
DNA Methylation Alterations in Blood Cells of Toddlers with Down Syndrome

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants
2.2. DNA Methylation Profiling and Data Processing
2.3. Differential Methylation Analysis
2.4. Analysis of Differential Methylation across Age Groups
3. Results
3.1. Blood Cell-Type Count in DS vs. TD Toddler Groups
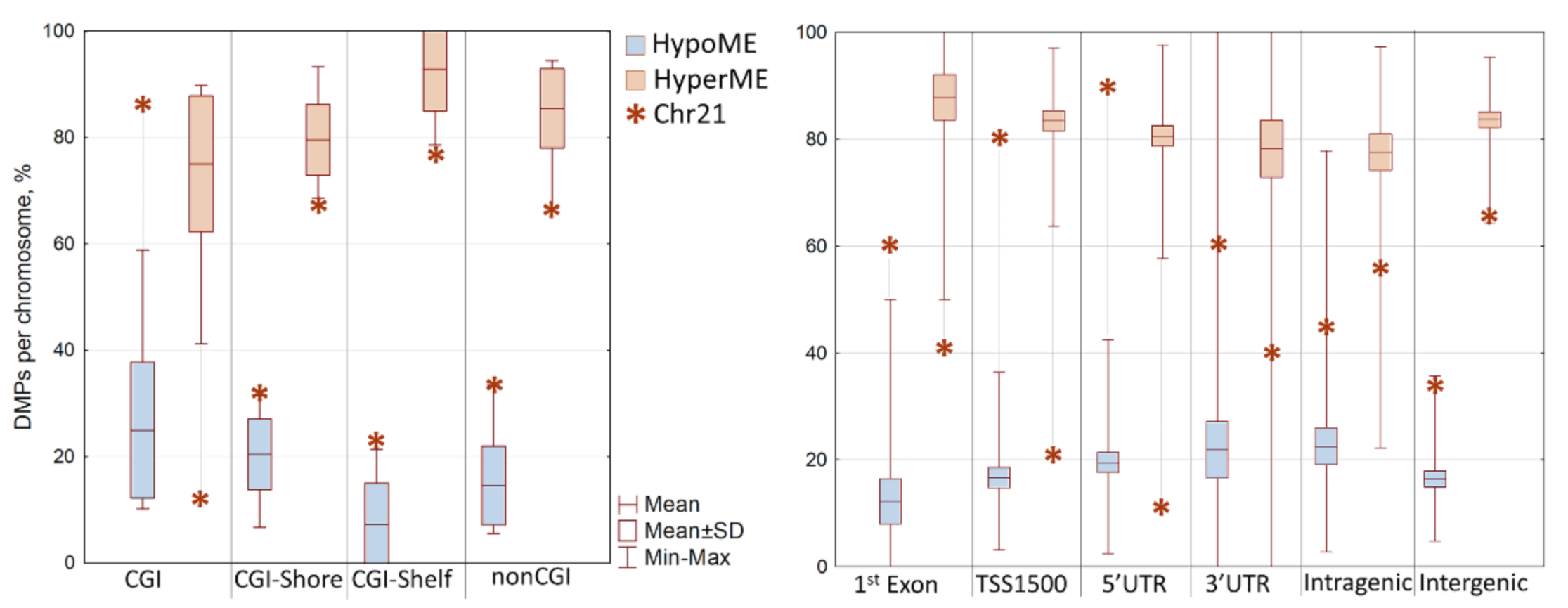
3.2. Differentially Methylated Positions (DMPs) in DS vs. TD Toddler Groups
3.3. Differentially Methylated Genes (DMGs) in DS vs. TD Toddler Groups
3.4. Differentially Methylated Regions (DMRs) in DS vs. TD Toddler Groups
3.5. DS-Specific DNA Methylation Pattern in Blood Cells throughout the Lifespan
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Letourneau, A.; Santoni, F.A.; Bonilla, X.; Sailani, M.R.; Gonzalez, D.; Kind, J.; Chevalier, C.; Thurman, R.; Sandstrom, R.S.; Hibaoui, Y.; et al. Domains of genome-wide gene expression dysregulation in Down’s syndrome. Nature 2014, 508, 345–350. [Google Scholar] [CrossRef]
- Laufer, B.I.; Gomez, J.A.; Jianu, J.M.; LaSalle, J.M. Stable DNMT3L overexpression in SH-SY5Y neurons recreates a facet of the genome-wide Down syndrome DNA methylation signature. Epigenetics Chromatin 2021, 14, 13. [Google Scholar] [CrossRef]
- Do, C.; Xing, Z.; Yu, Y.E.; Tycko, B. Trans-acting epigenetic effects of chromosomal aneuploidies: Lessons from Down syndrome and mouse models. Epigenomics 2017, 9, 189–207. [Google Scholar] [CrossRef] [Green Version]
- Mendioroz, M.; Do, C.; Jiang, X.; Liu, C.; Darbary, H.K.; Lang, C.F.; Lin, J.; Thomas, A.; Abu-Amero, S.; Stanier, P.; et al. Trans effects of chromosome aneuploidies on DNA methylation patterns in human Down syndrome and mouse models. Genome Biol. 2015, 16, 263. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.; Lee, Y.K.; Lim, Y.C.; Zheng, Z.; Lin, X.M.; Ng, D.P.; Holbrook, J.D.; Law, H.Y.; Kwek, K.Y.; Yeo, G.S.; et al. Global DNA hypermethylation in down syndrome placenta. PLoS Genet. 2013, 9, e1003515. [Google Scholar] [CrossRef] [Green Version]
- Bacalini, M.G.; Gentilini, D.; Boattini, A.; Giampieri, E.; Pirazzini, C.; Giuliani, C.; Fontanesi, E.; Scurti, M.; Remondini, D.; Capri, M.; et al. Identification of a DNA methylation signature in blood cells from persons with Down Syndrome. Aging 2015, 7, 82–93. [Google Scholar] [CrossRef] [Green Version]
- Henneman, P.; Bouman, A.; Mul, A.; Knegt, L.; van der Kevie-Kersemaekers, A.-M.; Zwaveling-Soonawala, N.; Meijers-Heijboer, H.E.J.; van Trotsenburg, A.S.P.; Mannens, M.M. Widespread domain-like perturbations of DNA methylation in whole blood of Down syndrome neonates. PLoS ONE 2018, 13, e0194938. [Google Scholar] [CrossRef] [Green Version]
- Muskens, I.S.; Li, S.; Jackson, T.; Elliot, N.; Hansen, H.M.; Myint, S.S.; Pandey, P.; Schraw, J.M.; Roy, R.; Anguiano, J.; et al. The genome-wide impact of trisomy 21 on DNA methylation and its implications for hematopoiesis. Nat. Commun. 2021, 12, 821. [Google Scholar] [CrossRef]
- Laufer, B.I.; Hwang, H.; Jianu, J.M.; Mordaunt, C.E.; Korf, I.F.; Hertz-Picciotto, I.; LaSalle, J.M. Low-pass whole genome bisulfite sequencing of neonatal dried blood spots identifies a role for RUNX1 in Down syndrome DNA methylation profiles. Hum. Mol. Genet. 2020, 29, 3465–3476. [Google Scholar] [CrossRef]
- Jones, M.J.; Farré, P.; McEwen, L.M.; Macisaac, J.L.; Watt, K.; Neumann, S.M.; Emberly, E.; Cynader, M.S.; Virji-Babul, N.; Kobor, M.S. Distinct DNA methylation patterns of cognitive impairment and trisomy 21 in Down syndrome. BMC Med. Genom. 2013, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Hajj, N.; Dittrich, M.; Böck, J.; Kraus, T.F.; Nanda, I.; Müller, T.; Seidmann, L.; Tralau, T.; Galetzka, D.; Schneider, E.; et al. Epigenetic dysregulation in the developing Down syndrome cortex. Epigenetics 2016, 11, 563–578. [Google Scholar] [CrossRef] [Green Version]
- Laufer, B.I.; Hwang, H.; Vogel Ciernia, A.; Mordaunt, C.E.; LaSalle, J.M. Whole genome bisulfite sequencing of Down syndrome brain reveals regional DNA hypermethylation and novel disorder insights. Epigenetics 2019, 14, 672–684. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Sheen, V. Chapter: 12. Genetic and Epigenetic Mechanisms in Down Syndrome Brain. In Down Syndrome; Dey, S., Ed.; InTech: London, UK, 2013; pp. 237–261. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.H.; Kang, Y.J.; Lee, B.Y.; Han, Y.J.; Chung, J.H.; Kim, M.Y.; Kim, M.H.; Kim, J.W.; Cho, Y.H.; Ryu, H.M. Epigenome-wide base-resolution profiling of DNA methylation in chorionic villi of fetuses with Down syndrome by methyl-capture sequencing. Clin. Epigenet. 2019, 11, 180. [Google Scholar] [CrossRef] [Green Version]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA Methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [Green Version]
- Reinius, L.E.; Acevedo, N.; Joerink, M.; Pershagen, G.; Dahlén, S.E.; Greco, D.; Söderhäll, C.; Scheynius, A.; Kere, J. Differential DNA methylation in purified human blood cells: Implications for cell lineage and studies on disease susceptibility. PLoS ONE 2012, 7, e41361. [Google Scholar] [CrossRef]
- Jaffe, A.E.; Irizarry, R.A. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol 2013, 15, R31. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Bibikova, M.; Barnes, B.; Tsan, C.; Ho, V.; Klotzle, B.; Le, J.M.; Delano, D.; Zhang, L.; Schroth, G.P.; Gunderson, K.L.; et al. High density DNA methylation array with single CpG site resolution. Genomics 2011, 98, 288–295. [Google Scholar] [CrossRef] [Green Version]
- Jaffe, A.E.; Murakami, P.; Lee, H.; Leek, J.T.; Fallin, M.D.; Feinberg, A.P.; Irizarry, R.A. Bump hunting to identify differentially methylated regions in epigenetic epidemiology studies. Int. J. Epidemiol. 2012, 41, 200–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casper, J.; Zweig, A.S.; Villarreal, C.; Tyner, C.; Speir, M.L.; Rosenbloom, K.R.; Raney, B.J.; Lee, C.M.; Lee, B.T.; Karolchik, D.; et al. The UCSC Genome Browser database: 2018 update. Nucleic Acids Res. 2018, 46, D762–D769. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Gene Ontology Consortium, The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [CrossRef]
- Robinson, P.N.; Köhler, S.; Bauer, S.; Seelow, D.; Horn, D.; Mundlos, S. The Human Phenotype Ontology: A tool for annotating and analyzing human hereditary disease. Am. J. Hum. Genet. 2008, 83, 610–615. [Google Scholar] [CrossRef] [Green Version]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef] [PubMed]
- Peters, T.J.; Buckley, M.J.; Statham, A.L.; Pidsley, R.; Samaras, K.; Lord, R.V.; Clark, S.J.; Molloy, P.L. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin 2015, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, B.S.; Schwartz, D.A.; Yang, I.V.; Kechris, K.J. Comb-p: Software for combining, analyzing, grouping and correcting spatially correlated P-values. Bioinformatics 2012, 28, 2986–2988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cossarizza, A.; Monti, D.; Montagnani, G.; Ortolani, C.; Masi, M.; Zannotti, M.; Franceschi, C. Precocious aging of the immune system in Down syndrome: Alteration of B lymphocytes, T-lymphocyte subsets, and cells with natural killer markers. Am. J. Med. Genet. Suppl. 1990, 7, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Kusters, M.A.; Verstegen, R.H.; Gemen, E.F.; de Vries, E. Intrinsic defect of the immune system in children with Down syndrome: A review. Clin. Exp. Immunol. 2009, 156, 189–193. [Google Scholar] [CrossRef]
- Webb, D.; Roberts, I.; Vyas, P. Haematology of Down syndrome. Arch. Dis. Child. Fetal. Neonatal. Ed. 2007, 92, F503–F507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012, 8, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgio, G.R.; Lanzavecchia, A.; Maccario, R.; Vitiello, A.; Plebani, A.; Ugazio, A.G. Immunodeficiency in Down’s syndrome: T-lymphocyte subset imbalance in trisomic children. Clin. Exp. Immunol. 1978, 33, 298–301. [Google Scholar]
- Carsetti, R.; Valentini, D.; Marcellini, V.; Scarsella, M.; Marasco, E.; Giustini, F.; Bartuli, A.; Villani, A.; Ugazio, A.G. Reduced numbers of switched memory B cells with high terminal differentiation potential in Down syndrome. Eur. J. Immunol. 2015, 45, 903–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Hingh, Y.C.; van der Vossen, P.W.; Gemen, E.F.; Mulder, A.B.; Hop, W.C.; Brus, F.; de Vries, E. Intrinsic abnormalities of lymphocyte counts in children with down syndrome. J. Pediatr. 2005, 147, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Bitar, M.; Hara, Y.; Sethi, D.; Couser, N.L. Chapter 5—Genetic Abnormalities of the Cornea. In Ophthalmic Genetic Diseases; Couser, N.L., Ed.; Elsevier: Philadelphia, PA, USA, 2019; pp. 61–80. [Google Scholar]
- Brandt, M.L. Pediatric hernias. Surg. Clin. N. Am. 2008, 88, 27–43. [Google Scholar] [CrossRef] [PubMed]
- Irving, C.A.; Chaudhari, M.P. Cardiovascular abnormalities in Down syndrome: Spectrum, management and survival over 22 years. Arch. Dis. Child. 2012, 97, 326. [Google Scholar] [CrossRef] [Green Version]
- Rappaport, N.; Nativ, N.; Stelzer, G.; Twik, M.; Guan-Golan, Y.; Stein, T.I.; Bahir, I.; Belinky, F.; Morrey, C.P.; Safran, M.; et al. MalaCards: An integrated compendium for diseases and their annotation. Database 2013, 2013, bat018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; McCarter, M.; Lian, G.; Esposito, G.; Capoccia, E.; Delli-Bovi, L.C.; Hecht, J.; Sheen, V. Global hypermethylation in fetal cortex of Down syndrome due to DNMT3L overexpression. Hum. Mol. Genet. 2016, 25, 1714–1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laan, L.; Klar, J.; Sobol, M.; Hoeber, J.; Shahsavani, M.; Kele, M.; Fatima, A.; Zakaria, M.; Annerén, G.; Falk, A.; et al. DNA methylation changes in Down syndrome derived neural iPSCs uncover co-dysregulation of ZNF and HOX3 families of transcription factors. Clin. Epigenet 2020, 12, 9. [Google Scholar] [CrossRef]
- Wiemels, J.L.; Muskens, I.S.; Li, S.; Pandey, P.; Roy, R.; Hansen, H.M.; Siegmund, K.D.; Mueller, B.A.; Ma, X.; Metayer, C.; et al. The Genome-Wide Impact of Trisomy 21 on DNA Methylation and Its Implications for Hematologic Malignancies. Blood 2019, 134 (Suppl. 1), 2510. [Google Scholar] [CrossRef]
- Horvath, S.; Garagnani, P.; Bacalini, M.G.; Pirazzini, C.; Salvioli, S.; Gentilini, D.; Di Blasio, A.M.; Giuliani, C.; Tung, S.; Vinters, H.V.; et al. Accelerated epigenetic aging in Down syndrome. Aging Cell 2015, 14, 491–495. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Muskens et al. 2021 [8] | Current Study | Bacalini et al. 2015 [6] | |
|---|---|---|---|
| Age Group | Newborns | Toddlers | Adults |
| Age, y (range) | 0 | 2.8 ± 1.4 (0.5–4.5) | 26.3 ± 9.5 (12–43) |
| Ethnicity | Mixed: Whites, Blacks, Asians | Whites; East Slavs | Whites; Italians |
| DS Sample Size, n | 198 | 17 | 29 |
| Study Design | Case-Control | Case-Control | Family-based Case-Control |
| Methylation profiling | EPIC microarray | HME450 microarray | HME450 microarray |
| Differential methylation analysis | DMRcate [26] and comb-p [27]; EWAS correction for cell counts, sex, and ancestry | Minfi [15] and bump-hunting [20]; EWAS correction for cell counts and batch | MANOVA and ANOVA of the pre-clustered blocks of probes; EWAS correction for cell counts, sex, and batch |
| Cell Type | Group | Mean | SD | Welch’s t-Test | Mann–Whitney U Test | ||||
|---|---|---|---|---|---|---|---|---|---|
| t-Value | df | p-Value | U-Value | Z-Score | p-Value | ||||
| T cells CD8+ | DS | 0.1903 | 0.0287 | 0.234 | 30.64 | 0.817 | 128.5 | 0.534 | 0.596 |
| TD | 0.1877 | 0.0356 | |||||||
| T cells CD4+ | DS | 0.1673 | 0.0448 | −1.305 | 29.02 | 0.202 | 112.5 | −1.09 | 0.281 |
| TD | 0.1917 | 0.0625 | |||||||
| NK cells | DS | 0.0919 | 0.0361 | 2.406 | 31.78 | 0.022 * | 85.0 | 2.03 | 0.042 * |
| TD | 0.0608 | 0.0392 | |||||||
| B cells | DS | 0.1371 | 0.0227 | −1.970 | 25.72 | 0.059 | 84.0 | −2.07 | 0.039 * |
| TD | 0.1587 | 0.0391 | |||||||
| Monocytes | DS | 0.0610 | 0.0192 | −0.918 | 29.08 | 0.366 | 113.0 | −1.07 | 0.255 |
| TD | 0.0683 | 0.0265 | |||||||
| Granulocytes | DS | 0.3665 | 0.0490 | 0.983 | 25.85 | 0.335 | 113.5 | 1.05 | 0.294 |
| TD | 0.3433 | 0.0836 | |||||||
| DMR Position (GRCh37/hg19) | CpGs, n (Cluster, n) | Mean Delta-Beta | padj | Gene Symbol | Gene Name | Gene Function and Associated Phenotype |
|---|---|---|---|---|---|---|
| chr21:36258423-36259797 | 7 (7) | 0.2812 | 1.03 × 10−4 | RUNX1 | Runt-related transcription factor 1 | Transcription factor; Hematopoiesis; Hemorrhagic diseases; Blood platelet diseases |
| chr14:45431685-45432516 | 6 (16) | 0.2053 | 8.57 × 10−3 | FAM179B | TOG array regulator of axonemal microtubules protein 1 | Primary cilia organization; Joubert syndrome, Spinocerebellar ataxia |
| chr16:89690088-89690262 | 2 (9) | 0.1897 | 2.40 × 10−3 | DPEP1 | Dipeptidase 1 | Kidney membrane enzyme; Glutathione metabolism; Blau syndrome, Glutamate-cysteine ligase deficiency |
| chr1:201618030-201619787 | 8 (16) | 0.1827 | 1.71 × 10−3 | NAV1 | Neuron navigator 1 | Neuronal migration and axon guidance; Episodic pain syndrome, Long qt syndrome |
| chr4:186732837-186733060 | 7 (9) | −0.1810 | 4.57 × 10−3 | SORBS2 | Sorbin and SH3 domain-containing protein 2 | Adapter protein; Signaling complexes assembling; Hypotrichosis-13, Spheroid body myopathy |
| chr7:43803803-43804002 | 2 (2) | 0.1794 | 2.86 × 10−3 | BLVRA | Biliverdin reductase A | Catalyze; Biliverdin to bilirubin conversion; Hyperbiliverdinemia, Cholestasis |
| chr19:55549590-55549746 | 3 (10) | −0.1767 | 1.37 × 10−2 | GP6 | Platelet glycoprotein VI | Collagen-induced platelet adhesion and activation; Bleeding disorder platelet-1 |
| chr5:176827082-176827697 | 5 (7) | 0.1747 | 1.37 × 10−2 | PFN3 | Profilin-3 | Regulation of actin cytoskeleton, Ras signaling pathway |
| chr21:44898090-44898206 | 3 (7) | −0.1699 | 1.49 × 10−2 | C21orf84 | Long Intergenic Non-Protein Coding RNA 313 | Long noncoding RNA; Lung cancer, Brain glioma |
| chr1:170115042-170115351 | 3 (7) | 0.1696 | 1.49 × 10−2 | METTL11B | α N-terminal protein methyltransferase 1B | Proteins methylation |
| chr12:119772354-119772577 | 5 (5) | 0.1682 | 1.49 × 10−2 | CCDC60 | Coiled-coil domain-containing protein 60 | Muscular dystrophy type A6, Neuronitis |
| chr4:81117647-81119473 | 14 (20) | 0.1638 | 2.86 × 10−3 | PRDM8 | PR domain zinc finger protein 8 | Transcription regulation, Histone methyltransferase; Progressive myoclonic epilepsy-10 |
| chr2:159651813-159651918 | 2 (4) | 0.1618 | 3.09 × 10−2 | DAPL1 | Death-associated protein-like 1 | Apoptosis, Early epithelial differentiation |
| chr11:128554939-128557589 | 13 (19) | 0.1618 | 8.00 × 10−3 | FLI1 | Friend leukemia integration 1 transcription factor | Transcription factor; Hematopoiesis; Hemorrhagic diseases, Bleeding disorder platelet-21 |
| chr6:33043868-33044510 | 5 (13) | 0.1564 | 9.71 × 10−3 | HLA-DPB1 | HLA class II histocompatibility antigen | Peptide antigen binding; Berylliosis, Granulomatosis with polyangiitis, Juvenile idiopathic arthritis |
| chr17:56744332-56744490 | 3 (3) | 0.1551 | 1.49 × 10−2 | TEX14 | Inactive serine/threonine-protein kinase TEX14 | Mitosis; Spermatogenesis; Spermatogenic failure; Azoospermia; Infertility |
| chr5:178422071-178422415 | 6 (11) | 0.1546 | 3.54 × 10−2 | GRM6 | Metabotropic glutamate receptor 6 | Signal transduction; Retinal dystrophy, Night blindness |
| chr17:7832680-7833237 | 9 (11) | 0.1546 | 9.14 × 10−3 | KCNAB3 | Voltage-gated potassium channel subunit β-3 | Signal transmission, Potassium ion transport; Cone-rod dystrophy-6 |
| chr22:51016501-51017166 | 13 (16) | 0.1527 | 1.49 × 10−2 | CPT1B | Carnitine O-palmitoyltransferase 1, muscle isoform | β-oxidation pathway in muscle mitochondria; CPT I deficiency, Visceral steatosis |
| Newborns | Toddlers | Adults | ||
|---|---|---|---|---|
| (Muskens et al. 2021 [8]) | (Current Study) | (Bacalini et al. 2015 [6]) | ||
| DMPs | Total, n | 652 | 4806 | 18,573 |
| Hypermethylated, % | 48.9 | 82.0 | 65.0 | |
| DMRs | Total, n | 1052 | 115 | 66 |
| Hypermethylated, % | 48.0 | 83.5 | 73.0 |
| DMR Position (GRCh37/hg19) | CGI Relation | Gene Name | Gene Region | DNAME Difference in DS (Mean Delta-Beta) | ||
|---|---|---|---|---|---|---|
| Newborns | Toddlers | Adults | ||||
| chr1:36786285-36787932 | CGI | SH3D21; FAM176B | Gene Body | 0.0843 | 0.1851 | 0.2014 |
| chr2:54086854-54087343 | CGI | ASB3; GPR75 | 5′UTR, TSS200 | −0.1151 | −0.1377 | −0.2035 |
| chr4:81117647-81119473 | CGI | PRDM8 | 5′UTR, TSS1500 | 0.1739 | 0.1638 | 0.1975 |
| chr4:145566200-145566903 | CGI | HHIP | TSS1500 | 0.079 | 0.1312 | 0.163 |
| chr5:176827082-176827697 | CGI | PFN3 | 1stExon, TSS200 | 0.0447 | 0.1747 | 0.3039 |
| chr6:31939106-31939546 | CGI Shore | STK19; DOM3Z | 5′UTR, TSS1500 | 0.0154 | 0.1315 | 0.1939 |
| chr6:33282628-33282997 | CGI | TAPBP; ZBTB22 | TSS1500 | 0.0355 | 0.1080 | 0.1845 |
| chr6:44243304-44243750 | CGI | TMEM151B | Gene Body | 0.117 | 0.1598 | 0.1782 |
| chr7:27142618-27143788 | CGI | HOXA2 | TSS1500 | −0.0408 | −0.1415 | −0.2116 |
| chr7:27169957-27171051 | CGI | HOXA4 | 1stExon, 5′UTR, TSS200 | 0.0811 | 0.1421 | 0.2069 |
| chr9:34370835-34371380 | CGI | MYORG | Gene Body | 0.0869 | 0.2016 | 0.2095 |
| chr10:70321668-70322874 | CGI Shore | TET1 | 5′UTR | −0.0449 | −0.1486 | −0.1820 |
| chr12:119772354-119772577 | CGI | CCDC60 | 1stExon, 5′UTR, TSS200 | 0.0721 | 0.1682 | 0.1778 |
| chr13:113689776-113689728 | CGI Shore | MCF2L | Gene Body | −0.1022 | −0.1722 | −0.2070 |
| chr16:979488-979898 | CGI | LMF1 | Gene Body | −0.1657 | −0.1698 | −0.3292 |
| chr16:2029256-2030892 | CGI | NOXO1 | Gene Body | 0.0492 | 0.1415 | 0.2216 |
| chr18:77905408-77905751 | CGI | PARD6G-AS1 | TSS200 | −0.0868 | −0.1317 | −0.1846 |
| chr21:36258423-36259797 | CGI | RUNX1 | 1stExon, 5′UTR | 0.2733 | 0.2812 | 0.3557 |
| chr22:51016501-51017166 | CGI | CPT1B | 1stExon, 5′UTR, TSS200 | 0.2096 | 0.1527 | 0.2586 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naumova, O.Y.; Lipschutz, R.; Rychkov, S.Y.; Zhukova, O.V.; Grigorenko, E.L. DNA Methylation Alterations in Blood Cells of Toddlers with Down Syndrome. Genes 2021, 12, 1115. https://doi.org/10.3390/genes12081115
Naumova OY, Lipschutz R, Rychkov SY, Zhukova OV, Grigorenko EL. DNA Methylation Alterations in Blood Cells of Toddlers with Down Syndrome. Genes. 2021; 12(8):1115. https://doi.org/10.3390/genes12081115
Chicago/Turabian StyleNaumova, Oxana Yu., Rebecca Lipschutz, Sergey Yu. Rychkov, Olga V. Zhukova, and Elena L. Grigorenko. 2021. "DNA Methylation Alterations in Blood Cells of Toddlers with Down Syndrome" Genes 12, no. 8: 1115. https://doi.org/10.3390/genes12081115
APA StyleNaumova, O. Y., Lipschutz, R., Rychkov, S. Y., Zhukova, O. V., & Grigorenko, E. L. (2021). DNA Methylation Alterations in Blood Cells of Toddlers with Down Syndrome. Genes, 12(8), 1115. https://doi.org/10.3390/genes12081115