D. Role of microRNAs in the age-related changes in skeletal muscle and diet or exercise interventions to promote healthy aging in humans. Ageing Res. Rev. 2014, 17, 25–33. [Google Scholar] [CrossRef]
- Sharma, M.; Juvvuna, P.K.; Kukreti, H.; McFarlane, C. Mega roles of microRNAs in regulation of skeletal muscle health and disease. Front. Physiol. 2014, 5. [Google Scholar] [CrossRef]
- Yin, J.; Qian, Z.; Chen, Y.; Li, Y.; Zhou, X. MicroRNA regulatory networks in the pathogenesis of sarcopenia. J. Cell. Mol. Med. 2020, 24, 4900–4912. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, B.; He, J.; Chen, D. From nutrient to MicroRNA: A novel insight into cell signaling involved in skeletal muscle development and disease. Int. J. Biol. Sci. 2016, 12, 1247–1261. [Google Scholar] [CrossRef]
- Valsecchi, V.; Anzilotti, S.; Serani, A.; Laudati, G.; Brancaccio, P.; Guida, N.; Cuomo, O.; Pignataro, G.; Annunziato, L. miR-206 Reduces the Severity of Motor Neuron Degeneration in the Facial Nuclei of the Brainstem in a Mouse Model of SMA. Mol. Ther. 2020, 28, 1154–1166. [Google Scholar] [CrossRef]
- Bruneteau, G.; Simonet, T.; Bauché, S.; Mandjee, N.; Malfatti, E.; Girard, E.; Tanguy, M.L.; Behin, A.; Khiami, F.; Sariali, E.; et al. Muscle histone deacetylase 4 upregulation in amyotrophic lateral sclerosis: Potential role in reinnervation ability and disease progression. Brain 2013, 136, 2359–2368. [Google Scholar] [CrossRef] [PubMed]
- Valdez, G.; Heyer, M.P.; Feng, G.; Sanes, J.R. The Role of Muscle microRNAs in Repairing the Neuromuscular Junction. PLoS ONE 2014, 9, e93140. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Xue, P.; Chen, H.; Yuan, M.; Kang, Y.; Duscher, D.; Machens, H.G.; Chen, Z. Denervation drives skeletal muscle atrophy and induces mitochondrial dysfunction, mitophagy and apoptosis via miR-142a-5p/MFN1 axis. Theranostics 2020, 10, 1415–1432. [Google Scholar] [CrossRef]
- Sison, S.L.; Patitucci, T.N.; Seminary, E.R.; Villalon, E.; Lorson, C.L.; Ebert, A.D. Astrocyte-produced miR-146a as a mediator of motor neuron loss in spinal muscular atrophy. Hum. Mol. Genet. 2017, 26, 3409–3420. [Google Scholar] [CrossRef] [PubMed]
- Snieckute, G.; Baltaci, O.; Liu, H.; Li, L.; Hu, Z.; Pocock, R. mir-234 controls neuropeptide release at the Caenorhabditis elegans neuromuscular junction. Mol. Cell. Neurosci. 2019, 98, 70–81. [Google Scholar] [CrossRef]
- Kim, K.C.; Choi, S.-W. Nutritional Epigenetics and Aging; Springer: Cham, Switzerland, 2015; pp. 1–28. [Google Scholar]
- Murphy, M.M.; Lawson, J.A.; Mathew, S.J.; Hutcheson, D.A.; Kardon, G. Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development 2011, 138, 3625–3637. [Google Scholar] [CrossRef] [PubMed]
- Lepper, C.; Partridge, T.A.; Fan, C.M. An absolute requirement for pax7-positive satellite cells in acute injury-induced skeletal muscle regeneration. Development 2011, 138, 3639–3646. [Google Scholar] [CrossRef] [PubMed]
- Dumont, N.A.; Bentzinger, C.F.; Sincennes, M.C.; Rudnicki, M.A. Satellite cells and skeletal muscle regeneration. Compr. Physiol. 2015, 5, 1027–1059. [Google Scholar]
- García-Prat, L.; Sousa-Victor, P.; Muñoz-Cánoves, P. Functional dysregulation of stem cells during aging: A focus on skeletal muscle stem cells. FEBS J. 2013, 280, 4051–4062. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.C.; Sinha, M.; Cerletti, M.; Dall’osso, C.; Wagers, A.J. Skeletal muscle stem cells: Effects of aging and metabolism on muscle regenerative function. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 101–111. [Google Scholar] [CrossRef]
- Wang, X.; Pickrell, A.M.; Rossi, S.G.; Pinto, M.; Dillon, L.M.; Hida, A.; Rotundo, R.L.; Moraes, C.T. Transient systemic mtDNA damage leads to muscle wasting by reducing the satellite cell pool. Hum. Mol. Genet. 2013, 22, 3976–3986. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Klose, A.; Forman, S.; Paris, N.D.; Wei-LaPierre, L.; Cortés-Lopéz, M.; Tan, A.; Flaherty, M.; Miura, P.; Dirksen, R.T.; et al. Loss of adult skeletal muscle stem cells drives age-related neuromuscular junction degeneration. Elife 2017, 6. [Google Scholar] [CrossRef]
- Liu, W.; Wei-LaPierre, L.; Klose, A.; Dirksen, R.T.; Chakkalakal, J.V. Inducible depletion of adult skeletal muscle stem cells impairs the regeneration of neuromuscular junctions. Elife 2015, 4, e09221. [Google Scholar] [CrossRef] [PubMed]
- Arentson-Lantz, E.J.; English, K.L.; Paddon-Jones, D.; Fry, C.S. Fourteen days of bed rest induces a decline in satellite cell content and robust atrophy of skeletal muscle fibers in middle-aged adults. J. Appl. Physiol. 2016, 120, 965–975. [Google Scholar] [CrossRef]
- Sousa-Victor, P.; Gutarra, S.; García-Prat, L.; Rodriguez-Ubreva, J.; Ortet, L.; Ruiz-Bonilla, V.; Jardí, M.; Ballestar, E.; González, S.; Serrano, A.L.; et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 2014, 506, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Scicchitano, B.M.; Sica, G.; Musarò, A. Stem Cells and Tissue Niche: Two Faces of the Same Coin of Muscle Regeneration. Eur. J. Transl. Myol. 2016, 26, 6125. [Google Scholar] [CrossRef] [PubMed]
- Barbiera, A.; Pelosi, L.; Sica, G.; Scicchitano, B.M. Nutrition and microRNAs: Novel insights to fight sarcopenia. Antioxidants 2020, 9, 951. [Google Scholar] [CrossRef] [PubMed]
- Conboy, I.M.; Conboy, M.J.; Wagers, A.J.; Girma, E.R.; Weismann, I.L.; Rando, T.A. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature 2005, 433, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Murphy, T.; Thuret, S. The systemic milieu as a mediator of dietary influence on stem cell function during ageing. Ageing Res. Rev. 2015, 19, 53–64. [Google Scholar] [CrossRef]
- Scicchitano, B.M.; Sica, G. The Beneficial Effects of Taurine to Counteract Sarcopenia. Curr. Protein Pept. Sci. 2018, 19, 673–680. [Google Scholar] [CrossRef]
- 164. Druz Aliaksandr, Betenbaugh Michael, S.J. Glucose Depletion Activates mmu-miR-466h-5p Expression Through Oxidative Stress and Inhibition of Histone Deacetylation. Nucleic Acids Res. 2012, 40, 7291–7302. [CrossRef]
- Drummond, M.J.; McCarthy, J.J.; Fry, C.S.; Esser, K.A.; Rasmussen, B.B. Aging differentially affects human skeletal muscle microRNA expression at rest and after an anabolic stimulus of resistance exercise and essential amino acids. Am. J. Physiol. Metab. 2008, 295. [Google Scholar] [CrossRef] [PubMed]
- Dey, N.; Das, F.; Mariappan, M.M.; Mandal, C.C.; Ghosh-Choudhury, N.; Kasinath, B.S.; Choudhury, G.G. MicroRNA-21 orchestrates high glucose-induced signals to TOR complex 1, resulting in renal cell pathology in diabetes. J. Biol. Chem. 2011, 286, 25586–25603. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Huang, Z.; Chen, D.; Yang, T.; Liu, G. MicroRNA-27a is induced by leucine and contributes to leucine-induced proliferation promotion in C2C12 cells. Int. J. Mol. Sci. 2013, 14, 14076–14084. [Google Scholar] [CrossRef] [PubMed]
- Iannone, F.; Montesanto, A.; Cione, E.; Crocco, P.; Caroleo, M.C.; Dato, S.; Rose, G.; Passarino, G. Expression patterns of muscle-specific miR-133b and miR-206 correlate with nutritional status and sarcopenia. Nutrients 2020, 12, 297. [Google Scholar] [CrossRef] [PubMed]
- Monroy, A.; Lithgow, G.J.; Alavez, S. Curcumin and neurodegenerative diseases. BioFactors 2013, 39, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Pellavio, G.; Rui, M.; Caliogna, L.; Martino, E.; Gastaldi, G.; Collina, S.; Laforenza, U. Regulation of aquaporin functional properties mediated by the antioxidant effects of natural compounds. Int. J. Mol. Sci. 2017, 18, 2665. [Google Scholar] [CrossRef] [PubMed]
- Boots, A.W.; Haenen, G.R.M.M.; Bast, A. Health effects of quercetin: From antioxidant to nutraceutical. Eur. J. Pharmacol. 2008, 585, 325–337. [Google Scholar] [CrossRef]
- Wu, Q.; Wang, X.; Nepovimova, E.; Wang, Y.; Yang, H.; Li, L.; Zhang, X.; Kuca, K. Antioxidant agents against trichothecenes: New hints for oxidative stress treatment. Oncotarget 2017, 8, 110708–110726. [Google Scholar] [CrossRef]
- Tresserra-Rimbau, A.; Arranz, S.; Vallverdu-Queralt, A. New Insights into the Benefits of Polyphenols in Chronic Diseases. Oxid. Med. Cell. Longev. 2017, 2017, 1–2. [Google Scholar] [CrossRef]
- Nieman, D.C.; Laupheimer, M.W.; Ranchordas, M.K.; Burke, L.M.; Stear, S.J.; Castell, L.M. A-Z of nutritional supplements: Dietary supplements, sports nutrition foods and ergogenic aids for health and performance—Part 33. Br. J. Sports Med. 2012, 46, 618–620. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Damiano, S.; Sasso, A.; De Felice, B.; Di Gregorio, I.; La Rosa, G.; Lupoli, G.A.; Belfiore, A.; Mondola, P.; Santillo, M. Quercetin increases MUC2 and MUC5AC gene expression and secretion in intestinal goblet cell-like LS174T via PLC/PKCα/ERK1-2 pathway. Front. Physiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Uchitomi, R.; Oyabu, M.; Kamei, Y. Vitamin d and sarcopenia: Potential of vitamin D supplementation in sarcopenia prevention and treatment. Nutrients 2020, 12, 3189. [Google Scholar] [CrossRef] [PubMed]
- Wintermeyer, E.; Ihle, C.; Ehnert, S.; Stöckle, U.; Ochs, G.; de Zwart, P.; Flesch, I.; Bahrs, C.; Nussler, A.K. Crucial role of vitamin D in the musculoskeletal system. Nutrients 2016, 8, 319. [Google Scholar] [CrossRef]
- Snijder, M.B.; Van Schoor, N.M.; Pluijm, S.M.F.; Van Dam, R.M.; Visser, M.; Lips, P. Vitamin D status in relation to one-year risk of recurrent falling in older men and women. J. Clin. Endocrinol. Metab. 2006, 91, 2980–2985. [Google Scholar] [CrossRef]
- Huo, Y.R.; Suriyaarachchi, P.; Gomez, F.; Curcio, C.L.; Boersma, D.; Muir, S.W.; Montero-Odasso, M.; Gunawardene, P.; Demontiero, O.; Duque, G. Phenotype of Osteosarcopenia in Older Individuals With a History of Falling. J. Am. Med. Dir. Assoc. 2015, 16, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Baek, K.H.; Song, K.H.; Il Kang, M.; Park, C.Y.; Lee, W.Y.; Won Oh, K. Vitamin D deficiency is associated with sarcopenia in older Koreans, regardless of obesity: The fourth Korea National Health and Nutrition Examination Surveys (KNHANES IV) 2009. J. Clin. Endocrinol. Metab. 2011, 96, 3250–3256. [Google Scholar] [CrossRef]
- Cummings, S.R.; Kiel, D.P.; Black, D.M. Vitamin D Supplementation and increased risk of falling: A cautionary tale of vitamin supplements retold. JAMA Intern. Med. 2016, 176, 171–172. [Google Scholar] [CrossRef]
- Gifondorwa, D.J.; Thompson, T.D.; Wiley, J.; Culver, A.E.; Shetler, P.K.; Rocha, G.V.; Ma, Y.L.; Krishnan, V.; Bryant, H.U. Vitamin D and/or calcium deficient diets may differentially affect muscle fiber neuromuscular junction innervation. Muscle Nerve 2016, 54, 1120–1132. [Google Scholar] [CrossRef]
- Szymańska, R.; Nowicka, B.; Kruk, J. Vitamin E—Occurrence, Biosynthesis by Plants and Functions in Human Nutrition. Mini-Reviews Med. Chem. 2016, 17, 1039–1052. [Google Scholar] [CrossRef]
- Zhang, Y.-G.; Wu, S.; Yi, J.; Xia, Y.; Jin, D.; Zhou, J.; Sun, J. Target Intestinal Microbiota to Alleviate Disease Progression in Amyotrophic Lateral Sclerosis. Clin. Ther. 2017, 39, 322–336. [Google Scholar] [CrossRef]
- Stockinger, J.; Maxwell, N.; Shapiro, D.; deCabo, R.; Valdez, G. Caloric Restriction Mimetics Slow Aging of Neuromuscular Synapses and Muscle Fibers. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 73, 21–28. [Google Scholar] [CrossRef]
- Bar, S.; Prasad, M.; Datta, R. Neuromuscular degeneration and locomotor deficit in a Drosophila model of mucopolysaccharidosis VII is attenuated by treatment with resveratrol. DMM Dis. Model. Mech. 2018, 11. [Google Scholar] [CrossRef]
- Han, S.; Choi, J.R.; Soon Shin, K.; Kang, S.J. Resveratrol upregulated heat shock proteins and extended the survival of G93A-SOD1 mice. Brain Res. 2012, 1483, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Nguyen, M.D.; Dobbin, M.M.; Fischer, A.; Sananbenesi, F.; Rodgers, J.T.; Delalle, I.; Baur, J.A.; Sui, G.; Armour, S.M.; et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J. 2007, 26, 3169–3179. [Google Scholar] [CrossRef]
- Mancuso, R.; del Valle, J.; Modol, L.; Martinez, A.; Granado-Serrano, A.B.; Ramirez-Núñez, O.; Pallás, M.; Portero-Otin, M.; Osta, R.; Navarro, X. Resveratrol Improves Motoneuron Function and Extends Survival in SOD1G93A ALS Mice. Neurotherapeutics 2014, 11, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Markert, C.D.; Kim, E.; Gifondorwa, D.J.; Childers, M.K.; Milligan, C.E. A single-dose resveratrol treatment in a mouse model of amyotrophic lateral sclerosis. J. Med. Food 2010, 13, 1081–1085. [Google Scholar] [CrossRef]
- Song, L.; Chen, L.; Zhang, X.; Li, J.; Le, W. Resveratrol ameliorates motor neuron degeneration and improves survival in SOD1G93A mouse model of amyotrophic lateral sclerosis. Biomed Res. Int. 2014. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.J.; Doherty, T.J. Sarcopenia: Prevalence, mechanisms, and functional consequences. Interdiscip. Top. Gerontol. 2010, 37, 94–114. [Google Scholar] [PubMed]
- Fahim, M.A. Endurance exercise modulates neuromuscular junction of C57BL/6NNia aging mice. J. Appl. Physiol. 1997, 83, 59–66. [Google Scholar] [CrossRef]
- Mammucari, C.; Milan, G.; Romanello, V.; Masiero, E.; Rudolf, R.; Del Piccolo, P.; Burden, S.J.; Di Lisi, R.; Sandri, C.; Zhao, J.; et al. FoxO3 Controls Autophagy in Skeletal Muscle In Vivo. Cell Metab. 2007, 6, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, S.; Mammucari, C.; Romanello, V.; Barberi, L.; Pietrangelo, L.; Fusella, A.; Mosole, S.; Gherardi, G.; Höfer, C.; Löfler, S.; et al. Physical exercise in aging human skeletal muscle increases mitochondrial calcium uniporter expression levels and affects mitochondria dynamics. Physiol. Rep. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Narici, M.V.; Monti, E.; Franchi, M.; Reggiani, C.; Toniolo, L.; Giacomello, E.; Zampieri, S.; Simunič, B.; Pisot, R. Early Biomarkers of Muscle Atrophy and of Neuromuscular Alterations During 10-Day Bed Rest. FASEB J. 2020, 34. [Google Scholar] [CrossRef]
- Harvey, J.A.; Chastin, S.F.M.; Skelton, D.A. Prevalence of sedentary behavior in older adults: A systematic review. Int. J. Environ. Res. Public Health 2013, 10, 6645–6661. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Zhang, H. Effects of hindlimb unloading on neurotrophins in the rat spinal cord and soleus muscle. Brain Res. 2016, 1630, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Karam, C.; Yi, J.; Xiao, Y.; Dhakal, K.; Zhang, L.; Li, X.; Manno, C.; Xu, J.; Li, K.; Cheng, H.; et al. Absence of physiological Ca2+ transients is an initial trigger for mitochondrial dysfunction in skeletal muscle following denervation. Skelet. Muscle 2017, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Giudice, J.; Taylor, J.M. Muscle as a paracrine and endocrine organ. Curr. Opin. Pharmacol. 2017, 34, 49–55. [Google Scholar] [CrossRef]
- Gomarasca, M.; Banfi, G.; Lombardi, G. Myokines: The endocrine coupling of skeletal muscle and bone. In International Review of Cytology; Elsevier BV: Amsterdam, The Netherlands, 2020; Volume 94, pp. 155–218. [Google Scholar]
- Sajer, S.; Guardiero, G.S.; Scicchitano, B.M. Myokines in Home-Based Functional Electrical Stimulation-Induced Recovery of Skeletal Muscle in Elderly and Permanent Denervation. Eur. J. Transl. Myol. 2018, 28, 7905. [Google Scholar] [CrossRef]
- Ji, L.L.; Zhang, Y. Antioxidant and anti-inflammatory effects of exercise: Role of redox signaling. Free Radic. Res. 2014, 48, 3–11. [Google Scholar] [CrossRef]
- Ghosh, S.; Karin, M. Missing pieces in the NF-κB puzzle. Cell 2002, 109, S81–S96. [Google Scholar] [CrossRef]
- Chambers, M.A.; Moylan, J.S.; Smith, J.D.; Goodyear, L.J.; Reid, M.B. Stretch-stimulated glucose uptake in skeletal muscle is mediated by reactive oxygen species and p38 MAP-kinase. J. Physiol. 2009, 587, 3363–3373. [Google Scholar] [CrossRef] [PubMed]
- Chakkalakal, J.V.; Nishimune, H.; Ruas, J.L.; Spiegelman, B.M.; Sanes, J.R. Retrograde influence of muscle fibers on their innervation revealed by a novel marker for slow motoneurons. Development 2010, 137, 3489–3499. [Google Scholar] [CrossRef] [PubMed]
- Handschin, C.; Kobayashi, Y.M.; Chin, S.; Seale, P.; Campbell, K.P.; Spiegelman, B.M. PGC-1α regulates the neuromuscular junction program and ameliorates Duchenne muscular dystrophy. Genes Dev. 2007, 21, 770–783. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Wu, H.; Tarr, P.T.; Zhang, C.Y.; Wu, Z.; Boss, O.; Michael, L.F.; Puigserver, P.; Isotani, E.; Olson, E.N.; et al. Transcriptional co-activator PGC-1α drives the formation of slow-twitch muscle fibres. Nature 2002, 418, 797–801. [Google Scholar] [CrossRef]
- Ascenzi, F.; Barberi, L.; Dobrowolny, G.; Villa Nova Bacurau, A.; Nicoletti, C.; Rizzuto, E.; Rosenthal, N.; Scicchitano, B.M.; Musarò, A. Effects of IGF-1 isoforms on muscle growth and sarcopenia. Aging Cell 2019, 18, e12954. [Google Scholar] [CrossRef]
- Musarò, A.; Scicchitano, B.M. Counteracting sarcopenia: The role of IGF-1 isoforms. Aging 2019, 11, 3410–3411. [Google Scholar] [CrossRef] [PubMed]
- Forcina, L.; Miano, C.; Scicchitano, B.; Musarò, A. Signals from the Niche: Insights into the Role of IGF-1 and IL-6 in Modulating Skeletal Muscle Fibrosis. Cells 2019, 8, 232. [Google Scholar] [CrossRef]
- Scicchitano, B.M.; Rizzuto, E.; Musarò, A. Counteracting muscle wasting in aging and neuromuscular diseases: The critical role of IGF-1. Aging 2009, 1, 451–457. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| miRNAs | Experimental Models | Role | References |
|---|---|---|---|
| miR-206 | Following nerve injury and in mouse models of muscular dystrophy and ALS. | Promotes the reinnervation process and counteracts the progression of the diseases. | [113,141,142] |
| miR-133b | Sciatic nerve excision and mouse model of ALS. | It is not required to maintain or restore NMJs following acute nerve injury or in a motor neuron disease. | [143] |
| miRNA-142a-5p | Sciatic nerve excision | It is an important regulator of denervation-induced skeletal muscle atrophy. | [144] |
| miR-146a | In a mouse model of spinal muscular atrophy. | Its overexpression results in loss of motoneurons while its abrogation prevents motoneurons loss. | [139,145] |
| miR-234 | In Caenorhabditis elegans | Its overexpression endows resistance to the Ach inhibitor suggesting modification of NMJ function. | [146] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dobrowolny, G.; Barbiera, A.; Sica, G.; Scicchitano, B.M. Age-Related Alterations at Neuromuscular Junction: Role of Oxidative Stress and Epigenetic Modifications. Cells 2021, 10, 1307. https://doi.org/10.3390/cells10061307
Dobrowolny G, Barbiera A, Sica G, Scicchitano BM. Age-Related Alterations at Neuromuscular Junction: Role of Oxidative Stress and Epigenetic Modifications. Cells. 2021; 10(6):1307. https://doi.org/10.3390/cells10061307
Chicago/Turabian StyleDobrowolny, Gabriella, Alessandra Barbiera, Gigliola Sica, and Bianca Maria Scicchitano. 2021. "Age-Related Alterations at Neuromuscular Junction: Role of Oxidative Stress and Epigenetic Modifications" Cells 10, no. 6: 1307. https://doi.org/10.3390/cells10061307
APA StyleDobrowolny, G., Barbiera, A., Sica, G., & Scicchitano, B. M. (2021). Age-Related Alterations at Neuromuscular Junction: Role of Oxidative Stress and Epigenetic Modifications. Cells, 10(6), 1307. https://doi.org/10.3390/cells10061307