
Oligomerization of Selective Autophagy Receptors for the Targeting and Degradation of Protein Aggregates


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. A Brief History of Selective Autophagy
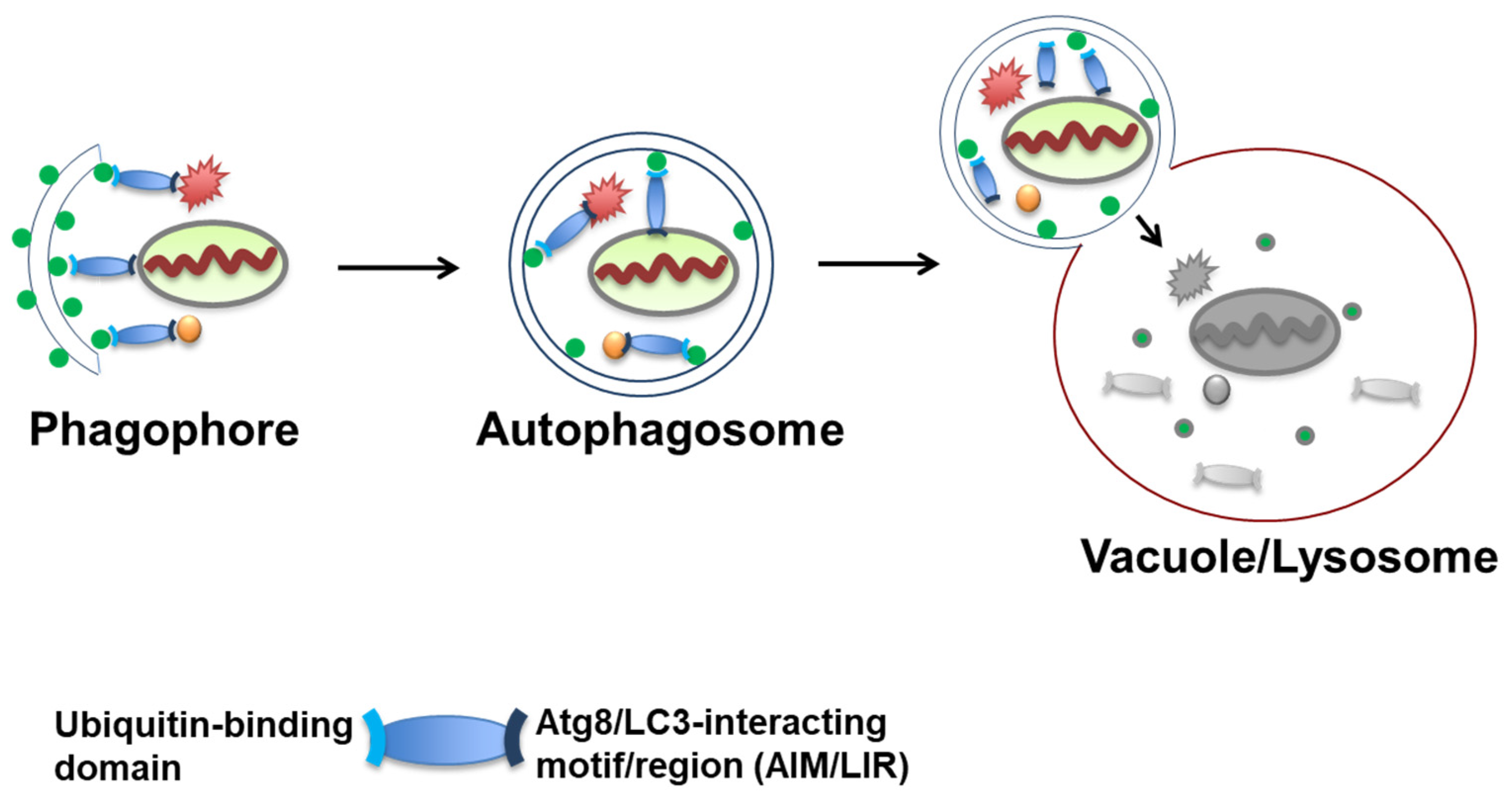
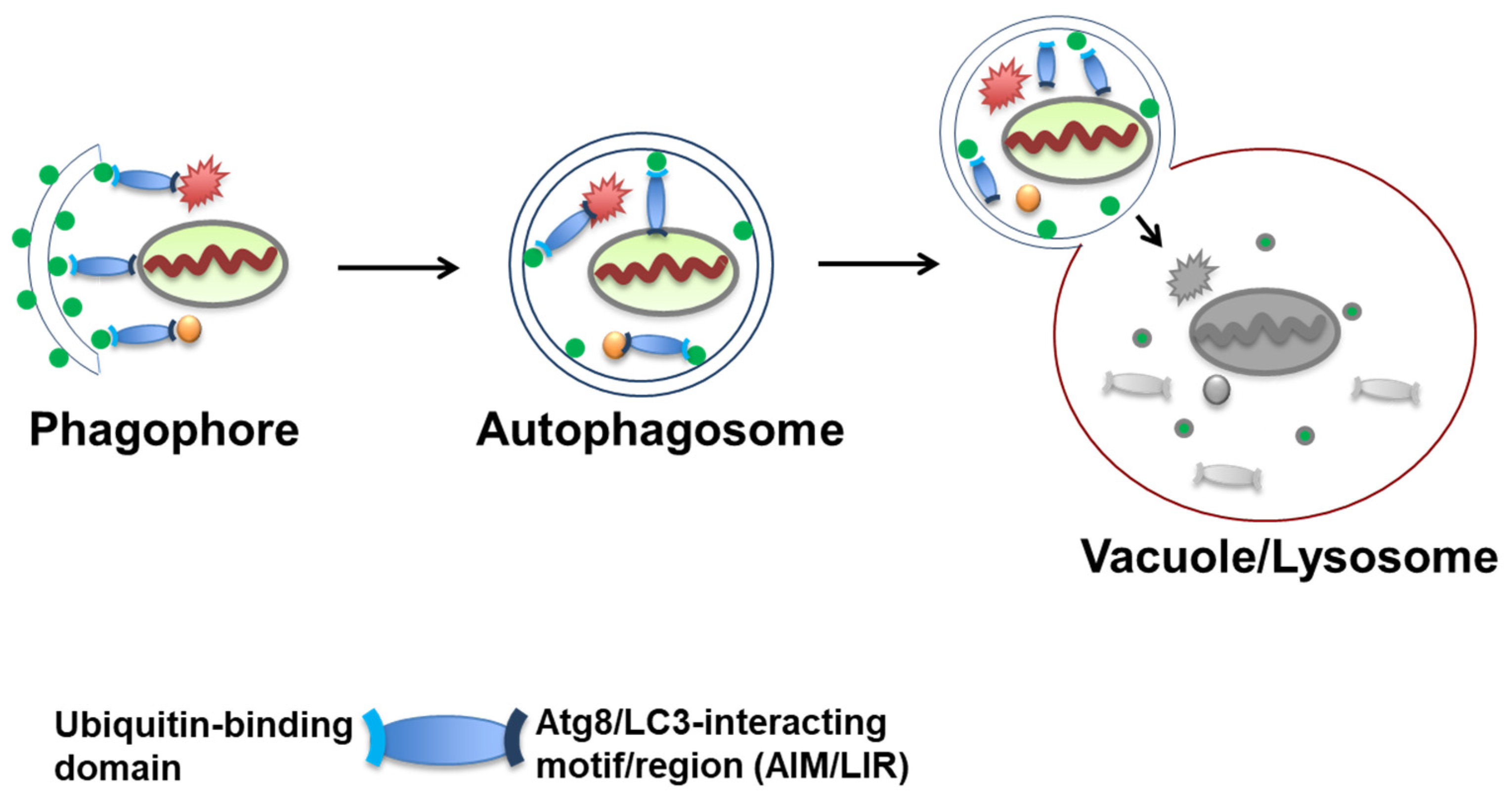
3. Autophagosome Formation and Atg8/LC3 Scaffolds as Platforms for Receptor Cargo Recruitment
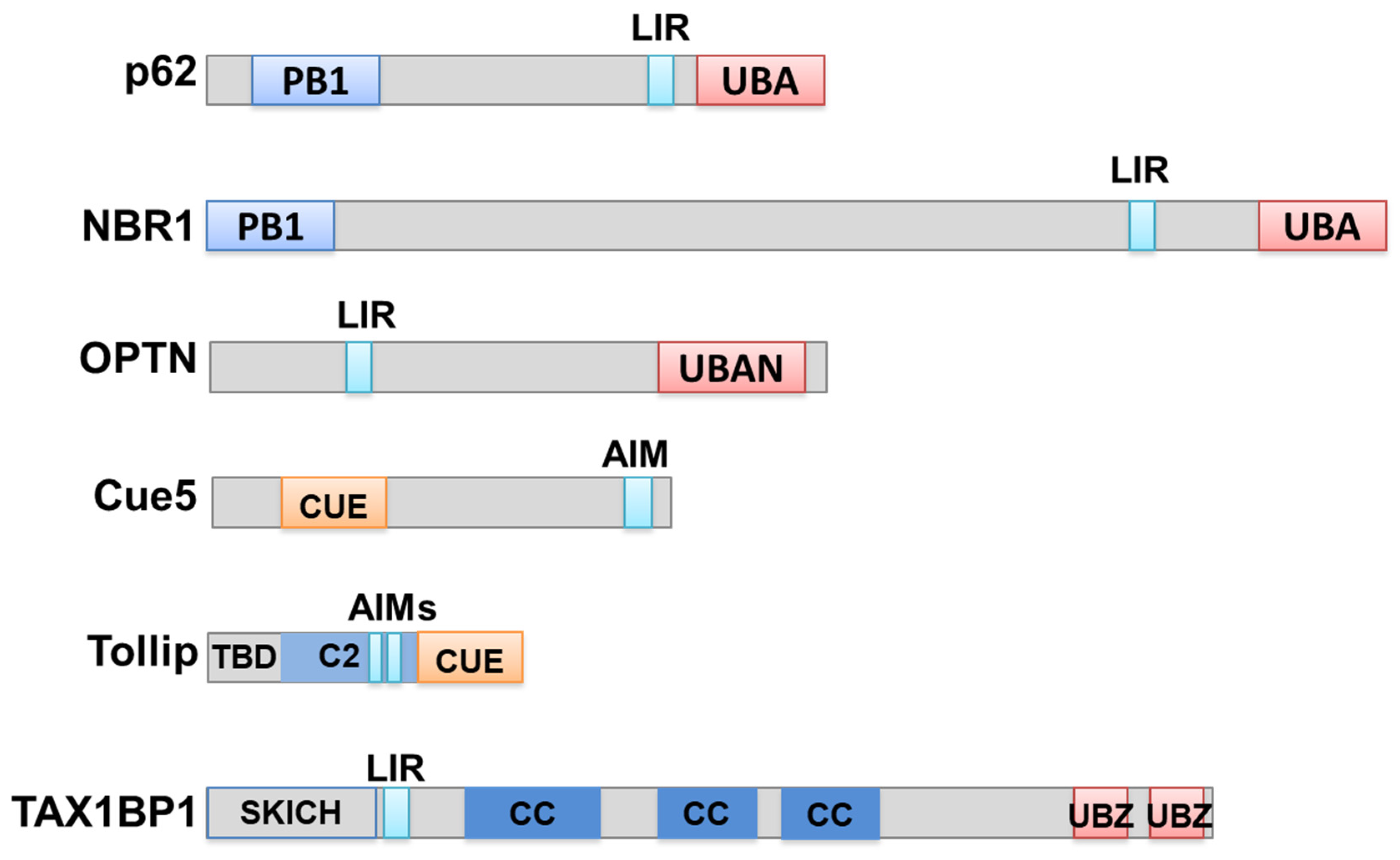
4. Autophagy Receptors for the Degradation of Protein Aggregates
5. Pivotal Role of Oligomerization in Autophagy Receptor Function
6. Role of Oligomerization in Autophagy Substrates
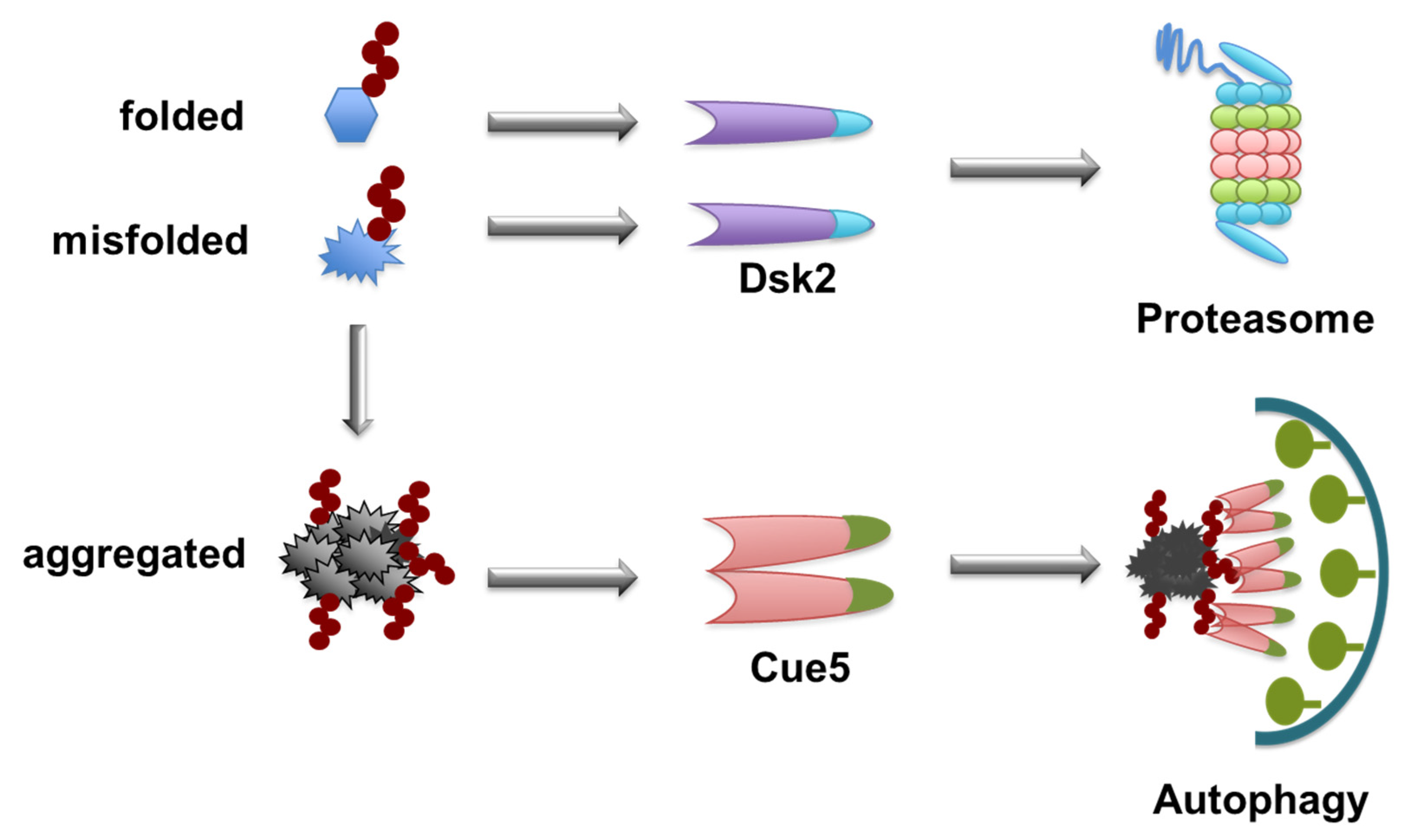
7. Segregation of Aggrephagy Cargos
8. Monomer and Oligomer Status of the Same Receptors Confers Dual Functions in Two Degradation Pathways
9. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hartl, F.U.; Hayer-Hartl, M. Converging concepts of protein folding in vitro and in vivo. Nat. Struct. Mol. Biol. 2009, 16, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Knowles, T.P.; Vendruscolo, M.; Dobson, C.M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Park, S.H.; Hartl, F.U. Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol. 2014, 24, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Schulman, B.A. Dynamic regulation of macroautophagy by distinctive ubiquitin-like proteins. Nat. Struct. Mol. Biol. 2014, 21, 336–345. [Google Scholar] [CrossRef]
- Khaminets, A.; Behl, C.; Dikic, I. Ubiquitin-Dependent And Independent Signals In Selective Autophagy. Trends Cell Biol. 2016, 26, 6–16. [Google Scholar] [CrossRef]
- Kirkin, V.; McEwan, D.G.; Novak, I.; Dikic, I. A role for ubiquitin in selective autophagy. Mol. Cell 2009, 34, 259–269. [Google Scholar] [CrossRef]
- Lippai, M.; Low, P. The role of the selective adaptor p62 and ubiquitin-like proteins in autophagy. Biomed. Res. Int. 2014, 2014, 832704. [Google Scholar] [CrossRef] [Green Version]
- Kraft, C.; Peter, M.; Hofmann, K. Selective autophagy: Ubiquitin-mediated recognition and beyond. Nat. Cell Biol. 2010, 12, 836–841. [Google Scholar] [CrossRef]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Overvatn, A.; Bjorkoy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [Green Version]
- Kirkin, V.; Lamark, T.; Sou, Y.S.; Bjorkoy, G.; Nunn, J.L.; Bruun, J.A.; Shvets, E.; McEwan, D.G.; Clausen, T.H.; Wild, P.; et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell 2009, 33, 505–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, K.; Psakhye, I.; Jentsch, S. Autophagic clearance of polyQ proteins mediated by ubiquitin-Atg8 adaptors of the conserved CUET protein family. Cell 2014, 158, 549–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Randles, L.; Shi, K.; Tarasov, S.G.; Aihara, H.; Walters, K.J. Structures of Rpn1 T1:Rad23 and hRpn13:hPLIC2 Reveal Distinct Binding Mechanisms between Substrate Receptors and Shuttle Factors of the Proteasome. Structure 2016, 24, 1257–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, R.Y.; Chen, L.; Ko, B.T.; Shen, Y.H.; Li, Y.T.; Chen, B.R.; Lin, K.T.; Madura, K.; Chuang, S.M. Rad23 interaction with the proteasome is regulated by phosphorylation of its ubiquitin-like (UbL) domain. J. Mol. Biol. 2014, 426, 4049–4060. [Google Scholar] [CrossRef] [Green Version]
- Lambertson, D.; Chen, L.; Madura, K. Investigating the importance of proteasome-interaction for Rad23 function. Curr. Genet. 2003, 42, 199–208. [Google Scholar] [CrossRef]
- Davies, S.E.; Hallett, P.J.; Moens, T.; Smith, G.; Mangano, E.; Kim, H.T.; Goldberg, A.L.; Liu, J.L.; Isacson, O.; Tofaris, G.K. Enhanced ubiquitin-dependent degradation by Nedd4 protects against alpha-synuclein accumulation and toxicity in animal models of Parkinson’s disease. Neurobiol. Dis. 2014, 64, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Kim, H.R.; Quinley, C.; Kim, J.; Gonzalez-Navajas, J.; Xavier, R.; Raz, E. Autophagy suppresses interleukin-1beta (IL-1beta) signaling by activation of p62 degradation via lysosomal and proteasomal pathways. J. Biol. Chem. 2012, 287, 4033–4040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stolz, A.; Ernst, A.; Dikic, I. Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 2014, 16, 495–501. [Google Scholar] [CrossRef]
- Tan, J.M.; Wong, E.S.; Kirkpatrick, D.S.; Pletnikova, O.; Ko, H.S.; Tay, S.P.; Ho, M.W.; Troncoso, J.; Gygi, S.P.; Lee, M.K.; et al. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum. Mol. Genet. 2008, 17, 431–439. [Google Scholar] [CrossRef]
- Lim, G.G.; Chew, K.C.; Ng, X.H.; Henry-Basil, A.; Sim, R.W.; Tan, J.M.; Chai, C.; Lim, K.L. Proteasome inhibition promotes Parkin-Ubc13 interaction and lysine 63-linked ubiquitination. PLoS ONE 2013, 8, e73235. [Google Scholar] [CrossRef] [Green Version]
- Riley, B.E.; Kaiser, S.E.; Shaler, T.A.; Ng, A.C.; Hara, T.; Hipp, M.S.; Lage, K.; Xavier, R.J.; Ryu, K.Y.; Taguchi, K.; et al. Ubiquitin accumulation in autophagy-deficient mice is dependent on the Nrf2-mediated stress response pathway: A potential role for protein aggregation in autophagic substrate selection. J. Cell Biol. 2010, 191, 537–552. [Google Scholar] [CrossRef] [Green Version]
- Ben Yehuda, A.; Risheq, M.; Novoplansky, O.; Bersuker, K.; Kopito, R.R.; Goldberg, M.; Brandeis, M. Ubiquitin Accumulation on Disease Associated Protein Aggregates is Correlated with Nuclear Ubiquitin Depletion, Histone De-Ubiquitination and Impaired DNA Damage Response. PLoS ONE 2017, 12, e0169054. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, P.; Gao, H.; Gu, Y.; Yang, J.; Peng, H.; Xu, X.; Wang, H.; Yang, M.; Liu, X.; et al. Ubiquitylation of autophagy receptor Optineurin by HACE1 activates selective autophagy for tumor suppression. Cancer Cell 2014, 26, 106–120. [Google Scholar] [CrossRef] [Green Version]
- Ohsumi, Y. Historical landmarks of autophagy research. Cell Res. 2014, 24, 9–23. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J. Autophagy revisited: A conversation with Christian de Duve. Autophagy 2008, 4, 740–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filimonenko, M.; Isakson, P.; Finley, K.D.; Anderson, M.; Jeong, H.; Melia, T.J.; Bartlett, B.J.; Myers, K.M.; Birkeland, H.C.; Lamark, T.; et al. The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol. Cell 2010, 38, 265–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolender, R.P.; Weibel, E.R. A morphometric study of the removal of phenobarbital-induced membranes from hepatocytes after cessation of threatment. J. Cell Biol. 1973, 56, 746–761. [Google Scholar] [CrossRef] [PubMed]
- Veenhuis, M.; Douma, A.; Harder, W.; Osumi, M. Degradation and turnover of peroxisomes in the yeast Hansenula polymorpha induced by selective inactivation of peroxisomal enzymes. Arch. Microbiol. 1983, 134, 193–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McElligott, M.A.; Dice, J.F. Degradation of microinjected ribonuclease A and ribonuclease S-protein by lysosomal pathways. Prog. Clin. Biol. Res. 1985, 180, 471–473. [Google Scholar] [PubMed]
- Mortimore, G.E.; Poso, A.R.; Kadowaki, M.; Wert, J.J., Jr. Multiphasic control of hepatic protein degradation by regulatory amino acids. General features and hormonal modulation. J. Biol. Chem. 1987, 262, 16322–16327. [Google Scholar] [CrossRef]
- Lemasters, J.J. Variants of mitochondrial autophagy: Types 1 and 2 mitophagy and micromitophagy (Type 3). Redox Biol. 2014, 2, 749–754. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, I.; Amano, A.; Mizushima, N.; Yamamoto, A.; Yamaguchi, H.; Kamimoto, T.; Nara, A.; Funao, J.; Nakata, M.; Tsuda, K.; et al. Autophagy defends cells against invading group A Streptococcus. Science 2004, 306, 1037–1040. [Google Scholar] [CrossRef]
- Singh, S.B.; Davis, A.S.; Taylor, G.A.; Deretic, V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 2006, 313, 1438–1441. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Kondo, C.; Morimoto, M.; Ohsumi, Y. Selective transport of alpha-mannosidase by autophagic pathways: Identification of a novel receptor, Atg34p. J. Biol. Chem. 2010, 285, 30019–30025. [Google Scholar] [CrossRef] [Green Version]
- Kirkin, V. History of the Selective Autophagy Research: How Did It Begin and Where Does It Stand Today? J. Mol. Biol. 2020, 432, 3–27. [Google Scholar] [CrossRef] [PubMed]
- Vadlamudi, R.K.; Shin, J. Genomic structure and promoter analysis of the p62 gene encoding a non-proteasomal multiubiquitin chain binding protein. FEBS Lett. 1998, 435, 138–142. [Google Scholar] [CrossRef] [Green Version]
- Fujioka, Y.; Suzuki, S.W.; Yamamoto, H.; Kondo-Kakuta, C.; Kimura, Y.; Hirano, H.; Akada, R.; Inagaki, F.; Ohsumi, Y.; Noda, N.N. Structural basis of starvation-induced assembly of the autophagy initiation complex. Nat. Struct. Mol. Biol. 2014, 21, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V. Links between autophagy, innate immunity, inflammation and Crohn’s disease. Dig. Dis. 2009, 27, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Brest, P.; Corcelle, E.A.; Cesaro, A.; Chargui, A.; Belaid, A.; Klionsky, D.J.; Vouret-Craviari, V.; Hebuterne, X.; Hofman, P.; Mograbi, B. Autophagy and Crohn′s disease: At the crossroads of infection, inflammation, immunity, and cancer. Curr. Mol. Med. 2010, 10, 486–502. [Google Scholar] [CrossRef] [Green Version]
- Cappelletti, C.; Galbardi, B.; Kapetis, D.; Vattemi, G.; Guglielmi, V.; Tonin, P.; Salerno, F.; Morandi, L.; Tomelleri, G.; Mantegazza, R.; et al. Autophagy, inflammation and innate immunity in inflammatory myopathies. PLoS ONE 2014, 9, e111490. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Z.; Sanchez-Lopez, E.; Karin, M. Autophagy, Inflammation, and Immunity: A Troika Governing Cancer and Its Treatment. Cell 2016, 166, 288–298. [Google Scholar] [CrossRef] [Green Version]
- Nakatogawa, H.; Suzuki, K.; Kamada, Y.; Ohsumi, Y. Dynamics and diversity in autophagy mechanisms: Lessons from yeast. Nat. Rev. Mol. Cell Biol. 2009, 10, 458–467. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Osawa, T.; Fujioka, Y.; Noda, N.N. Structural biology of the core autophagy machinery. Curr. Opin. Struct. Biol. 2017, 43, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Wen, X.; Klionsky, D.J. An overview of macroautophagy in yeast. J. Mol. Biol. 2016, 428, 1681–1699. [Google Scholar] [CrossRef] [Green Version]
- Matoba, K.; Kotani, T.; Tsutsumi, A.; Tsuji, T.; Mori, T.; Noshiro, D.; Sugita, Y.; Nomura, N.; Iwata, S.; Ohsumi, Y.; et al. Atg9 is a lipid scramblase that mediates autophagosomal membrane expansion. Nat. Struct. Mol. Biol. 2020, 27, 1185–1193. [Google Scholar] [CrossRef]
- Yin, Z.; Pascual, C.; Klionsky, D.J. Autophagy: Machinery and regulation. Microb. Cell 2016, 3, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yang, Q.; Mao, Z. Chaperone-mediated autophagy: Machinery, regulation and biological consequences. Cell Mol. Life Sci. 2011, 68, 749–763. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Periyasamy-Thandavan, S.; Jiang, M.; Schoenlein, P.; Dong, Z. Autophagy: Molecular machinery, regulation, and implications for renal pathophysiology. Am. J. Physiol. Renal. Physiol. 2009, 297, F244–F256. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Noda, T.; Yoshimori, T.; Tanaka, Y.; Ishii, T.; George, M.D.; Klionsky, D.J.; Ohsumi, M.; Ohsumi, Y. A protein conjugation system essential for autophagy. Nature 1998, 395, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Martens, S.; Fracchiolla, D. Activation and targeting of ATG8 protein lipidation. Cell Discov. 2020, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Collier, J.J.; Guissart, C.; Oláhová, M.; Sasorith, S.; Piron-Prunier, F.; Suomi, F.; Zhang, D.; Martinez-Lopez, N.; Leboucq, N.; Bahr, A.; et al. Developmental Consequences of Defective ATG7-Mediated Autophagy in Humans. N. Engl. J. Med. 2021, 384, 2406–2417. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Padman, B.S.; Usher, J.; Oorschot, V.; Ramm, G.; Lazarou, M. Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J. Cell Biol. 2016, 215, 857–874. [Google Scholar] [CrossRef] [PubMed]
- Ohnstad, A.E.; Delgado, J.M.; North, B.J.; Nasa, I.; Kettenbach, A.N.; Schultz, S.W.; Shoemaker, C.J. Receptor-mediated clustering of FIP200 bypasses the role of LC3 lipidation in autophagy. EMBO J. 2020, 39, e104948. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, C.; Legouis, R.; Culetto, E. ESCRT and autophagies: Endosomal functions and beyond. Semin. Cell Dev. Biol. 2018, 74, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Wu, Z.; Zhao, M.; Murtazina, R.; Cai, J.; Zhang, A.; Li, R.; Sun, D.; Li, W.; Zhao, L.; et al. Rab5-dependent autophagosome closure by ESCRT. J. Cell Biol. 2019, 218, 1908–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, F.; Wu, Z.; Zhao, M.; Segev, N.; Liang, Y. Autophagosome closure by ESCRT: Vps21/RAB5-regulated ESCRT recruitment via an Atg17-Snf7 interaction. Autophagy 2019, 15, 1653–1654. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Y.; Radulovic, M.; Vietri, M.; Stenmark, H. Sealing holes in cellular membranes. EMBO J. 2021, 40, e106922. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; He, H.; Tang, Z.; Hattori, T.; Liu, Y.; Young, M.M.; Serfass, J.M.; Chen, L.; Gebru, M.; Chen, C.; et al. An autophagy assay reveals the ESCRT-III component CHMP2A as a regulator of phagophore closure. Nat. Commun. 2018, 9, 2855. [Google Scholar] [CrossRef] [Green Version]
- Zhen, Y.; Spangenberg, H.; Munson, M.J.; Brech, A.; Schink, K.O.; Tan, K.W.; Sørensen, V.; Wenzel, E.M.; Radulovic, M.; Engedal, N.; et al. ESCRT-mediated phagophore sealing during mitophagy. Autophagy 2020, 16, 826–841. [Google Scholar] [CrossRef] [Green Version]
- Fatyol, K.; Grummt, I. Proteasomal ATPases are associated with rDNA: The ubiquitin proteasome system plays a direct role in RNA polymerase I transcription. Biochim. Biophys. Acta 2008, 1779, 850–859. [Google Scholar] [CrossRef]
- Domingues, A.F.; Arduino, D.M.; Esteves, A.R.; Swerdlow, R.H.; Oliveira, C.R.; Cardoso, S.M. Mitochondria and ubiquitin-proteasomal system interplay: Relevance to Parkinson′s disease. Free Radic. Biol. Med. 2008, 45, 820–825. [Google Scholar] [CrossRef]
- Taylor, J.M.; Song, Y.J.; Huang, Y.; Farrer, M.J.; Delatycki, M.B.; Halliday, G.M.; Lockhart, P.J. Parkin Co-Regulated Gene (PACRG) is regulated by the ubiquitin-proteasomal system and is present in the pathological features of Parkinsonian diseases. NeuroBiol. Dis. 2007, 27, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Pfirrmann, T.; Jandt, E.; Ranft, S.; Lokapally, A.; Neuhaus, H.; Perron, M.; Hollemann, T. Hedgehog-dependent E3-ligase Midline1 regulates ubiquitin-mediated proteasomal degradation of Pax6 during visual system development. Proc. Natl. Acad. Sci. USA 2016, 113, 10103–10108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadhave, K.; Bolshette, N.; Ahire, A.; Pardeshi, R.; Thakur, K.; Trandafir, C.; Istrate, A.; Ahmed, S.; Lahkar, M.; Muresanu, D.F.; et al. The ubiquitin proteasomal system: A potential target for the management of Alzheimer′s disease. J. Cell Mol. Med. 2016, 20, 1392–1407. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, J.; Rajamma, U.; Jana, N.; Mohanakumar, K.P. Quercetin improves the activity of the ubiquitin-proteasomal system in 150Q mutated huntingtin-expressing cells but exerts detrimental effects on neuronal survivability. J. Neurosci. Res. 2015, 93, 1581–1591. [Google Scholar] [CrossRef] [PubMed]
- Kastle, M.; Grune, T. Protein oxidative modification in the aging organism and the role of the ubiquitin proteasomal system. Curr. Pharm. Des. 2011, 17, 4007–4022. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Mao, X. The ubiquitin-proteasomal system is critical for multiple myeloma: Implications in drug discovery. Am. J. Blood Res. 2011, 1, 46–56. [Google Scholar]
- Ryabovol, V.V.; Minibayeva, F.V. Molecular Mechanisms of Autophagy in Plants: Role of ATG8 Proteins in Formation and Functioning of Autophagosomes. Biochemistry 2016, 81, 348–363. [Google Scholar] [CrossRef]
- Kellner, R.; De la Concepcion, J.C.; Maqbool, A.; Kamoun, S.; Dagdas, Y.F. ATG8 Expansion: A Driver of Selective Autophagy Diversification? Trends Plant. Sci. 2017, 22, 204–214. [Google Scholar] [CrossRef]
- Fracchiolla, D.; Zens, B.; Martens, S. In Vitro Reconstitution of Atg8 Conjugation and Deconjugation. Methods Enzymol. 2017, 587, 377–390. [Google Scholar] [CrossRef]
- Abreu, S.; Kriegenburg, F.; Gomez-Sanchez, R.; Mari, M.; Sanchez-Wandelmer, J.; Skytte Rasmussen, M.; Soares Guimaraes, R.; Zens, B.; Schuschnig, M.; Hardenberg, R.; et al. Conserved Atg8 recognition sites mediate Atg4 association with autophagosomal membranes and Atg8 deconjugation. EMBO Rep. 2017, 18, 765–780. [Google Scholar] [CrossRef] [Green Version]
- Voss, C.; Ehrenman, K.; Mlambo, G.; Mishra, S.; Kumar, K.A.; Sacci, J.B., Jr.; Sinnis, P.; Coppens, I. Overexpression of Plasmodium berghei ATG8 by Liver Forms Leads to Cumulative Defects in Organelle Dynamics and to Generation of Noninfectious Merozoites. MBio 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjorkoy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Waguri, S.; Koike, M.; Sou, Y.S.; Ueno, T.; Hara, T.; Mizushima, N.; Iwata, J.; Ezaki, J.; Murata, S.; et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007, 131, 1149–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gal, J.; Strom, A.L.; Kilty, R.; Zhang, F.; Zhu, H. p62 accumulates and enhances aggregate formation in model systems of familial amyotrophic lateral sclerosis. J. Biol. Chem. 2007, 282, 11068–11077. [Google Scholar] [CrossRef] [Green Version]
- Slowicka, K.; Vereecke, L.; Mc Guire, C.; Sze, M.; Maelfait, J.; Kolpe, A.; Saelens, X.; Beyaert, R.; van Loo, G. Optineurin deficiency in mice is associated with increased sensitivity to Salmonella but does not affect proinflammatory NF-kappaB signaling. Eur. J. Immunol. 2016, 46, 971–980. [Google Scholar] [CrossRef] [Green Version]
- Chew, T.S.; O’Shea, N.R.; Sewell, G.W.; Oehlers, S.H.; Mulvey, C.M.; Crosier, P.S.; Godovac-Zimmermann, J.; Bloom, S.L.; Smith, A.M.; Segal, A.W. Optineurin deficiency in mice contributes to impaired cytokine secretion and neutrophil recruitment in bacteria-driven colitis. Dis. Model. Mech. 2015, 8, 817–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.W.; Hong, S.B.; Kim, J.H.; Kwon, D.H.; Song, H.K. Structural basis for recognition of autophagic receptor NDP52 by the sugar receptor galectin-8. Nat. Commun. 2013, 4, 1613. [Google Scholar] [CrossRef] [Green Version]
- Lu, K.; Psakhye, I.; Jentsch, S. A new class of ubiquitin-Atg8 receptors involved in selective autophagy and polyQ protein clearance. Autophagy 2014, 10, 2381–2382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, K.; den Brave, F.; Jentsch, S. Receptor oligomerization guides pathway choice between proteasomal and autophagic degradation. Nat. Cell Biol. 2017, 19, 732–739. [Google Scholar] [CrossRef]
- Sarraf, S.A.; Shah, H.V.; Kanfer, G.; Pickrell, A.M.; Holtzclaw, L.A.; Ward, M.E.; Youle, R.J. Loss of TAX1BP1-Directed Autophagy Results in Protein Aggregate Accumulation in the Brain. Mol. Cell 2020, 80, 779–795 e710. [Google Scholar] [CrossRef]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewitt, G.; Carroll, B.; Sarallah, R.; Correia-Melo, C.; Ogrodnik, M.; Nelson, G.; Otten, E.G.; Manni, D.; Antrobus, R.; Morgan, B.A.; et al. SQSTM1/p62 mediates crosstalk between autophagy and the UPS in DNA repair. Autophagy 2016, 12, 1917–1930. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Purtell, K.; Lachance, V.; Wold, M.S.; Chen, S.; Yue, Z. Autophagy Receptors and Neurodegenerative Diseases. Trends Cell Biol. 2017, 27, 491–504. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, G.; Wada, K.; Okuno, M.; Kurosawa, M.; Nukina, N. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol. Cell 2011, 44, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A.; et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. USA 2016, 113, 4039–4044. [Google Scholar] [CrossRef] [Green Version]
- Wild, P.; Farhan, H.; McEwan, D.G.; Wagner, S.; Rogov, V.V.; Brady, N.R.; Richter, B.; Korac, J.; Waidmann, O.; Choudhary, C.; et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 2011, 333, 228–233. [Google Scholar] [CrossRef] [Green Version]
- O’Loughlin, T.; Kruppa, A.J.; Ribeiro, A.L.R.; Edgar, J.R.; Ghannam, A.; Smith, A.M.; Buss, F. OPTN recruitment to a Golgi-proximal compartment regulates immune signalling and cytokine secretion. J. Cell Sci. 2020, 133, 1–15. [Google Scholar] [CrossRef]
- Moore, A.S.; Holzbaur, E.L. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc. Natl. Acad. Sci. USA 2016, 113, E3349–E3358. [Google Scholar] [CrossRef] [Green Version]
- Tumbarello, D.A.; Manna, P.T.; Allen, M.; Bycroft, M.; Arden, S.D.; Kendrick-Jones, J.; Buss, F. The Autophagy Receptor TAX1BP1 and the Molecular Motor Myosin VI Are Required for Clearance of Salmonella Typhimurium by Autophagy. PLoS Pathog. 2015, 11, e1005174. [Google Scholar] [CrossRef] [Green Version]
- Verstrepen, L.; Verhelst, K.; Carpentier, I.; Beyaert, R. TAX1BP1, a ubiquitin-binding adaptor protein in innate immunity and beyond. Trends Biochem. Sci. 2011, 36, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Gurung, R.; Tan, A.; Ooms, L.M.; McGrath, M.J.; Huysmans, R.D.; Munday, A.D.; Prescott, M.; Whisstock, J.C.; Mitchell, C.A. Identification of a novel domain in two mammalian inositol-polyphosphate 5-phosphatases that mediates membrane ruffle localization. The inositol 5-phosphatase skip localizes to the endoplasmic reticulum and translocates to membrane ruffles following epidermal growth factor stimulation. J. Biol. Chem. 2003, 278, 11376–11385. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Wang, G.; Huang, X.; Du, Z. Expression, purification and crystallization of the SKICH domain of human TAX1BP1. Acta Crystallogr. F Struct. Biol. Commun. 2014, 70, 619–623. [Google Scholar] [CrossRef] [Green Version]
- Iha, H.; Peloponese, J.M.; Verstrepen, L.; Zapart, G.; Ikeda, F.; Smith, C.D.; Starost, M.F.; Yedavalli, V.; Heyninck, K.; Dikic, I.; et al. Inflammatory cardiac valvulitis in TAX1BP1-deficient mice through selective NF-kappaB activation. EMBO J. 2008, 27, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Ceregido, M.A.; Spinola Amilibia, M.; Buts, L.; Rivera-Torres, J.; Garcia-Pino, A.; Bravo, J.; van Nuland, N.A. The structure of TAX1BP1 UBZ1+2 provides insight into target specificity and adaptability. J. Mol. Biol. 2014, 426, 674–690. [Google Scholar] [CrossRef]
- De Valck, D.; Jin, D.Y.; Heyninck, K.; Van de Craen, M.; Contreras, R.; Fiers, W.; Jeang, K.T.; Beyaert, R. The zinc finger protein A20 interacts with a novel anti-apoptotic protein which is cleaved by specific caspases. Oncogene 1999, 18, 4182–4190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toruń, A.; Szymańska, E.; Castanon, I.; Wolińska-Nizioł, L.; Bartosik, A.; Jastrzębski, K.; Miętkowska, M.; Gonzalez-Gaitan, M.; Miaczynska, M. Endocytic Adaptor Protein Tollip Inhibits Canonical Wnt Signaling. PLoS ONE 2015, 10, e0130818. [Google Scholar]
- Burns, K.; Clatworthy, J.; Martin, L.; Martinon, F.; Plumpton, C.; Maschera, B.; Lewis, A.; Ray, K.; Tschopp, J.; Volpe, F. Tollip, a new component of the IL-1RI pathway, links IRAK to the IL-1 receptor. Nat. Cell Biol. 2000, 2, 346–351. [Google Scholar] [CrossRef]
- Prag, G.; Misra, S.; Jones, E.A.; Ghirlando, R.; Davies, B.A.; Horazdovsky, B.F.; Hurley, J.H. Mechanism of ubiquitin recognition by the CUE domain of Vps9p. Cell 2003, 113, 609–620. [Google Scholar] [CrossRef] [Green Version]
- Carroll, B.; Otten, E.G.; Manni, D.; Stefanatos, R.; Menzies, F.M.; Smith, G.R.; Jurk, D.; Kenneth, N.; Wilkinson, S.; Passos, J.F.; et al. Oxidation of SQSTM1/p62 mediates the link between redox state and protein homeostasis. Nat. Commun. 2018, 9, 256. [Google Scholar] [CrossRef] [Green Version]
- Svenning, S.; Lamark, T.; Krause, K.; Johansen, T. Plant NBR1 is a selective autophagy substrate and a functional hybrid of the mammalian autophagic adapters NBR1 and p62/SQSTM1. Autophagy 2011, 7, 993–1010. [Google Scholar] [CrossRef]
- Hirano, S.; Uemura, T.; Annoh, H.; Fujita, N.; Waguri, S.; Itoh, T.; Fukuda, M. Differing susceptibility to autophagic degradation of two LC3-binding proteins: SQSTM1/p62 and TBC1D25/OATL1. Autophagy 2016, 12, 312–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamasaki, A.; Watanabe, Y.; Adachi, W.; Suzuki, K.; Matoba, K.; Kirisako, H.; Kumeta, H.; Nakatogawa, H.; Ohsumi, Y.; Inagaki, F.; et al. Structural Basis for Receptor-Mediated Selective Autophagy of Aminopeptidase I Aggregates. Cell Rep. 2016, 16, 19–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertipaglia, C.; Schneider, S.; Jakobi, A.J.; Tarafder, A.K.; Bykov, Y.S.; Picco, A.; Kukulski, W.; Kosinski, J.; Hagen, W.J.; Ravichandran, A.C.; et al. Higher-order assemblies of oligomeric cargo receptor complexes form the membrane scaffold of the Cvt vesicle. EMBO Rep. 2016, 17, 1044–1060. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, A.; Noda, N.N. Structural Biology of the Cvt Pathway. J. Mol. Biol. 2017, 429, 531–542. [Google Scholar] [CrossRef] [PubMed]
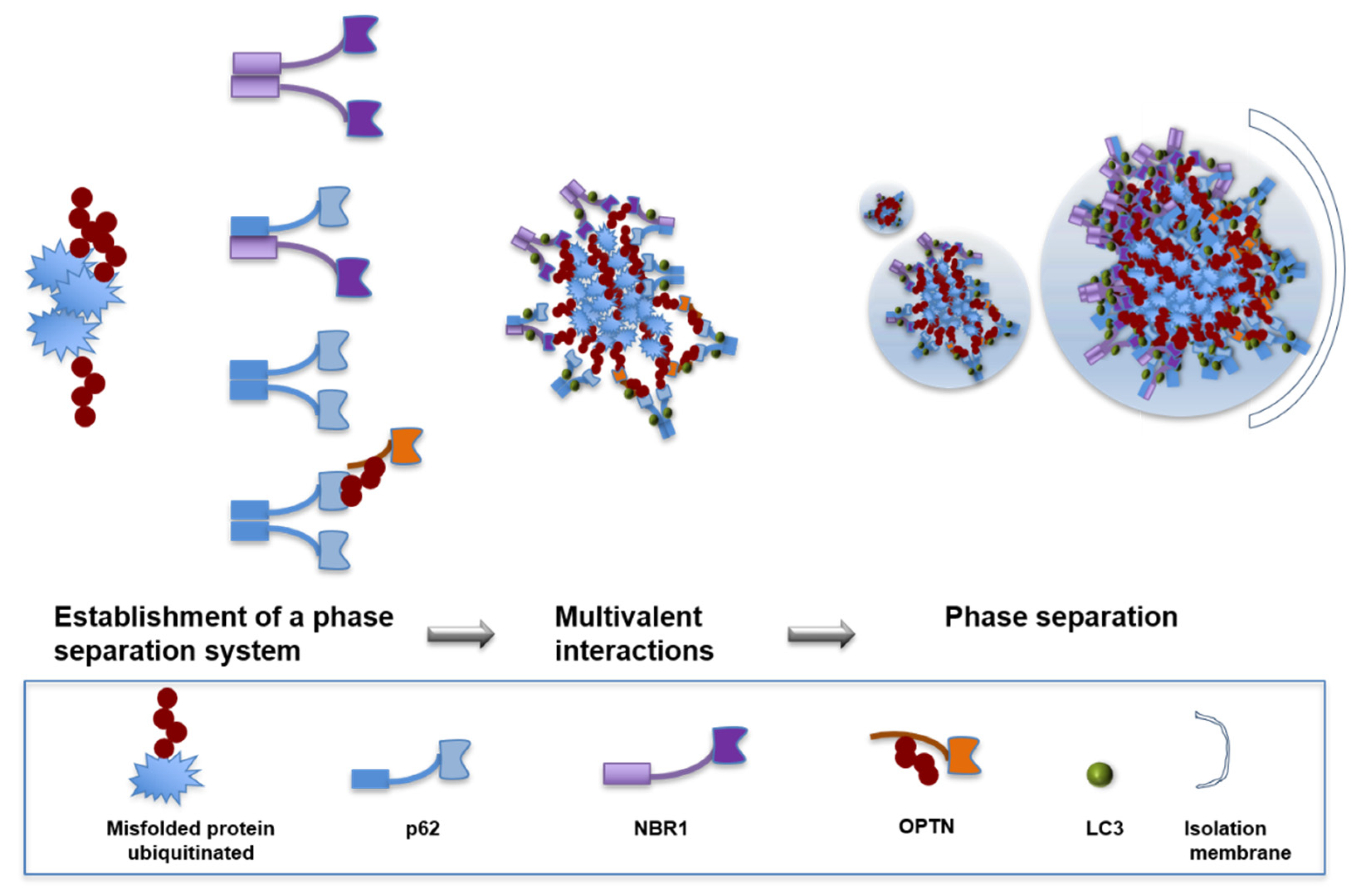
- Sun, D.; Wu, R.; Li, P.; Yu, L. Phase Separation in Regulation of Aggrephagy. J. Mol. Biol. 2020, 432, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef]
- Li, P.; Banjade, S.; Cheng, H.C.; Kim, S.; Chen, B.; Guo, L.; Llaguno, M.; Hollingsworth, J.V.; King, D.S.; Banani, S.F.; et al. Phase transitions in the assembly of multivalent signalling proteins. Nature 2012, 483, 336–340. [Google Scholar] [CrossRef]
- Wilson, M.I.; Gill, D.J.; Perisic, O.; Quinn, M.T.; Williams, R.L. PB1 domain-mediated heterodimerization in NADPH oxidase and signaling complexes of atypical protein kinase C with Par6 and p62. Mol. Cell 2003, 12, 39–50. [Google Scholar] [CrossRef]
- Sun, D.; Wu, R.; Zheng, J.; Li, P.; Yu, L. Polyubiquitin chain-induced p62 phase separation drives autophagic cargo segregation. Cell Res. 2018, 28, 405–415. [Google Scholar] [CrossRef] [Green Version]
- Altmeyer, M.; Neelsen, K.J.; Teloni, F.; Pozdnyakova, I.; Pellegrino, S.; Grøfte, M.; Rask, M.B.D.; Streicher, W.; Jungmichel, S.; Nielsen, M.L. Liquid demixing of intrinsically disordered proteins is seeded by poly(ADP-ribose). Nat. Commun. 2015, 6, 8088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agudo-Canalejo, J.; Schultz, S.W.; Chino, H.; Migliano, S.M.; Saito, C.; Koyama-Honda, I.; Stenmark, H.; Brech, A.; May, A.I.; Mizushima, N.; et al. Wetting regulates autophagy of phase-separated compartments and the cytosol. Nature 2021, 591, 142–146. [Google Scholar] [CrossRef]
- Kirkin, V.; Lamark, T.; Johansen, T.; Dikic, I. NBR1 cooperates with p62 in selective autophagy of ubiquitinated targets. Autophagy 2009, 5, 732–733. [Google Scholar] [CrossRef] [Green Version]
- Itakura, E.; Mizushima, N. p62 Targeting to the autophagosome formation site requires self-oligomerization but not LC3 binding. J. Cell Biol. 2011, 192, 17–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
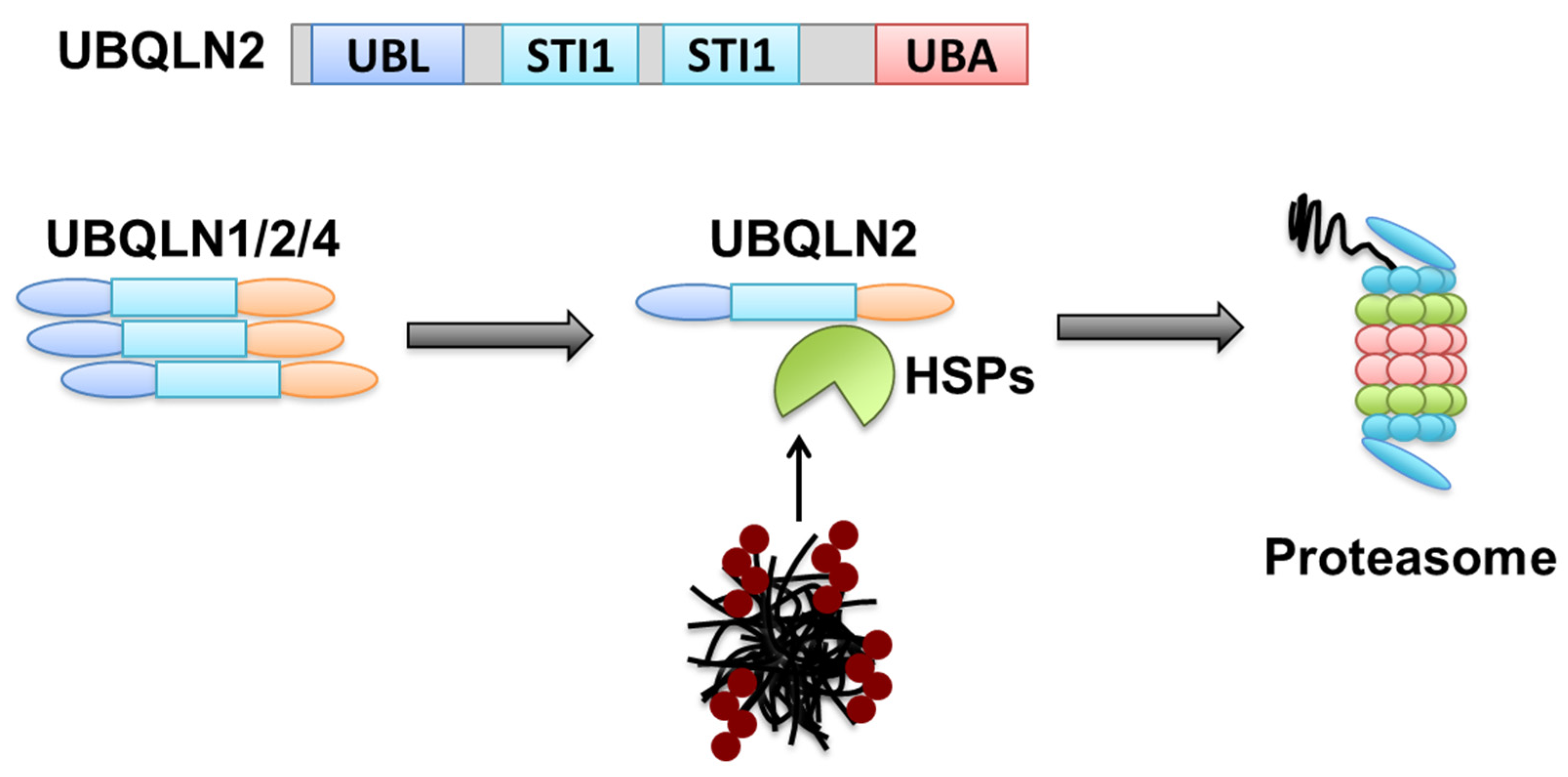
- Hjerpe, R.; Bett, J.S.; Keuss, M.J.; Solovyova, A.; McWilliams, T.G.; Johnson, C.; Sahu, I.; Varghese, J.; Wood, N.; Wightman, M.; et al. UBQLN2 Mediates Autophagy-Independent Protein Aggregate Clearance by the Proteasome. Cell 2016, 166, 935–949. [Google Scholar] [CrossRef] [Green Version]
- Deng, H.X.; Chen, W.; Hong, S.T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.Y.; Yang, S.; Warraich, S.T.; Blair, I.P. Ubiquilin 2: A component of the ubiquitin-proteasome system with an emerging role in neurodegeneration. Int. J. Biochem. Cell Biol. 2014, 50, 123–126. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, W.; Shen, T.; Wang, L.; Lu, K. Oligomerization of Selective Autophagy Receptors for the Targeting and Degradation of Protein Aggregates. Cells 2021, 10, 1989. https://doi.org/10.3390/cells10081989
Chen W, Shen T, Wang L, Lu K. Oligomerization of Selective Autophagy Receptors for the Targeting and Degradation of Protein Aggregates. Cells. 2021; 10(8):1989. https://doi.org/10.3390/cells10081989
Chicago/Turabian StyleChen, Wenjun, Tianyun Shen, Lijun Wang, and Kefeng Lu. 2021. "Oligomerization of Selective Autophagy Receptors for the Targeting and Degradation of Protein Aggregates" Cells 10, no. 8: 1989. https://doi.org/10.3390/cells10081989
APA StyleChen, W., Shen, T., Wang, L., & Lu, K. (2021). Oligomerization of Selective Autophagy Receptors for the Targeting and Degradation of Protein Aggregates. Cells, 10(8), 1989. https://doi.org/10.3390/cells10081989