
A Homozygous Deletion of Exon 5 of KYNU Resulting from a Maternal Chromosome 2 Isodisomy (UPD2) Causes Catel-Manzke-Syndrome/VCRL Syndrome


 and
and
Abstract
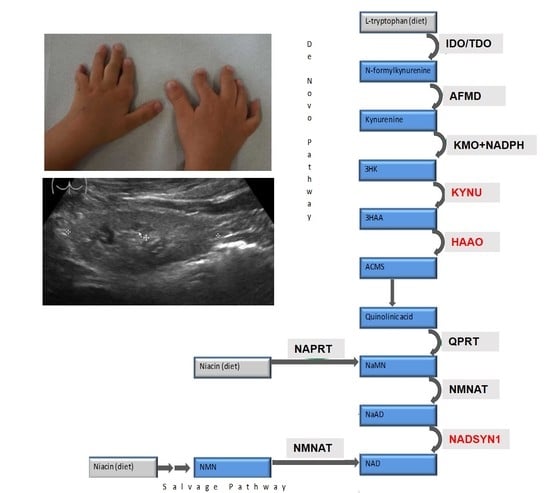
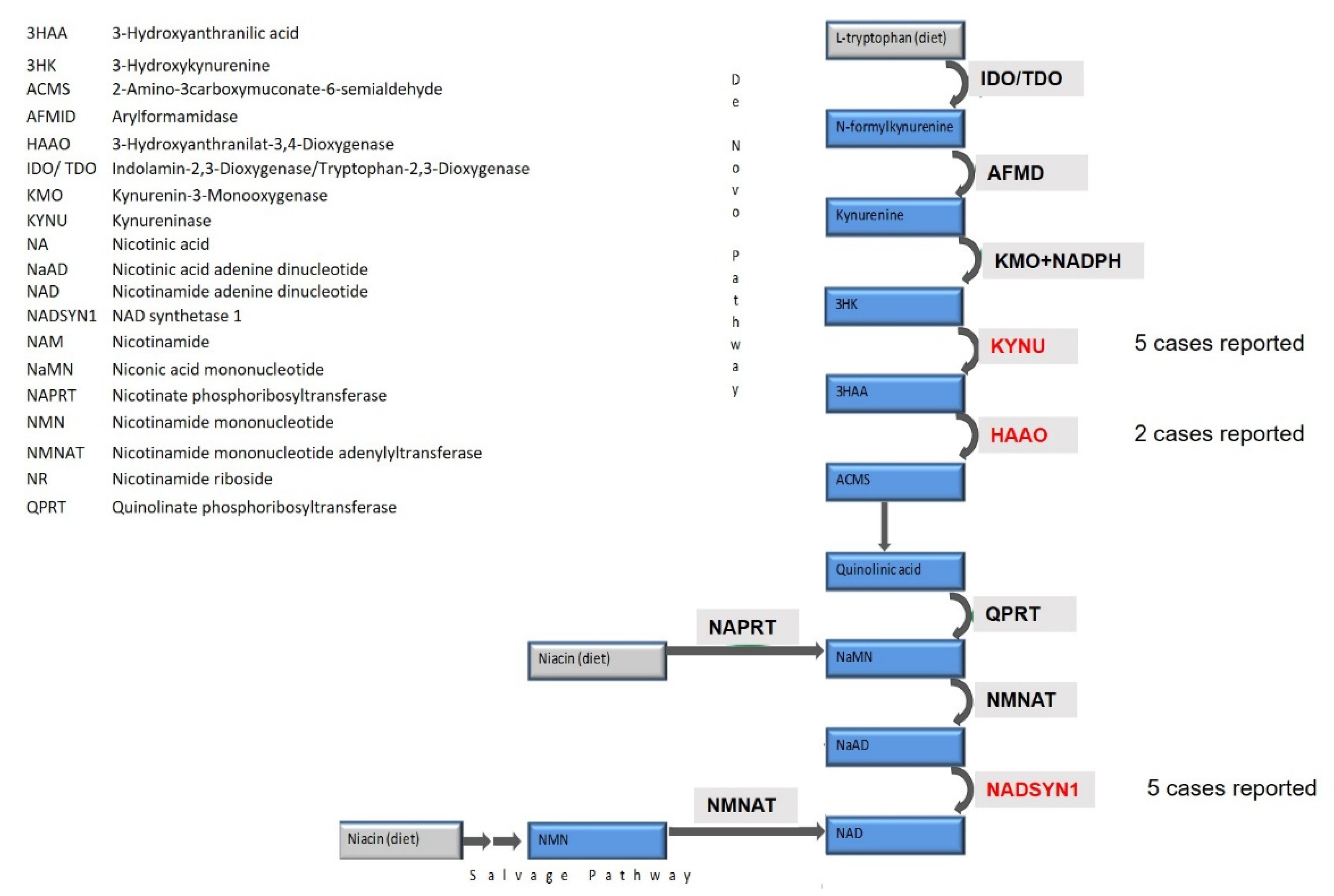
:
1. Introduction
2. Materials and Methods
2.1. Whole Exome Sequencing
2.2. CGH Array
2.3. SNP Array
2.4. Long Range Polymerase Chain Reactions (LRPCR)
2.5. Sager Sequencing
2.6. Urine Organic Acid Analysis
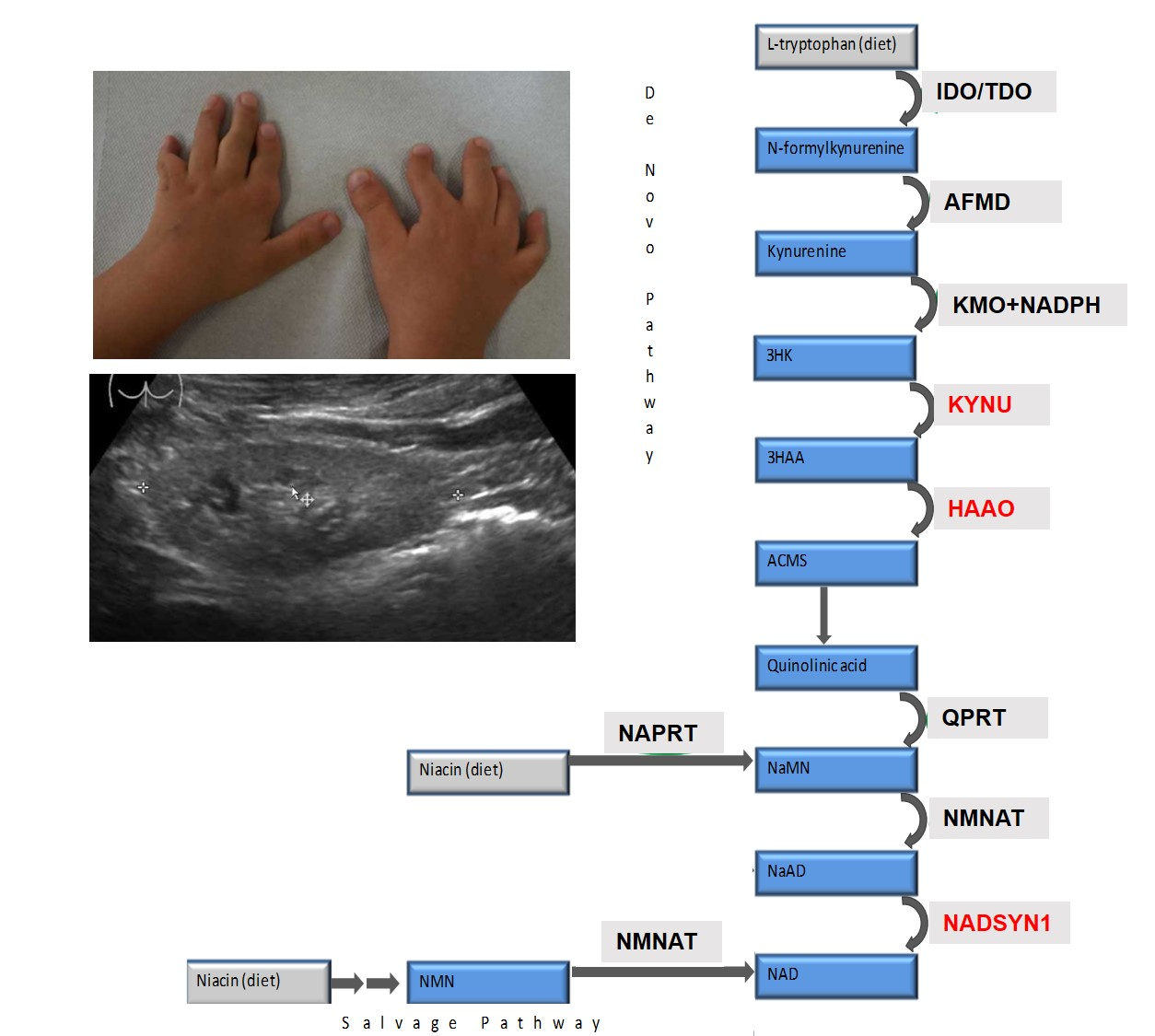
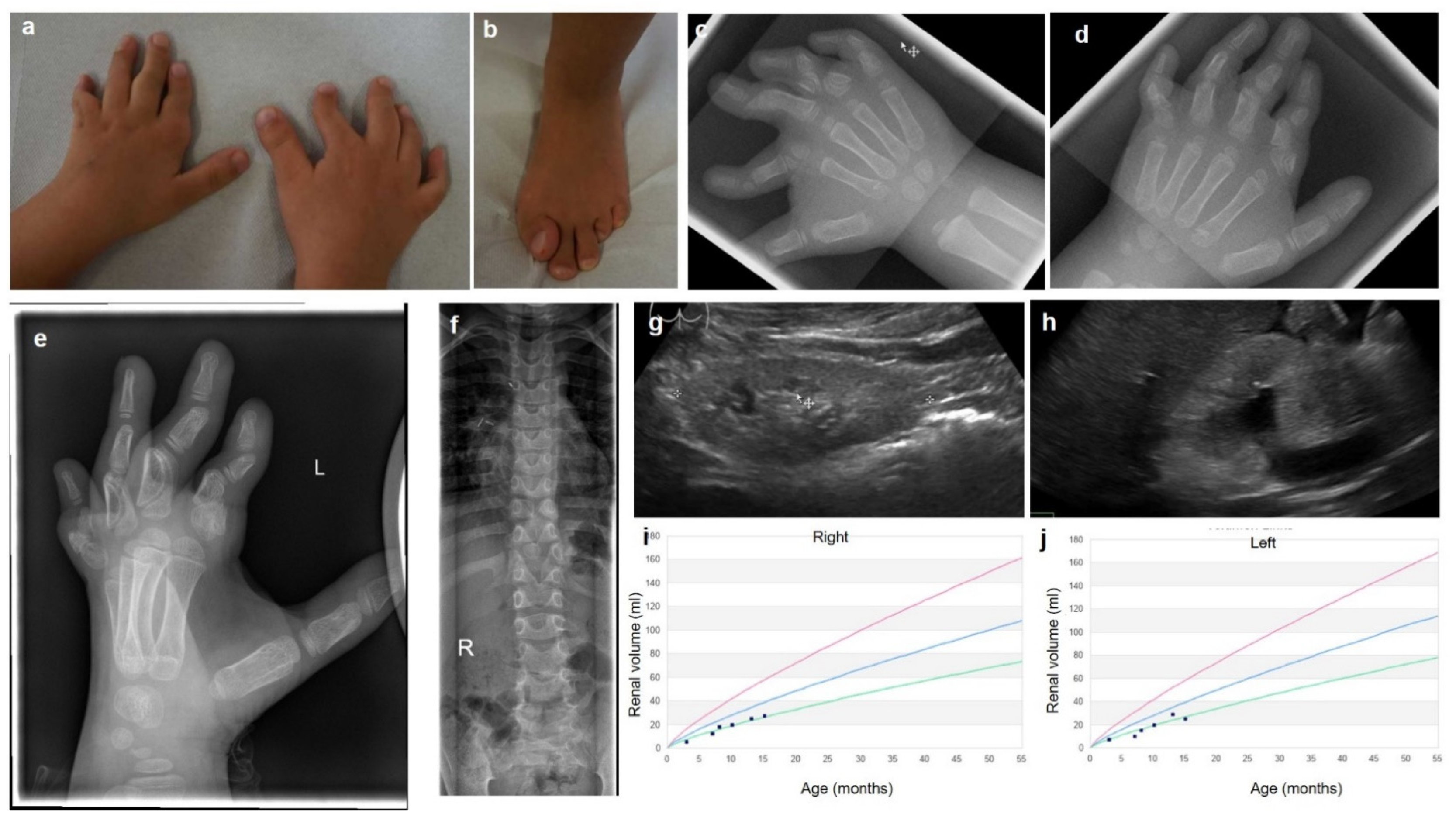
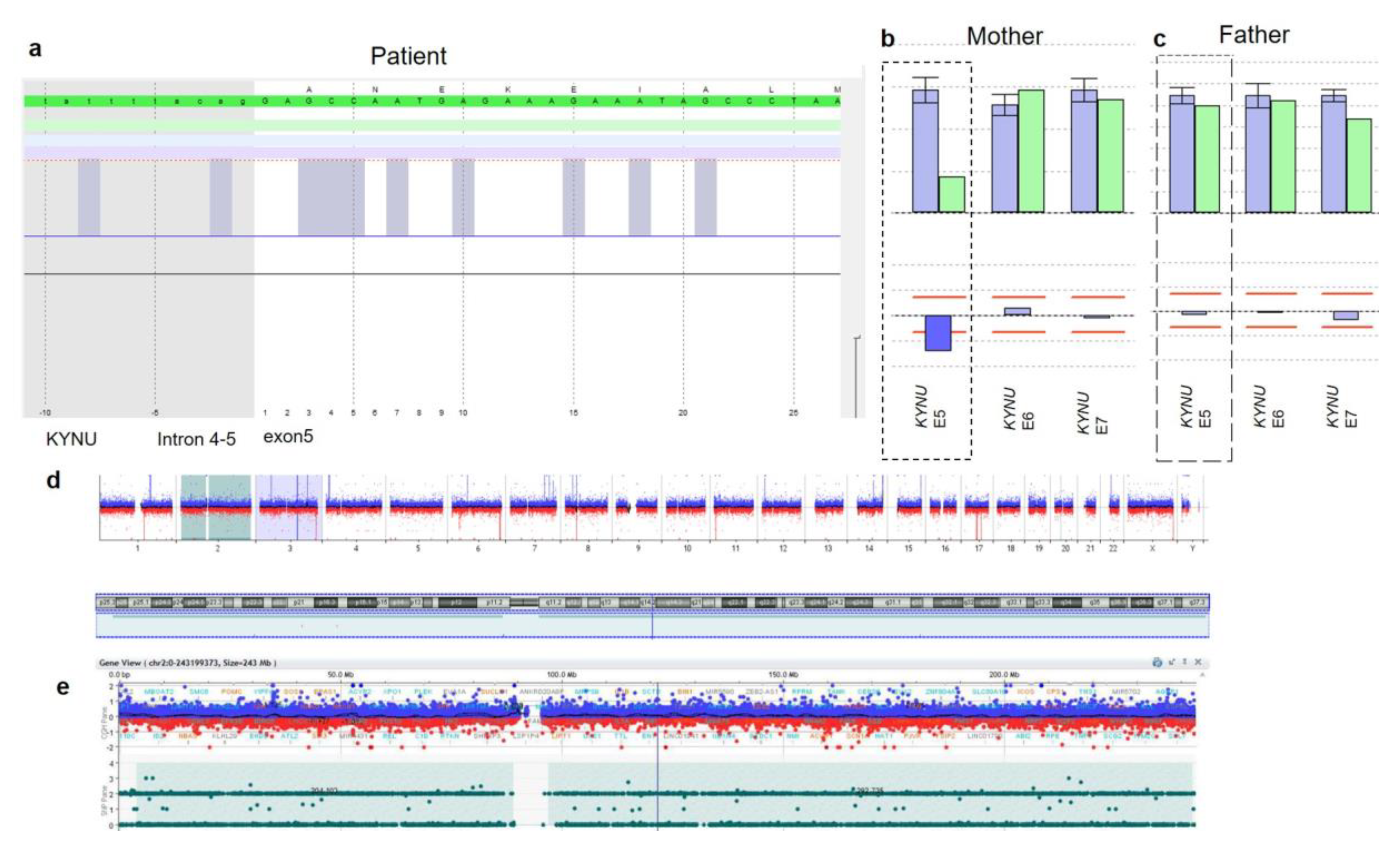
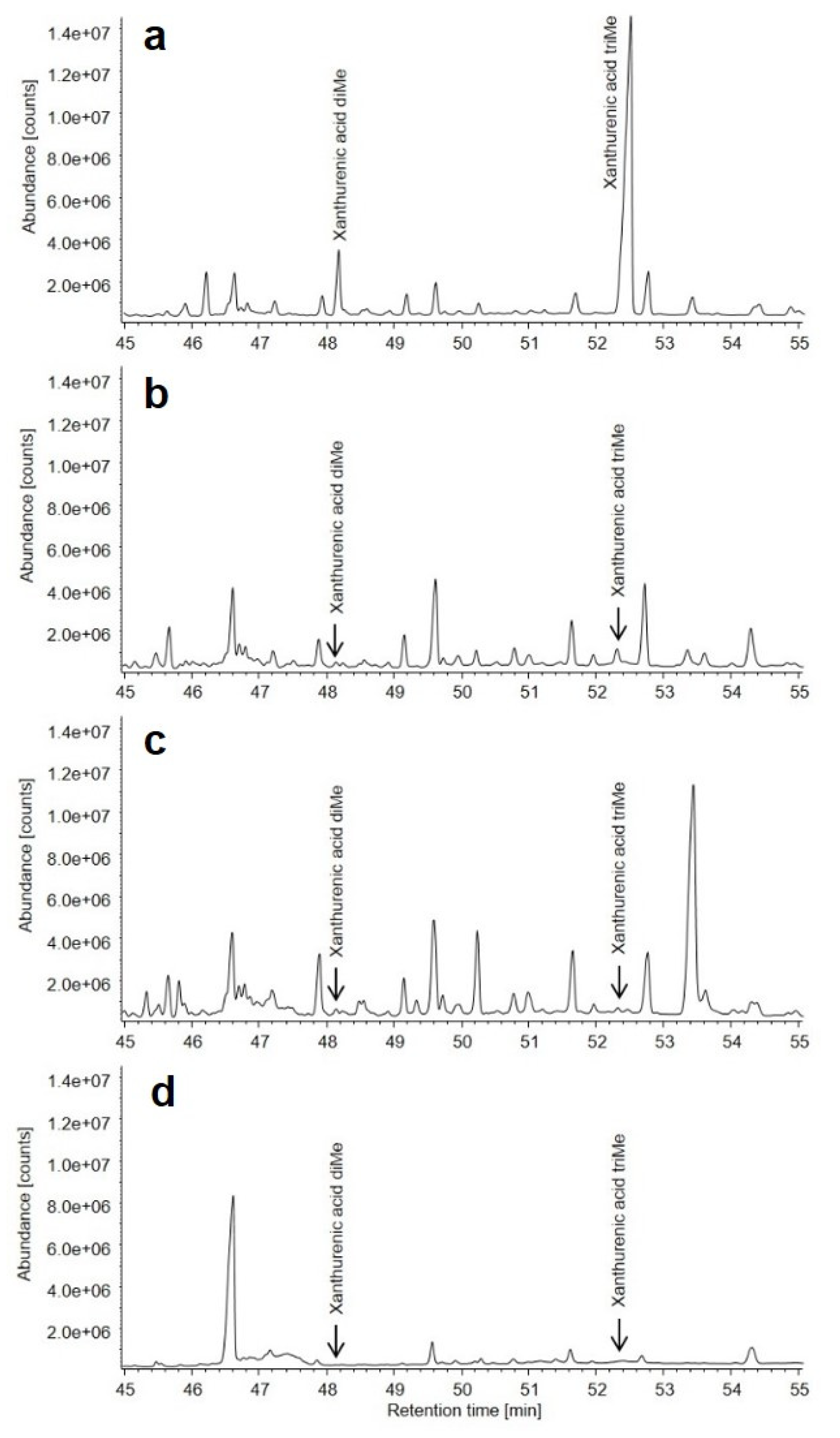
3. Results
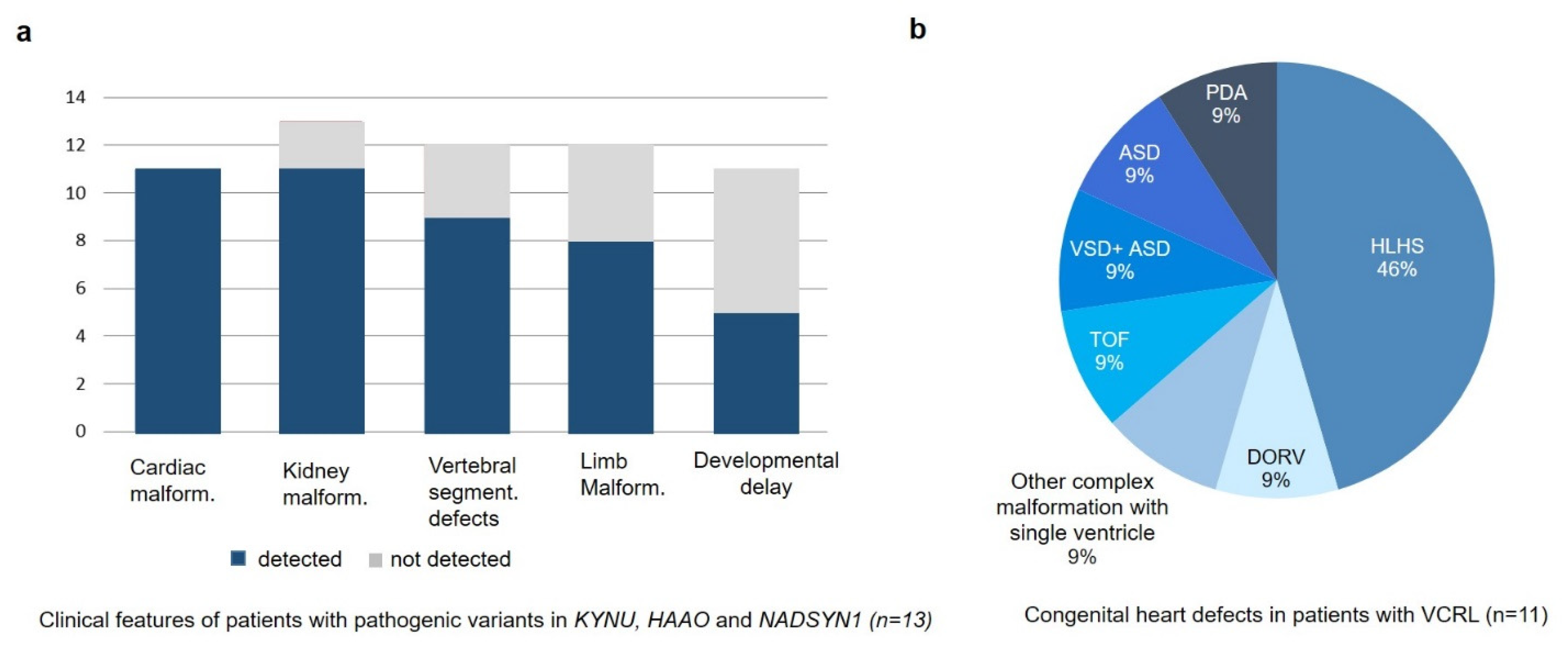
3.1. Clinical Description
3.2. Genetic Findings
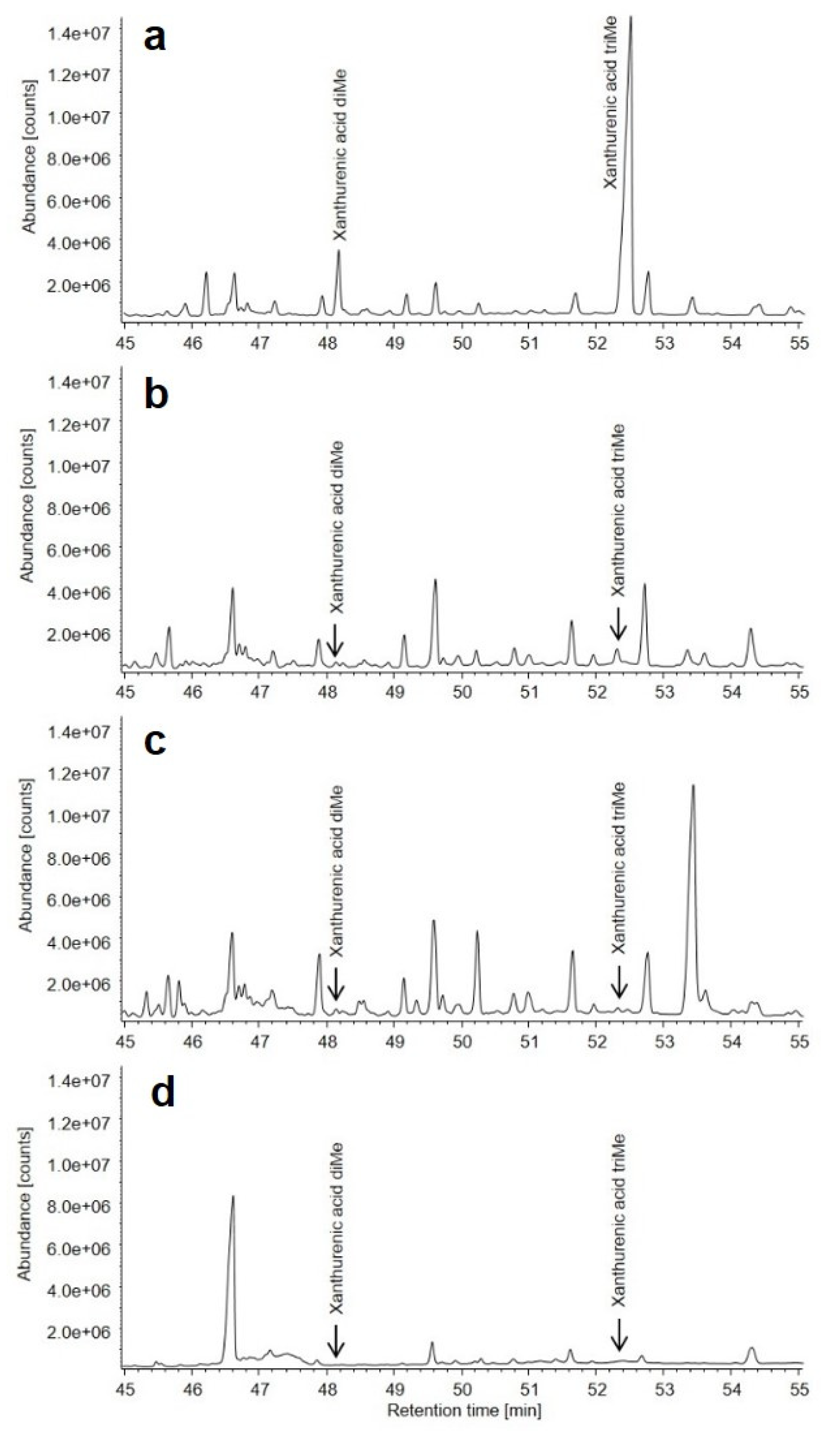
3.3. Metabolic Findings
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shi, H.; Enriquez, A.; Rapadas, M.; Martin, E.; Wang, R.; Moreau, J.; Lim, C.K.; Szot, J.O.; Ip, E.; Hughes, J.N.; et al. NAD Deficiency, Congenital Malformations, and Niacin Supplementation. N. Engl. J. Med. 2017, 377, 544–552. [Google Scholar] [CrossRef]
- Szot, J.O.; Campagnolo, C.; Cao, Y.; Iyer, K.R.; Cuny, H.; Drysdale, T.; Flores-Daboub, J.A.; Bi, W.; Westerfield, L.; Liu, P.; et al. Bi-allelic Mutations in NADSYN1 Cause Multiple Organ Defects and Expand the Genotypic Spectrum of Congenital NAD Deficiency Disorders. Am. J. Hum. Genet. 2020, 106, 129–136. [Google Scholar] [CrossRef]
- Ehmke, N.; Cusmano-Ozog, K.; Koenig, R.; Holtgrewe, M.; Nur, B.; Mihci, E.; Babcock, H.; Gonzaga-Jauregui, C.; Overton, J.D.; Xiao, J.; et al. Biallelic variants in KYNU cause a multisystemic syndrome with hand hyperphalangism. Bone 2020, 133, 115219. [Google Scholar] [CrossRef]
- Catel, W.; Heintzen, P.H. Differentialdiagnose von Krankheitssymptomen bei Kindern und Jugendlichen: Krankheiten der Thorax- und Bauchorgane; Catel, W., Heintzen, P., Eds.; Bearb ThiemeMedical Publishers: Stuttgart, Germany, 1963; Available online: https://booksgooglede/books?id=ktNtjwEACAAJ (accessed on 1 May 2021).
- Manzke, H.; Lehmann, K.; Klopocki, E.; Caliebe, A. Catel-Manzke syndrome: Two new patients and a critical review of the literature. Eur. J. Med. Genet. 2008, 51, 452–465. [Google Scholar] [CrossRef]
- Ehmke, N.; Caliebe, A.; Koenig, R.; Kant, S.G.; Stark, Z.; Cormier-Daire, V.; Wieczorek, D.; Gillessen-Kaesbach, G.; Hoff, K.; Kawalia, A.; et al. Homozygous and compound-heterozygous mutations in TGDS cause Catel-Manzke syndrome. Am. J. Hum. Genet. 2014, 95, 763–770. [Google Scholar] [CrossRef] [Green Version]
- Pferdehirt, R.; Jain, M.; Blazo, M.A.; Lee, B.; Burrage, L.C. Catel-Manzke Syndrome: Further Delineation of the Phenotype Associated with Pathogenic Variants in TGDS. Mol. Genet. Metab. Rep. 2015, 4, 89–91. [Google Scholar] [CrossRef]
- Schoner, K.; Bald, R.; Horn, D.; Rehder, H.; Kornak, U.; Ehmke, N. Mutations in TGDS associated with additional malformations of the middle fingers and halluces: Atypical Catel-Manzke syndrome in a fetus. Am. J. Med. Genet. Part 2017, 173, 1694–1697. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.E.; Chow, P.; Gallagher, E.R.; Perkins, J.A.; Wenger, T.L. Catel-Manzke syndrome without Manzke dysostosis. Am. J. Med. Genet. Part 2020, 182, 437–440. [Google Scholar] [CrossRef]
- Boschann, F.; Stuurman, K.E.; de Bruin, C.; van Slegtenhorst, M.; van Duyvenvoorde, H.A.; Kant, S.G.; Ehmke, N. TGDS pathogenic variants cause Catel-Manzke syndrome without hyperphalangy. Am. J. Med. Genet. Part 2020, 182, 431–436. [Google Scholar] [CrossRef]
- Latronico, A.C.; Billerbeck, A.E.; Pinto, E.M.; Brazil, D.; Alva, C.; Arnhold, I.J.; Mendonca, B.B. Maternal isodisomy causing homozygosity for a dominant activating mutation of the luteinizing hormone receptor gene in a boy with familial male-limited precocious puberty. Clin. Endocrinol. 2003, 59, 533–534. [Google Scholar] [CrossRef]
- Chevalier-Porst, F.; Rolland, M.O.; Cochat, P.; Bozon, D. Maternal isodisomy of the telomeric end of chromosome 2 is responsible for a case of primary hyperoxaluria type 1. Am. J. Med. Genet. Part 2005, 132, 80–83. [Google Scholar] [CrossRef]
- Thompson, D.A.; McHenry, C.L.; Li, Y.; Richards, J.E.; Othman, M.I.; Schwinger, E.; Vollrath, D.; Jacobson, S.G.; Gal, A. Retinal dystrophy due to paternal isodisomy for chromosome 1 or chromosome 2, with homoallelism for mutations in RPE65 or MERTK, respectively. Am. J. Hum. Genet. 2002, 70, 224–229. [Google Scholar] [CrossRef] [Green Version]
- Giovannoni, I.; Terracciano, A.; Gennari, F.; David, E.; Francalanci, P.; Santorelli, F.M. Paternal isodisomy of chromosome 2 in a child with bile salt export pump deficiency. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2012, 42, 327–331. [Google Scholar] [CrossRef]
- Kirkland, J.B. Niacin requirements for genomic stability. Mutat. Res. 2012, 733, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic Potential of NAD-Boosting Molecules: The In Vivo Evidence. Cell Metab. 2018, 27, 529–547. [Google Scholar] [CrossRef] [Green Version]
- Forbes, J.M. Mitochondria-Power Players in Kidney Function? Metab. TEM 2016, 27, 441–442. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Lin, S.J. Regulation of NAD+ metabolism, signaling and compartmentalization in the yeast Saccharomyces cerevisiae. DNA Repair 2014, 23, 49–58. [Google Scholar] [CrossRef] [Green Version]
- Yoshino, J.; Baur, J.A.; Imai, S.I. NAD(+) Intermediates: The Biology and Therapeutic Potential of NMN and NR. Cell Metab. 2018, 27, 513–528. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Newman, J.C.; Wang, M.Z.; Ho, L.; Verdin, E. Mitochondrial sirtuins: Regulators of protein acylation and metabolism. Trends Endocrinol. Metab. TEM 2012, 23, 467–476. [Google Scholar] [CrossRef]
- Hershberger, K.A.; Martin, A.S.; Hirschey, M.D. Role of NAD(+) and mitochondrial sirtuins in cardiac and renal diseases. Nat. Rev. Nephrol. 2017, 13, 213–225. [Google Scholar] [CrossRef] [Green Version]
- Christensen, M.; Duno, M.; Lund, A.M.; Skovby, F.; Christensen, E. Xanthurenic aciduria due to a mutation in KYNU encoding kynureninase. J. Inherit. Metab. Dis. 2007, 30, 248–255. [Google Scholar] [CrossRef]
- Oxenkrug, G. Insulin resistance and dysregulation of tryptophan-kynurenine and kynurenine-nicotinamide adenine dinucleotide metabolic pathways. Mol. Neurobiol. 2013, 48, 294–301. [Google Scholar] [CrossRef] [Green Version]
- Benson, D.W.; Martin, L.J.; Lo, C.W. Genetics of Hypoplastic Left Heart Syndrome. J. Pediatrics 2016, 173, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Natowicz, M.; Chatten, J.; Clancy, R.; Conard, K.; Glauser, T.; Huff, D.; Lin, A.; Norwood, W.; Rorke, L.B.; Uri, A.; et al. Genetic disorders and major extracardiac anomalies associated with the hypoplastic left heart syndrome. Pediatrics 1988, 82, 698–706. [Google Scholar] [PubMed]
- Genetic and environmental risk factors of major cardiovascular malformations. In the Baltimore-Washington Infant Study 1981–1989; Ferencz, C.L.C.; Correa-Villasenor, A.; Wilson, P.D. (Eds.) Futura Publishing Company Inc.: Armonk, NY, USA, 1997. [Google Scholar]
- Hinton, R.B., Jr.; Martin, L.J.; Tabangin, M.E.; Mazwi, M.L.; Cripe, L.H.; Benson, D.W. Hypoplastic left heart syndrome is heritable. J. Am. Coll. Cardiol. 2007, 50, 1590–1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
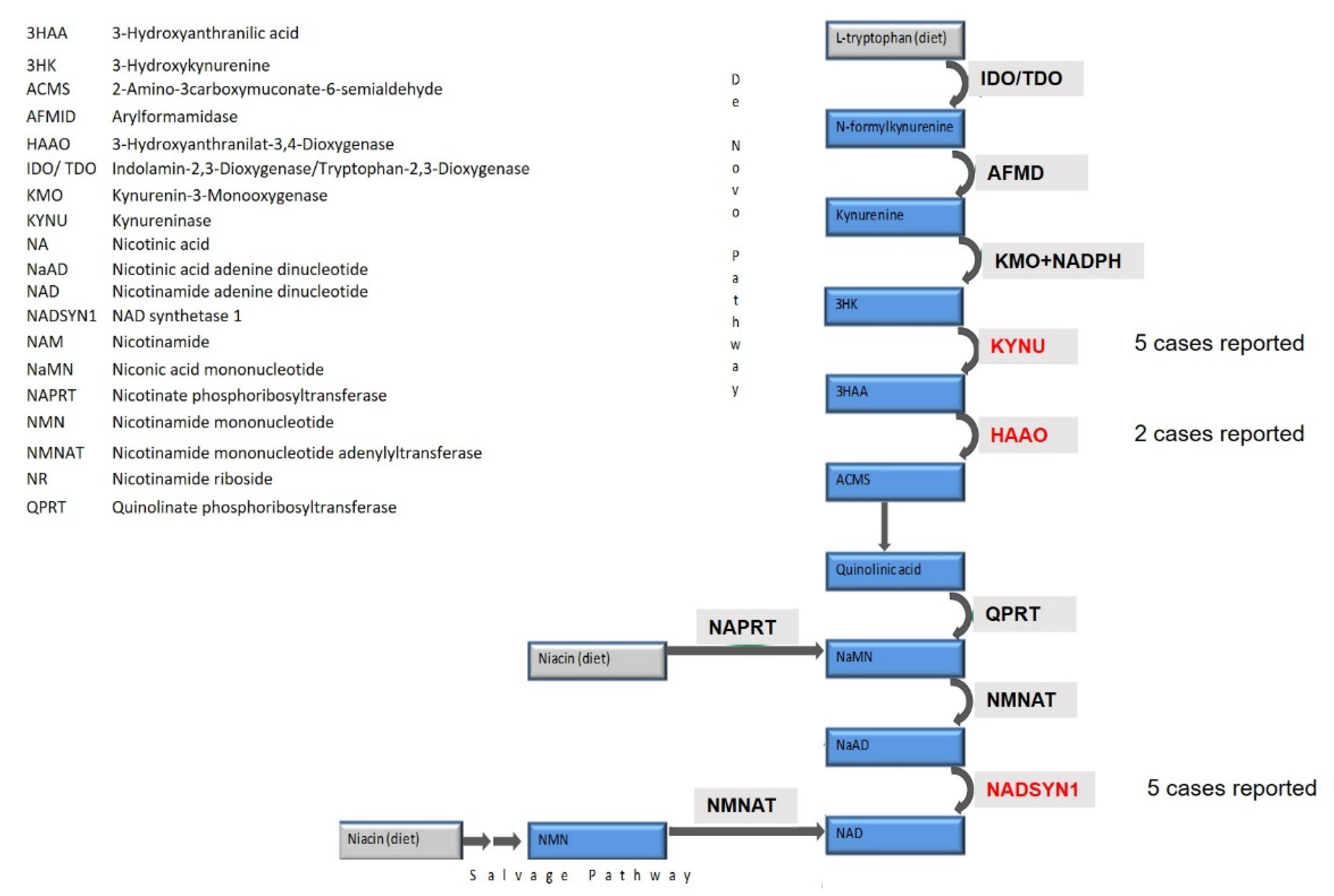{kind=link}
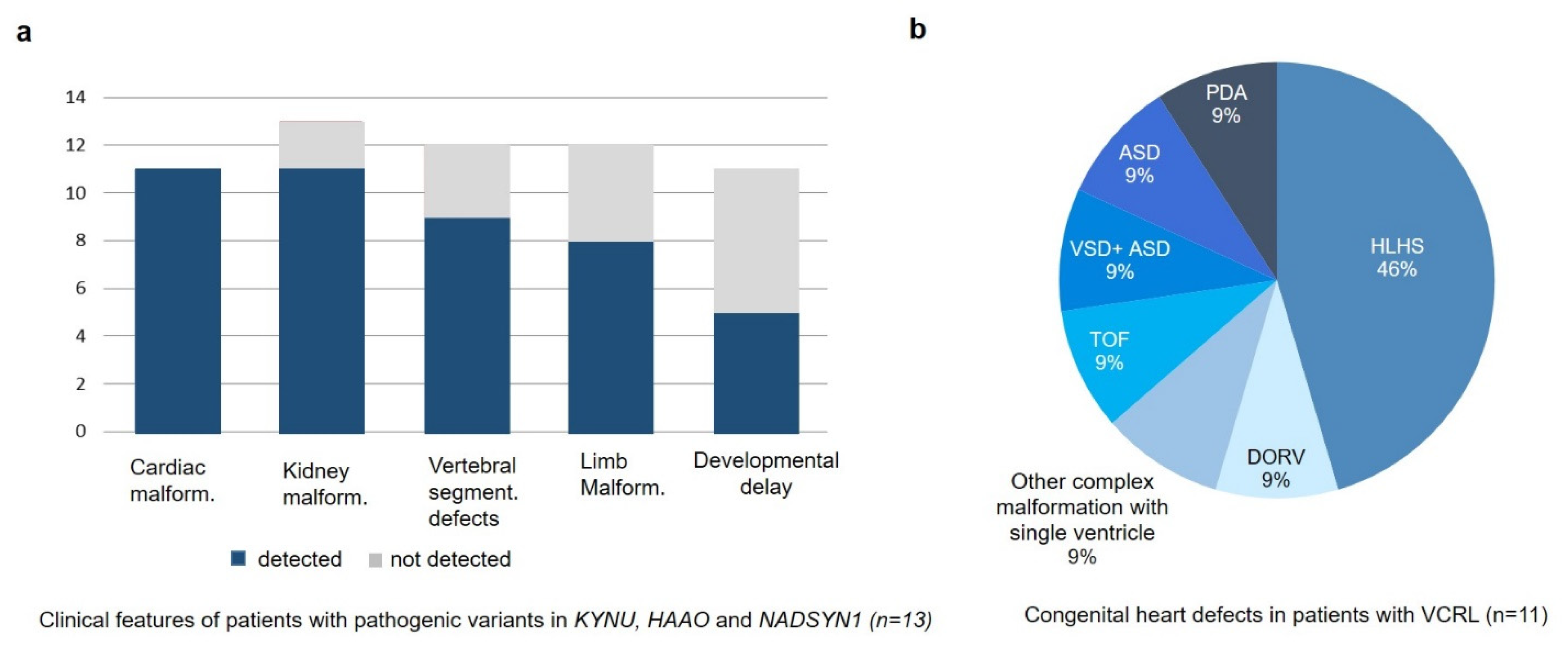{kind=link}
| Literature | Gene | Genotype | Cardiac Phenotype | Renal Phenotype | Skeletal Phenotype | Neurological Features | Additional Features | Metabolic Findings | NAD+ Level |
|---|---|---|---|---|---|---|---|---|---|
| Patient reported in this report | KYNU | 863 bp deletion (g.del142954376-g.142955239): loss of exon 5 Hom | HLHS | Bilateral hypoplasia | DVS clinodactily of 4th and 5th fingers, right-sided polydactily, feet: Shortening of the metatarsalia IV, hallux valgus, shortening of the upper arms | Normal develop-ment | Sacral hyperpegmentation | Increased Xanthenu-renic acid excretion in the urine | n.a. |
| Shi et al., 2017 | KYNU | c.170-1G>T (p.V57Efs*21) Hom | PDA | Bilateral hypoplasia | DVS, Talipes, syndactyly, rhizomelia | - | Low-set ears, anterior anus | n.a. | n.a. |
| Shi et al., 2017 | KYNU | c.468T > A (p.Y156*) het + c.1045_1051delTTTAAGC (p.F349Kfs*4) het | HLHS | Solitary kidney, CKD | DVS, Short Stature, bilateral shortening of humeri and femora | Speech delay | - | * 3HK, 161 times the mean | * NAD(H), 1/7th of the mean |
| Ehmke et al., 2019 | KYNU | delEx1-8 het + c.1282C > T; (p.Arg428Trp) het | HLHS | - | DVS, MD | Mild DD | Hepato-megaly, microretro-gnathia, facial dysmor-phism | ** 3HK, 56 times the mean | |
| Ehmke et al., 2019 | KYNU | c.989G > A (p.Arg330Gln) Hom | Tetralogy of Fallot, ALCAPA | - | DVS, MD, long thumbs | DD, muscular hypotonia, microcephaly | Senso-neuronal hearing loss, facial dysmor-phism | ** 3HK, 45 times the mean | |
| Ehmke et al., 2019 | KYNU | c.326G > C (p.Trp109Ser) Hom | Secundum ASD, subaortic VSD | Unilateral renal agenesis | Brachydac-tyly, Clinodactyly, 2nd/3rd-toe syndactyly, MD | Mild DD, microcephaly | Bilateral single transverse palmar crease, joint hypermo-bility, facial dysmor-phism | n.a | n.a |
| Shi et al., 2017 | HAAO | c.483dupT (p.D162*) Hom | ASD | Hypoplasiavesicoure-teral reflux | DVS Short stature, talipes | DD | Sensorineural hearing loss, laryngo-malacia | * 3HAA, 64 times the mean | * NAD+, 1/3rd of the mean |
| Shi et al., 2017 | HAAO | c.558G > A (p.W186*) Hom | HLHS | Hypodys- plasia | DVS - | Palsy of left vocal cord | Sensori-neural hearing loss on left side | * 3HAA, 385 times the mean | * NAD(H), 1/4th of the mean |
| Szot et al., 2020 | NADSYN1 | c.1717G > A (p.Ala573Thr) Hom | Borderline HLHS hypoplastic mitral valve, small bicuspid aortic valve, hyperplasia/coarctation of the aortic arch ALCAPA | Absent left kidney | Thoracic vertebral defect, bilateral shortening of humeri and femora | - | Sacral dimple | n.a. | n.a. |
| Szot et al., 2020 | NADSYN1 | c.1717G > A (p.Ala573Thr) Hom | Absent left ventricle and pulmonary trunk, right ventricular outlet to the aorta | Bilateral hypoplastic kidneys | DVS, bilateral shortening of humeri and femora | - | - | n.a. | n.a. |
| Szot et al., 2020 | NADSYN1 | c.1717G > A (p.Ala573Thr) c.1819del (p.Val607Trpfs*30) | DORV, TGA in side by side orientation, VSD, PDA, left aortic arch | Mild hyperecho-genic renal cortex | DVS, scoliosis, rib abnormalities, bilateral shortening of humeri and femora, bowing of lower extremities | n.a. | n.a. | n.a. | n.a |
| Szot et al., 2020 | NADSYN1 | c.145T > C (p.Cys49Arg) c.395G > T (p.Trp132Leu) | n.a. termination of pregnancy at 16 w | Oligohy-dramnion, bilateral renal agenesis | n.a. | n.a. | n.a. | n.a. | n.a |
| Szot et al., 2020 | NADSYN1 | c.735T > A (p.Cys245*) c.1839C > G (p.Tyr613*) | n.a. termination of pregnancy at 16 w | Left renal and ureter agenesis, edema | Small thorax, microme-lia, bilateral club feet | Hydrocephalus | Facial dysmor-phism echogenic bowel, polysplenia, pulmonary hypoplasia | n.a. | n.a |
| Literature | Number of Patients | Cardiac Malformations | Kidney Malformations | Spine/Thorax | Manzke Dysostosis | Shortening of the Limbs | Clinodactily/Brachydactily | Pierre Robin Sequence | Neurological Features | Additional Features |
|---|---|---|---|---|---|---|---|---|---|---|
| Ehmke et al., 2014 | 7 | 2/7 (2VSD) | 0/7 | 1/7 (pectus deformity) | 7/7 | 1/7 | 5/7 | 7/7 | 0/7 | Hearing loss |
| Pferdehirt et al., 2015 | 1 | 1/1 (PDA) | 0/1 | 1/1 (pectus deformity) | 1/1 | 0/1 | 1/1 | 1/1 | 0/1 | Laryng-omalacia |
| Schoner et al., 2017 | 1 | 1/1 (VSD, coarcation of aorta) | 0/1 | 0/1 | 1/1 | 1/1 | 1/1 | 1/1 | n.a. | n.a |
| Miller et al., 2019 | 1 | 1/1 (ASD, VSD) | 0/1 | 1/1 (scoliosis) | 0/1 | 0/1 | 0/1 | 1/1 | 0/1 | n.a. |
| Boschann et al., 2020 | 2 | 0/2 | 0/2 | 1/2 (pectus deformity | 0/2 | 2/2 | 2/2 | 2/2 | 0/1 | Hip dysplasia |
| total | 12 | 5/12 | 0/12 | 4/12 | 9/12 | 4/12 | 9/12 | 12/12 | 0/11 | 3/10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schüle, I.; Berger, U.; Matysiak, U.; Ruzaike, G.; Stiller, B.; Pohl, M.; Spiekerkoetter, U.; Lausch, E.; Grünert, S.C.; Schmidts, M. A Homozygous Deletion of Exon 5 of KYNU Resulting from a Maternal Chromosome 2 Isodisomy (UPD2) Causes Catel-Manzke-Syndrome/VCRL Syndrome. Genes 2021, 12, 879. https://doi.org/10.3390/genes12060879
Schüle I, Berger U, Matysiak U, Ruzaike G, Stiller B, Pohl M, Spiekerkoetter U, Lausch E, Grünert SC, Schmidts M. A Homozygous Deletion of Exon 5 of KYNU Resulting from a Maternal Chromosome 2 Isodisomy (UPD2) Causes Catel-Manzke-Syndrome/VCRL Syndrome. Genes. 2021; 12(6):879. https://doi.org/10.3390/genes12060879
Chicago/Turabian StyleSchüle, Isabel, Urs Berger, Uta Matysiak, Gunda Ruzaike, Brigitte Stiller, Martin Pohl, Ute Spiekerkoetter, Ekkehart Lausch, Sarah C. Grünert, and Miriam Schmidts. 2021. "A Homozygous Deletion of Exon 5 of KYNU Resulting from a Maternal Chromosome 2 Isodisomy (UPD2) Causes Catel-Manzke-Syndrome/VCRL Syndrome" Genes 12, no. 6: 879. https://doi.org/10.3390/genes12060879
APA StyleSchüle, I., Berger, U., Matysiak, U., Ruzaike, G., Stiller, B., Pohl, M., Spiekerkoetter, U., Lausch, E., Grünert, S. C., & Schmidts, M. (2021). A Homozygous Deletion of Exon 5 of KYNU Resulting from a Maternal Chromosome 2 Isodisomy (UPD2) Causes Catel-Manzke-Syndrome/VCRL Syndrome. Genes, 12(6), 879. https://doi.org/10.3390/genes12060879