
Molecular Markers and Targets in Melanoma

 ,
,  ,
, 
Abstract
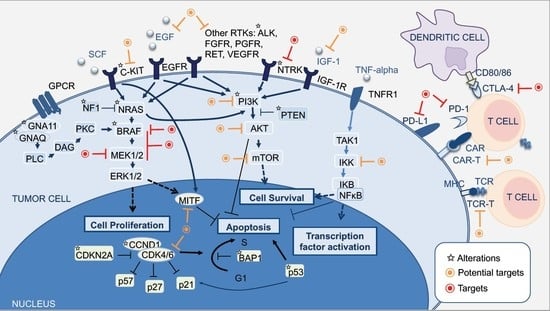
:
1. Introduction
1.1. Epidemiology
1.2. Risk Factors
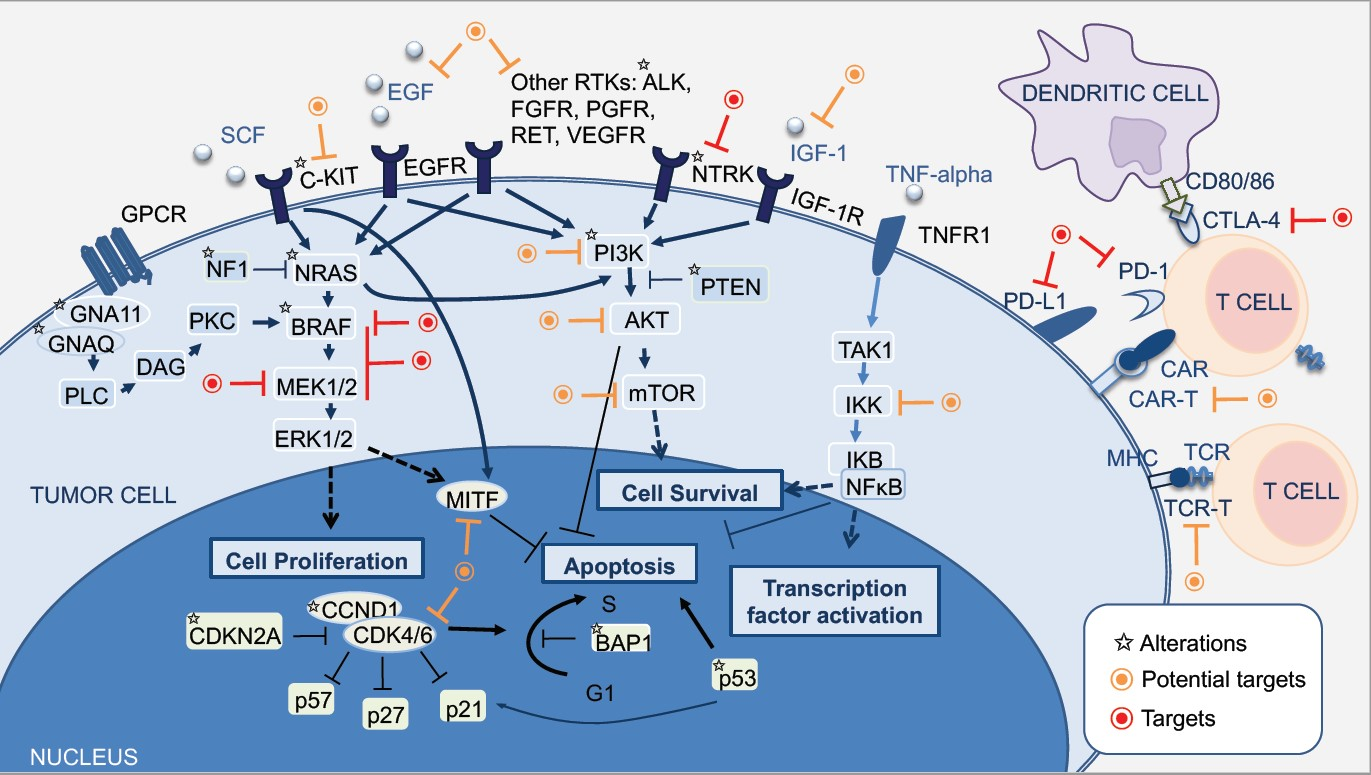
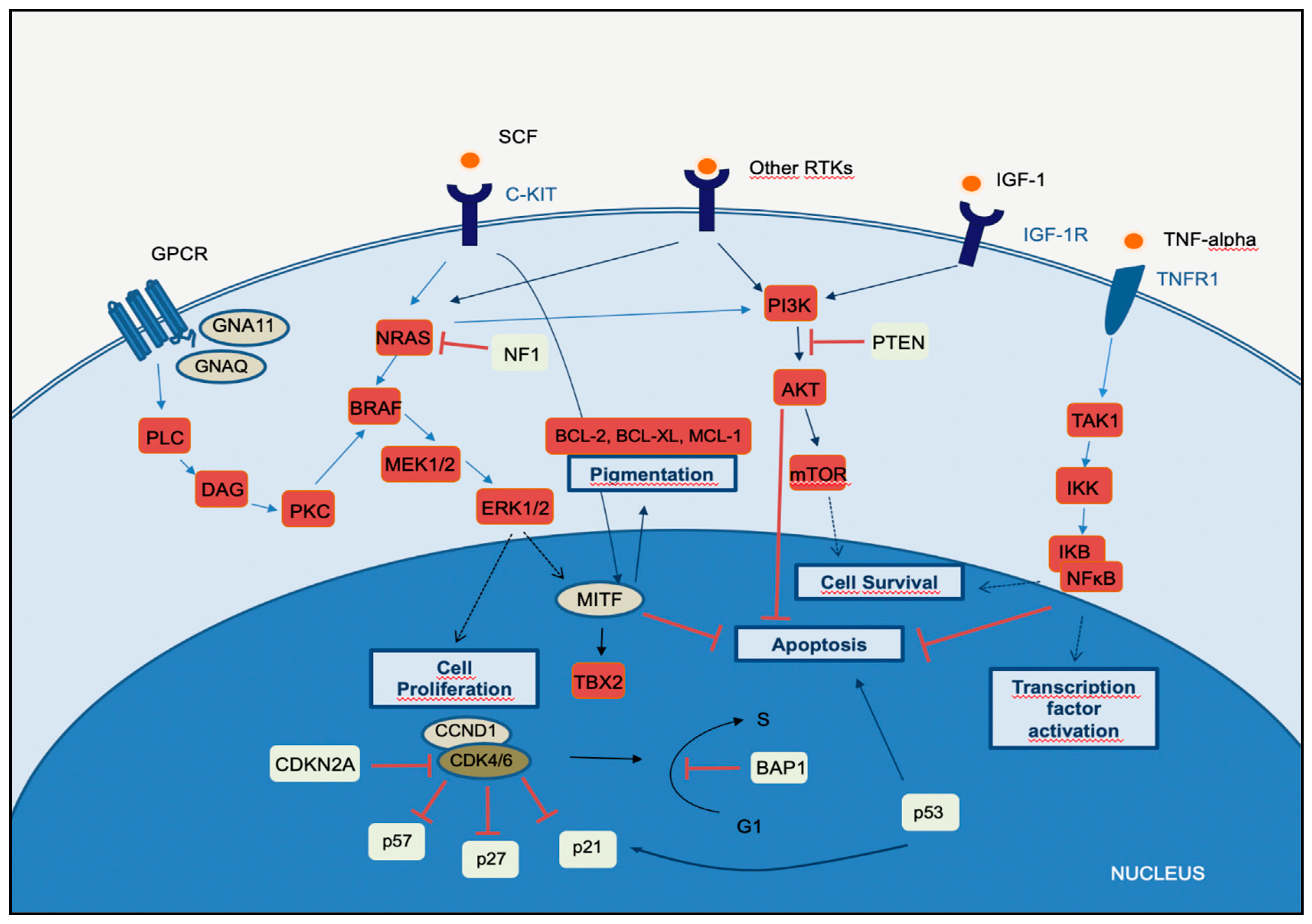
2. Molecular Pathways of Melanoma Development
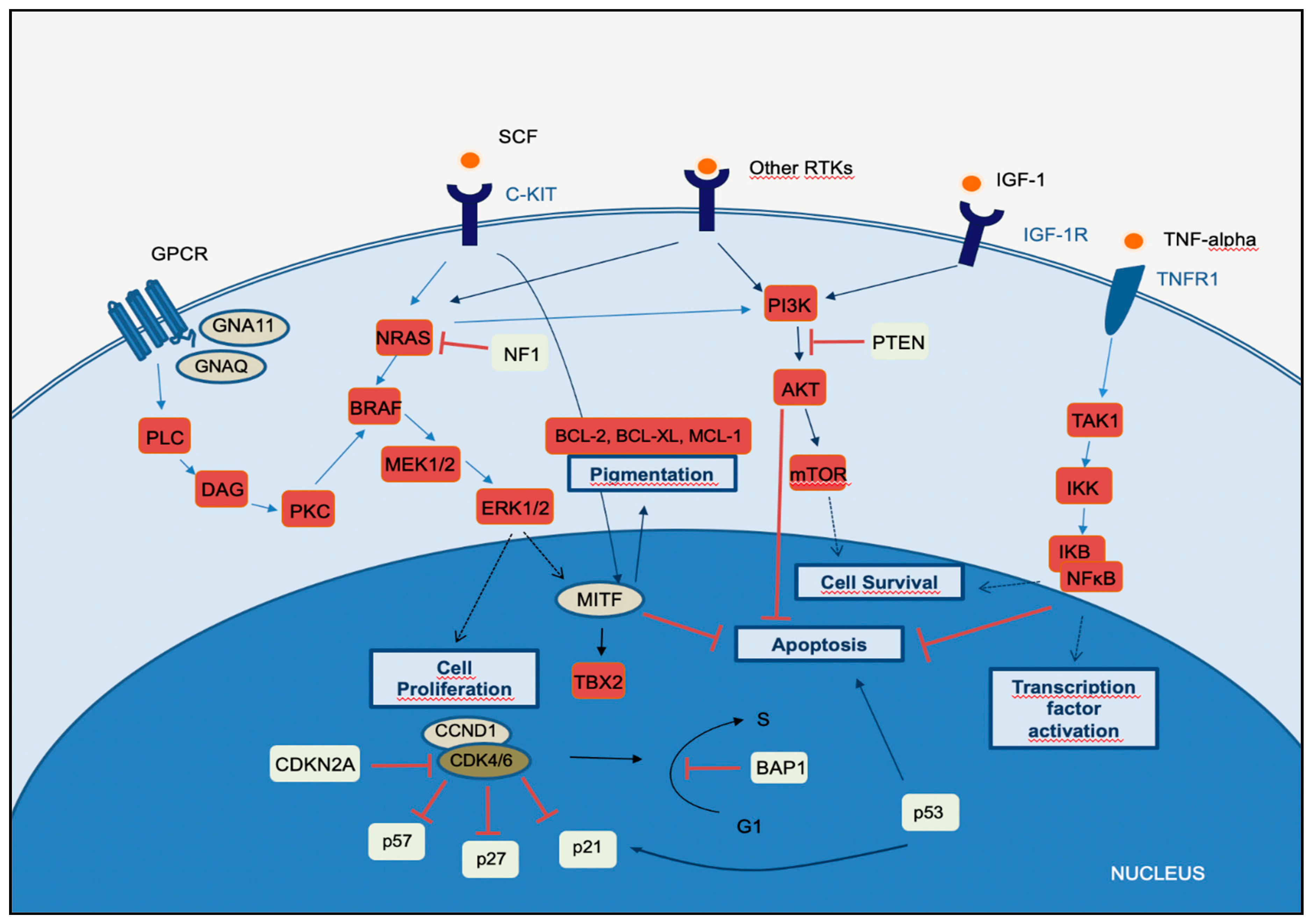
2.1. MAPK Pathway
2.2. PI3K-AKT Pathway
2.3. CDKN2A, Cell Cycle, and Apoptosis Regulation
2.4. MITF Pathway
2.5. NFκB Pathway
2.6. WNT Pathway
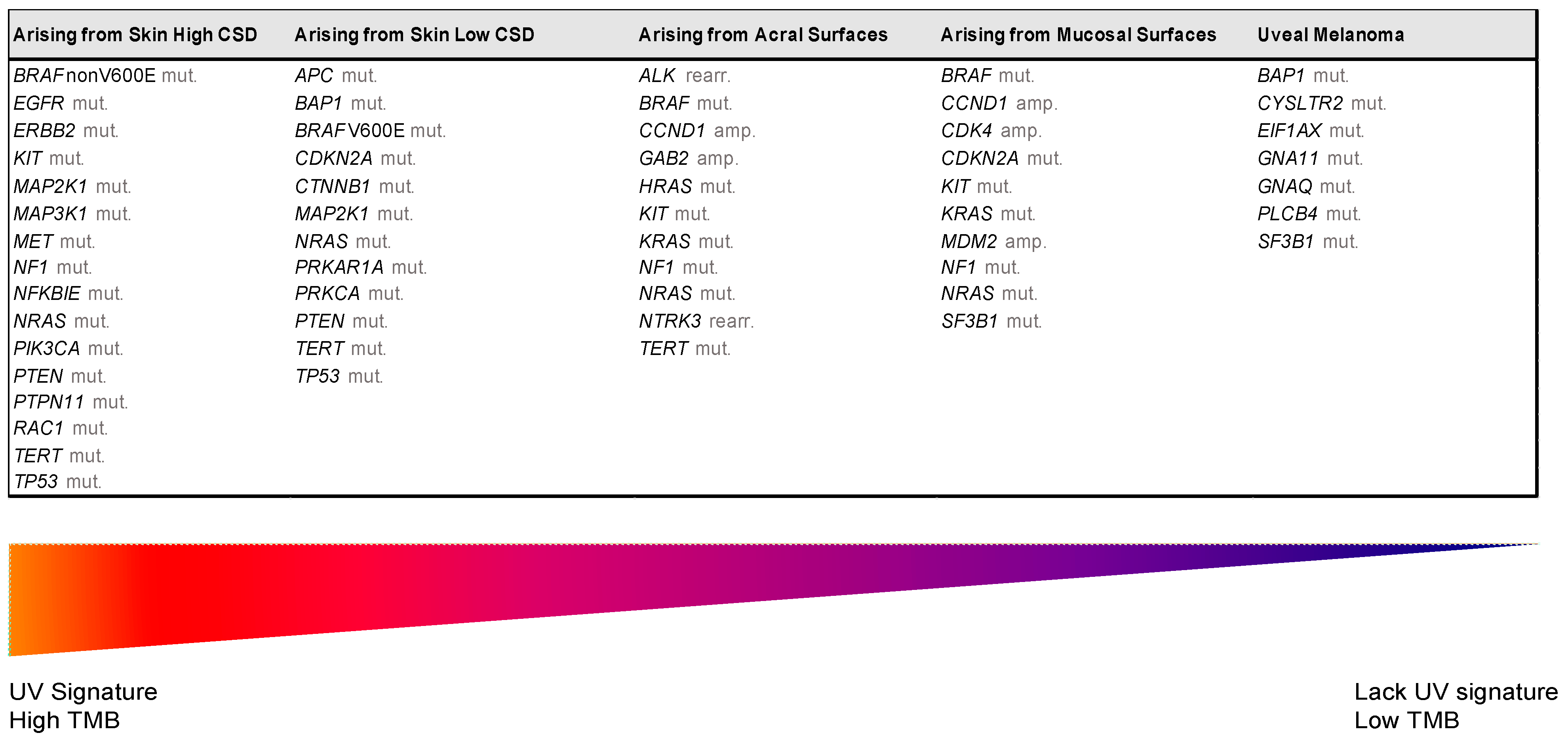
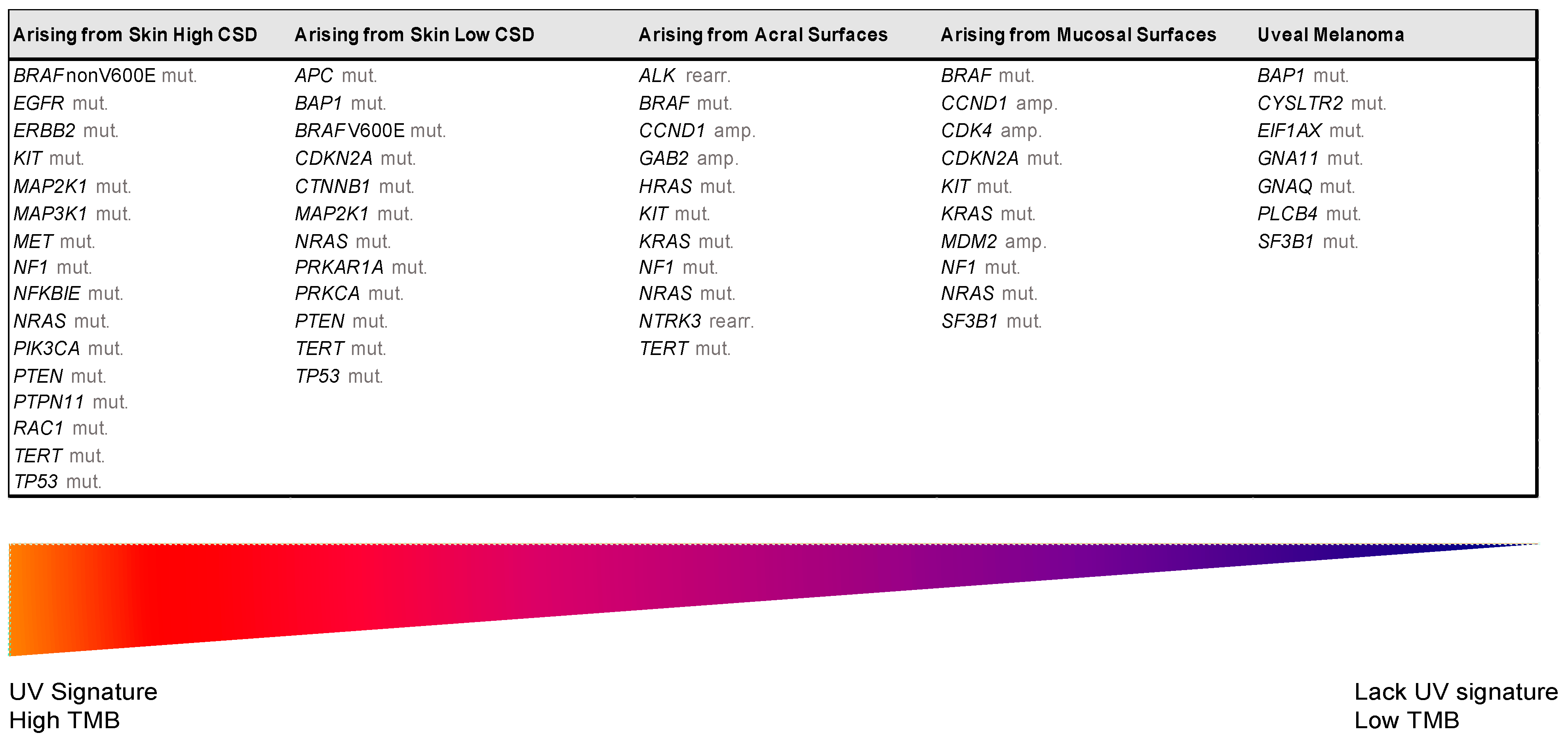
3. The Integration of Histology and Molecular Diagnostics of Melanoma
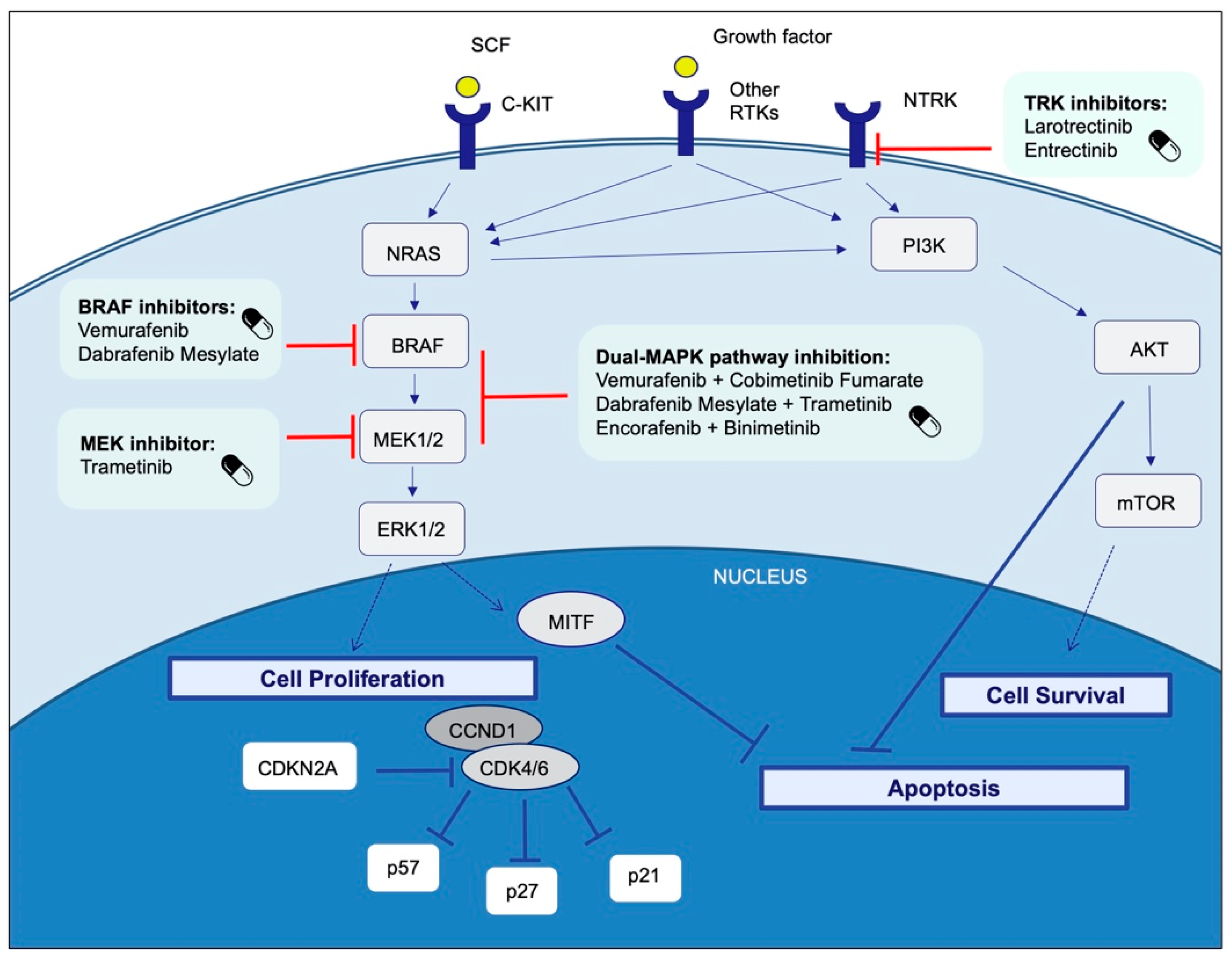
4. Therapeutic Targets and Current Treatment Strategies in Advanced Melanoma Patients
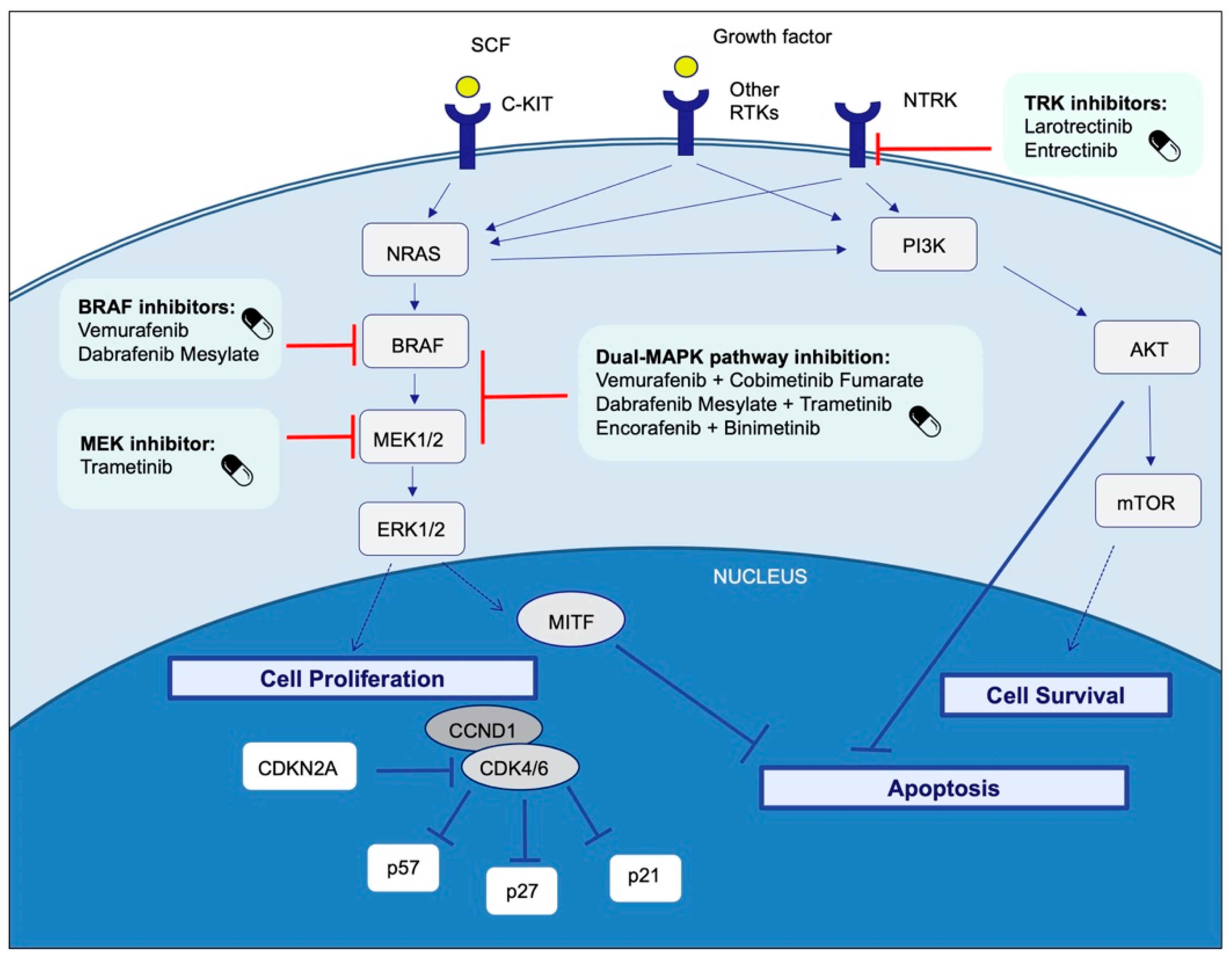
4.1. BRAF
4.1.1. BRAF and MEK Inhibitors
- Dabrafenib plus trametinib
- Cobimetinib plus vemurafenib
- Encorafenib plus binimetinib
4.1.2. Differences between BRAFV600E and BRAFV600K Mutations
4.1.3. Resistance Mechanisms to BRAF and MEK Inhibition
4.2. Immune Checkpoint Inhibitors: Anti-CTLA-4 and Anti-PD1
4.2.1. Anti-CTLA4
4.2.2. Anti-PD1-Based Therapies
- Combination of Anti-PD1 with Anti-CTLA-4 Monoclonal Antibodies
- Novel Combinations of BRAF/MEK Inhibitors and Immune Checkpoint Inhibitors
- Biomarkers of response and resistance mechanisms to immune checkpoint inhibitors
4.3. Other Immunotherapy Treatment Strategies: Adoptive Cell Therapy
4.3.1. Tumor-Infiltrating Lymphocytes
4.3.2. T Cell Receptor-Engineered T Cells
4.3.3. Chimeric Antigen Receptor T Cells
4.4. NRAS
4.5. C-KIT
4.6. GNAQ/GNA11
4.7. SF3B1
4.8. NTRK
4.9. Other Therapeutic Options
- NFκB pathway
- WNT pathway
5. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACT | adoptive cell therapy; |
| AE | adverse events; |
| ALK | Anaplastic lymphoma kinase; |
| BAD | BCL-2 antagonist of cell death |
| BRAF | v-raf murine sarcoma viral oncogene homolog B1; |
| BRAF | inhibitor (BRAFi) |
| CAR-T | chimeric antigen receptor T cells; |
| CCND1 | cyclin D1; |
| CDK4 | cyclin-dependent kinase 4; |
| CDKN2A | cyclin-dependent kinase inhibitor 2 receptor; |
| ChT | chemotherapy; |
| CSD | cumulative sun damage; |
| CTLA-4 | cytotoxic T-lymphocyte–associated antigen 4; |
| CTNNB1 | Catenin Beta 1; |
| cuSCC | cutaneous squamous cell carcinoma; |
| DVL | dishevelled; |
| ERK | extracellular signal-related kinase; |
| FAMMM | familial atypical multiple mole-melanoma; |
| FDA | US Food and Drug Administration; |
| FRZD | Frizzled receptors; |
| GEP | gene expression profiles; |
| GPCR | G-protein-coupled receptors; |
| GSK3β | glycogen synthase kinase 3β; |
| GTPase | guanosine triphosphatases; |
| HRAS | v-Ha-ras Harvey rat sarcoma viral oncogene homolog; |
| ICI | immune checkpoint inhibitor; |
| IFNG | interferon-gamma; |
| IL | interleukin; |
| iNOS | inducible nitric oxide synthase; |
| JNK | c-Jun N-terminal kinases; |
| KRAS | v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog; |
| LEF | lymphoid enhanced transcription factor; |
| LRP | lipoprotein receptor; |
| MAPK | mitogen-activated protein kinase; |
| MAP2K1 | mitogen-activated protein kinase kinase 1; |
| MAP3K1 | mitogen-activated protein kinase kinase kinase 1; |
| MC1R | melanocortin 1 receptor; |
| MDM2 | Mouse double minute 2 homolog; |
| MEK | inhibitors (MEKi) |
| MITF | microphthalmia-associated transcription factor; |
| mTOR | mammalian target of rapamycin; |
| NBD | NEMO-binding domain; |
| NF1 | neurofibromin 1; |
| NFκB | nuclear factor-kappaB; |
| NTRK | Neurotrophic Tyrosine Receptor Kinase; |
| NRAS | neuroblastoma ras viral oncogene homolog; |
| ORR | overall response rate; |
| OS | overall survival; |
| PD1 | programmed cell death-1; |
| PD-L1 | programmed cell death ligand-1; |
| PI3K | phosphatidylinositol-3-kinase; |
| PIP2 | phosphatidylinositol-4,5-diphosphate |
| PIP3 | phosphatidylinositol-3,4,5-trisphosphate |
| PTEN | phosphatase and tensin homolog; |
| RTK | receptor tyrosine kinase; |
| SF3B1 | Splicing Factor 3b Subunit 1; |
| TCR-T | T-cell receptor– engineered T cells; |
| TERT | Telomerase Reverse Transcriptase; |
| TILS | Tumor-infiltrating lymphocytes; |
| TCF | T-cell transcription factor; |
| TCGA | The Cancer Genome Atlas |
| TNF | tumor necrosis factor; |
| TRAE | treatment-related adverse event |
| TRK | tropomyosin receptor kinases; |
| UV | ultraviolet; |
| WHO | World Health Organization |
References
- Cress, R.D.; Holly, E.A. Incidence of cutaneous melanoma among non-Hispanic whites, Hispanics, Asians, and blacks: An analysis of california cancer registry data, 1988–1993. Cancer Causes Control. 1997, 8, 246–252. [Google Scholar] [CrossRef]
- Ridky, T.W. Nonmelanoma skin cancer. J. Am. Acad. Dermatol. 2007, 57, 484–501. [Google Scholar] [CrossRef]
- Jhappan, C.; Noonan, F.P.; Merlino, G. Ultraviolet radiation and cutaneous malignant melanoma. Oncogene 2003, 22, 3099–3112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA: A Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Rastrelli, M.; Tropea, S.; Rossi, C.R.; Alaibac, M. Melanoma: Epidemiology, risk factors, pathogenesis, diagnosis and classification. In Vivo (Athens, Greece) 2014, 28, 1005–1011. [Google Scholar]
- Michielin, O.; van Akkooi, A.C.J.; Ascierto, P.A.; Dummer, R.; Keilholz, U. Cutaneous melanoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2019, 30, 1884–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Fellner, C. Ipilimumab (yervoy) prolongs survival in advanced melanoma: Serious side effects and a hefty price tag may limit its use. P T 2012, 37, 503–530. [Google Scholar]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [Green Version]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N. Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef] [Green Version]
- Sample, A.; He, Y.-Y. Mechanisms and prevention of UV-induced melanoma. Photodermatol. Photoimmunol. Photomed. 2018, 34, 13–24. [Google Scholar] [CrossRef]
- D’Orazio, J.; Jarrett, S.; Amaro-Ortiz, A.; Scott, T. UV radiation and the skin. Int. J. Mol. Sci. 2013, 14, 12222–12248. [Google Scholar] [CrossRef] [Green Version]
- Bevona, C.; Goggins, W.; Quinn, T.; Fullerton, J.; Tsao, H. Cutaneous melanomas associated with nevi. Arch. Dermatol. 2003, 139, 1620–1624; discussion 1624. [Google Scholar] [CrossRef]
- Dessinioti, C.; Antoniou, C.; Katsambas, A.; Stratigos, A.J. Melanocortin 1 receptor variants: Functional role and pigmentary associations. Photochem. Photobiol. 2011, 87, 978–987. [Google Scholar] [CrossRef]
- Rossi, M.; Pellegrini, C.; Cardelli, L.; Ciciarelli, V.; Di Nardo, L.; Fargnoli, M.C. Familial Melanoma: Diagnostic and Management Implications. Dermatol. Pract. Concept 2019, 9, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, A.M.; Tucker, M.A. Genetic epidemiology of cutaneous melanoma: A global perspective. Arch. Dermatol. 2001, 137, 1493–1496. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Raman, M.; Chen, W.; Cobb, M.H. Differential regulation and properties of MAPKs. Oncogene 2007, 26, 3100–3112. [Google Scholar] [CrossRef] [Green Version]
- Eliceiri, B.P.; Klemke, R.; Strömblad, S.; Cheresh, D.A. Integrin alphavbeta3 requirement for sustained mitogen-activated protein kinase activity during angiogenesis. J. Cell Biol. 1998, 140, 1255–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Nissan, M.H.; Pratilas, C.A.; Jones, A.M.; Ramirez, R.; Won, H.; Liu, C.; Tiwari, S.; Kong, L.; Hanrahan, A.J.; Yao, Z.; et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res. 2014, 74, 2340–2350. [Google Scholar] [CrossRef] [Green Version]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [Green Version]
- Scheid, M.P.; Woodgett, J.R. PKB/AKT: Functional insights from genetic models. Nat. Rev. Mol. Cell Biol. 2001, 2, 760–768. [Google Scholar] [CrossRef]
- Stahl, J.M.; Cheung, M.; Sharma, A.; Trivedi, N.R.; Shanmugam, S.; Robertson, G.P. Loss of PTEN promotes tumor development in malignant melanoma. Cancer Res. 2003, 63, 2881–2890. [Google Scholar]
- Lito, P.; Pratilas, C.A.; Joseph, E.W.; Tadi, M.; Halilovic, E.; Zubrowski, M.; Huang, A.; Wong, W.L.; Callahan, M.K.; Merghoub, T.; et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell 2012, 22, 668–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, Y.; Si, L.; Li, Y.; Wu, X.; Xu, X.; Dai, J.; Tang, H.; Ma, M.; Chi, Z.; Sheng, X.; et al. Analysis of mTOR Gene Aberrations in Melanoma Patients and Evaluation of Their Sensitivity to PI3K-AKT-mTOR Pathway Inhibitors. Clin. Cancer Res. 2016, 22, 1018–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goel, V.K.; Lazar, A.J.F.; Warneke, C.L.; Redston, M.S.; Haluska, F.G. Examination of Mutations in BRAF, NRAS, and PTEN in Primary Cutaneous Melanoma. J. Invest. Dermatol. 2006, 126, 154–160. [Google Scholar] [CrossRef] [Green Version]
- Brown, V.L.; Harwood, C.A.; Crook, T.; Cronin, J.G.; Kelsell, D.P.; Proby, C.M. p16INK4a and p14ARF tumor suppressor genes are commonly inactivated in cutaneous squamous cell carcinoma. J. Invest. Dermatol 2004, 122, 1284–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alos, L.; Fuster, C.; Castillo, P.; Jares, P.; Garcia-Herrera, A.; Marginet, M.; Agreda, F.; Arance, A.; Gonzalvo, E.; Garcia, M.; et al. TP53 mutation and tumoral PD-L1 expression are associated with depth of invasion in desmoplastic melanomas. Ann. Transl. Med. 2020, 8, 1218. [Google Scholar] [CrossRef]
- Sini, M.C.; Manca, A.; Cossu, A.; Budroni, M.; Botti, G.; Ascierto, P.A.; Cremona, F.; Muggiano, A.; D’Atri, S.; Casula, M.; et al. Molecular alterations at chromosome 9p21 in melanocytic naevi and melanoma. Br. J. Dermatol. 2008, 158, 243–250. [Google Scholar] [CrossRef]
- Fargnoli, M.C.; Gandini, S.; Peris, K.; Maisonneuve, P.; Raimondi, S. MC1R variants increase melanoma risk in families with CDKN2A mutations: A meta-analysis. Eur. J. Cancer 2010, 46, 1413–1420. [Google Scholar] [CrossRef]
- Curtin, J.A.; Fridlyand, J.; Kageshita, T.; Patel, H.N.; Busam, K.J.; Kutzner, H.; Cho, K.-H.; Aiba, S.; Bröcker, E.-B.; LeBoit, P.E.; et al. Distinct Sets of Genetic Alterations in Melanoma. N. Engl. J. Med. 2005, 353, 2135–2147. [Google Scholar] [CrossRef] [PubMed]
- Garraway, L.A.; Widlund, H.R.; Rubin, M.A.; Getz, G.; Berger, A.J.; Ramaswamy, S.; Beroukhim, R.; Milner, D.A.; Granter, S.R.; Du, J.; et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 2005, 436, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Carreira, S.; Goodall, J.; Aksan, I.; La Rocca, S.A.; Galibert, M.D.; Denat, L.; Larue, L.; Goding, C.R. Mitf cooperates with Rb1 and activates p21Cip1 expression to regulate cell cycle progression. Nature 2005, 433, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Ben-Neriah, Y.; Karin, M. Inflammation meets cancer, with NF-κB as the matchmaker. Nature Immunol. 2011, 12, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.S. Control of oncogenesis and cancer therapy resistance by the transcription factor NF-κB. J. Clin. Invest. 2001, 107, 241–246. [Google Scholar] [CrossRef]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretić, L.; Kong, G.; Leenders, F.; Lu, X.; Fernández-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef]
- Dhawan, P.; Richmond, A. A novel NF-kappa B-inducing kinase-MAPK signaling pathway up-regulates NF-kappa B activity in melanoma cells. J. Biol. Chem. 2002, 277, 7920–7928. [Google Scholar] [CrossRef] [Green Version]
- Gajos-Michniewicz, A.; Czyz, M. WNT Signaling in Melanoma. Int. J. Mol. Sci. 2020, 21, 4852. [Google Scholar] [CrossRef]
- Semenov, M.V.; Habas, R.; MacDonald, B.T.; He, X. SnapShot: Noncanonical Wnt Signaling Pathways. Cell 2007, 131, 1378.e1371–1378.e1372. [Google Scholar] [CrossRef] [Green Version]
- Piepkorn, M.W.; Longton, G.M.; Reisch, L.M.; Elder, D.E.; Pepe, M.S.; Kerr, K.F.; Tosteson, A.N.A.; Nelson, H.D.; Knezevich, S.; Radick, A.; et al. Assessment of Second-Opinion Strategies for Diagnoses of Cutaneous Melanocytic Lesions. JAMA Netw Open 2019, 2, e1912597. [Google Scholar] [CrossRef]
- Ferrara, G.; Argenziano, G. The WHO 2018 Classification of Cutaneous Melanocytic Neoplasms: Suggestions From Routine Practice. Front. Oncol 2021, 11, 675296. [Google Scholar] [CrossRef]
- WHO. WHO Classification of Skin Tumours, 4th ed.; Elder, D.E., Massi, D., Scolyer, R.A., Willemze, R., Eds.; International Agency for Research on Cancer: Lyon, France, 2018; p. 470. [Google Scholar]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef]
- Castillo, P.; Marginet, M.; Jares, P.; García, M.; Gonzalvo, E.; Arance, A.; García, A.; Alos, L.; Teixidó, C. Implementation of an NGS panel for clinical practice in paraffin-embedded tissue samples from locally advanced and metastatic melanoma patients. Explor. Targeted Anti-tumor Ther. 2020. [Google Scholar] [CrossRef] [Green Version]
- Menzies, A.M.; Haydu, L.E.; Visintin, L.; Carlino, M.S.; Howle, J.R.; Thompson, J.F.; Kefford, R.F.; Scolyer, R.A.; Long, G.V. Distinguishing Clinicopathologic Features of Patients with V600E and V600K BRAF-Mutant Metastatic Melanoma. Clin. Cancer Res. 2012, 18, 3242–3249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollock, P.M.; Harper, U.L.; Hansen, K.S.; Yudt, L.M.; Stark, M.; Robbins, C.M.; Moses, T.Y.; Hostetter, G.; Wagner, U.; Kakareka, J.; et al. High frequency of BRAF mutations in nevi. Nature Genetics 2003, 33, 19–20. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.F.; Testori, A.; Grob, J.J.; et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [Green Version]
- Larkin, J.; Hodi, F.S.; Wolchok, J.D. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 1270–1271. [Google Scholar] [CrossRef] [Green Version]
- Lebbé, C.; Meyer, N.; Mortier, L.; Marquez-Rodas, I.; Robert, C.; Rutkowski, P.; Menzies, A.M.; Eigentler, T.; Ascierto, P.A.; Smylie, M.; et al. Evaluation of Two Dosing Regimens for Nivolumab in Combination With Ipilimumab in Patients With Advanced Melanoma: Results From the Phase IIIb/IV CheckMate 511 Trial. J. Clin. Oncol. 2019, 37, 867–875. [Google Scholar] [CrossRef]
- Schachter, J.; Ribas, A.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus ipilimumab for advanced melanoma: Final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet 2017, 390, 1853–1862. [Google Scholar] [CrossRef]
- Hauschild, A.; Grob, J.-J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. The Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, G.V.; Flaherty, K.T.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: Long-term survival and safety analysis of a phase 3 study. Ann. Oncol. 2017, 28, 1631–1639. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Ascierto, P.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF -mutant melanoma (COLUMBUS): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef] [Green Version]
- Gutzmer, R.; Stroyakovskiy, D.; Gogas, H.; Robert, C.; Lewis, K.; Protsenko, S.; Pereira, R.P.; Eigentler, T.; Rutkowski, P.; Demidov, L.; et al. Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAF. Lancet 2020, 395, 1835–1844. [Google Scholar] [CrossRef]
- Long, G.V.; Lebbe, C.; Atkinson, V.; Mandalà, M.; Nathan, P.D.; Arance, A.; Richtig, E.; Yamazaki, N.; Robert, C.; Schadendorf, D.; et al. The anti–PD-1 antibody spartalizumab in combination with dabrafenib and trametinib in advanced BRAF V600–mutant melanoma: Efficacy and safety findings from parts 1 and 2 of the Phase III COMBI-i trial. J. Clin. Oncol. 2020, 38, 10028. [Google Scholar] [CrossRef]
- Fruehauf, J.; Lutzky, J.; McDermott, D.; Brown, C.K.; Meric, J.B.; Rosbrook, B.; Shalinsky, D.R.; Liau, K.F.; Niethammer, A.G.; Kim, S.; et al. Multicenter, Phase II Study of Axitinib, a Selective Second-Generation Inhibitor of Vascular Endothelial Growth Factor Receptors 1, 2, and 3, in Patients with Metastatic Melanoma. Clin. Cancer Res. 2011, 17, 7462–7469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, X.; Yan, X.; Chi, Z.; Si, L.; Cui, C.; Tang, B.; Li, S.; Mao, L.; Lian, B.; Wang, X.; et al. Axitinib in Combination With Toripalimab, a Humanized Immunoglobulin G. J. Clin. Oncol. 2019, 37, 2987–2999. [Google Scholar] [CrossRef]
- Hong, D.S.; Kurzrock, R.; Wheler, J.J.; Naing, A.; Falchook, G.S.; Fu, S.; Kim, K.B.; Davies, M.A.; Nguyen, L.M.; George, G.C.; et al. Phase I Dose-Escalation Study of the Multikinase Inhibitor Lenvatinib in Patients with Advanced Solid Tumors and in an Expanded Cohort of Patients with Melanoma. Clin. Cancer Res. 2015, 21, 4801–4810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, M.H.; Lee, C.H.; Makker, V.; Rasco, D.; Dutcus, C.E.; Wu, J.; Stepan, D.E.; Shumaker, R.C.; Motzer, R.J. Phase IB/II Trial of Lenvatinib Plus Pembrolizumab in Patients With Advanced Renal Cell Carcinoma, Endometrial Cancer, and Other Selected Advanced Solid Tumors. J. Clin. Oncol. 2020, 38, 1154–1163. [Google Scholar] [CrossRef]
- Guo, J.; Si, L.; Kong, Y.; Flaherty, K.T.; Xu, X.; Zhu, Y.; Corless, C.L.; Li, L.; Li, H.; Sheng, X.; et al. Phase II, Open-Label, Single-Arm Trial of Imatinib Mesylate in Patients With Metastatic Melanoma Harboring c-Kit Mutation or Amplification. J. Clin. Oncol. 2011, 29, 2904–2909. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, R.D.; Antonescu, C.R.; Wolchok, J.D.; Chapman, P.B.; Roman, R.A.; Teitcher, J.; Panageas, K.S.; Busam, K.J.; Chmielowski, B.; Lutzky, J.; et al. KIT as a therapeutic target in metastatic melanoma. JAMA 2011, 305, 2327–2334. [Google Scholar] [CrossRef] [Green Version]
- Kalinsky, K.; Lee, S.; Rubin, K.M.; Lawrence, D.P.; Iafrarte, A.J.; Borger, D.R.; Margolin, K.A.; Leitao, M.M.; Tarhini, A.A.; Koon, H.B.; et al. A phase 2 trial of dasatinib in patients with locally advanced or stage IV mucosal, acral, or vulvovaginal melanoma: A trial of the ECOG-ACRIN Cancer Research Group (E2607): Dasatinib in Metastatic Melanoma. Cancer 2017, 123, 2688–2697. [Google Scholar] [CrossRef] [PubMed]
- Macaulay, V.M.; Middleton, M.R.; Eckhardt, S.G.; Rudin, C.M.; Juergens, R.A.; Gedrich, R.; Gogov, S.; McCarthy, S.; Poondru, S.; Stephens, A.W.; et al. Phase I Dose-Escalation Study of Linsitinib (OSI-906) and Erlotinib in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2016, 22, 2897–2907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leong, S.; Moss, R.A.; Bowles, D.W.; Ware, J.A.; Zhou, J.; Spoerke, J.M.; Lackner, M.R.; Shankar, G.; Schutzman, J.L.; van der Noll, R.; et al. A Phase I Dose-Escalation Study of the Safety and Pharmacokinetics of Pictilisib in Combination with Erlotinib in Patients with Advanced Solid Tumors. The Oncologist 2017, 22, 1491–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S.P.; Kim, K.B.; Papadopoulos, N.E.; Hwu, W.-J.; Hwu, P.; Prieto, V.G.; Bar-Eli, M.; Zigler, M.; Dobroff, A.; Bronstein, Y.; et al. A phase II study of gefitinib in patients with metastatic melanoma. Melanoma Res. 2011, 21, 357–363. [Google Scholar] [CrossRef] [Green Version]
- Ferrucci, P.F.; Minchella, I.; Mosconi, M.; Gandini, S.; Verrecchia, F.; Cocorocchio, E.; Passoni, C.; Pari, C.; Testori, A.; Coco, P.; et al. Dacarbazine in combination with bevacizumab for the treatment of unresectable/metastatic melanoma: A phase II study. Melanoma Res. 2015, 25, 239–245. [Google Scholar] [CrossRef]
- Piperno-Neumann, S.; Diallo, A.; Etienne-Grimaldi, M.-C.; Bidard, F.-C.; Rodrigues, M.; Plancher, C.; Mariani, P.; Cassoux, N.; Decaudin, D.; Asselain, B.; et al. Phase II Trial of Bevacizumab in Combination With Temozolomide as First-Line Treatment in Patients With Metastatic Uveal Melanoma. The Oncologist 2016, 21, 281–282. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.P.; Kim, K.B. Selumetinib (AZD6244; ARRY-142886) in the treatment of metastatic melanoma. Expert Opin. Invest. Drugs 2012, 21, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Lebbé, C.; Dutriaux, C.; Lesimple, T.; Kruit, W.; Kerger, J.; Thomas, L.; Guillot, B.; de Braud, F.; Garbe, C.; Grob, J.-J.; et al. Pimasertib Versus Dacarbazine in Patients With Unresectable NRAS-Mutated Cutaneous Melanoma: Phase II, Randomized, Controlled Trial with Crossover. Cancers 2020, 12, 1727. [Google Scholar] [CrossRef] [PubMed]
- Lebbé, C.; Italiano, A.; Houédé, N.; Awada, A.; Aftimos, P.; Lesimple, T.; Dinulescu, M.; Schellens, J.H.M.; Leijen, S.; Rottey, S.; et al. Selective Oral MEK1/2 Inhibitor Pimasertib in Metastatic Melanoma: Antitumor Activity in a Phase I, Dose-Escalation Trial. Target. Oncol. 2021, 16, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Schram, A.M.; Gandhi, L.; Mita, M.M.; Damstrup, L.; Campana, F.; Hidalgo, M.; Grande, E.; Hyman, D.M.; Heist, R.S. A phase Ib dose-escalation and expansion study of the oral MEK inhibitor pimasertib and PI3K/MTOR inhibitor voxtalisib in patients with advanced solid tumours. Br. J. Cancer 2018, 119, 1471–1476. [Google Scholar] [CrossRef]
- Sarker, D.; Ang, J.E.; Baird, R.; Kristeleit, R.; Shah, K.; Moreno, V.; Clarke, P.A.; Raynaud, F.I.; Levy, G.; Ware, J.A.; et al. First-in-Human Phase I Study of Pictilisib (GDC-0941), a Potent Pan–Class I Phosphatidylinositol-3-Kinase (PI3K) Inhibitor, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2015, 21, 77–86. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Molina, J.R.; Dy, G.K.; Croghan, G.A.; Qi, Y.; Glockner, J.; Hanson, L.J.; Roos, M.M.; Tan, A.D.; Adjei, A.A. A phase I study of the VEGFR kinase inhibitor vatalanib in combination with the mTOR inhibitor, everolimus, in patients with advanced solid tumors. Invest. N. Drugs 2020, 38, 1755–1762. [Google Scholar] [CrossRef] [PubMed]
- Margolin, K.A.; Moon, J.; Flaherty, L.E.; Lao, C.D.; Akerley, W.L.; Othus, M.; Sosman, J.A.; Kirkwood, J.M.; Sondak, V.K. Randomized phase II trial of sorafenib with temsirolimus or tipifarnib in untreated metastatic melanoma (S0438). Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 1129–1137. [Google Scholar] [CrossRef] [Green Version]
- Slingluff, C.L.; Petroni, G.R.; Molhoek, K.R.; Brautigan, D.L.; Chianese-Bullock, K.A.; Shada, A.L.; Smolkin, M.E.; Olson, W.C.; Gaucher, A.; Chase, C.M.; et al. Clinical activity and safety of combination therapy with temsirolimus and bevacizumab for advanced melanoma: A phase II trial (CTEP 7190/Mel47). Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 3611–3620. [Google Scholar] [CrossRef] [Green Version]
- Tolcher, A.W.; Kurzrock, R.; Valero, V.; Gonzalez, R.; Heist, R.S.; Tan, A.R.; Means-Powell, J.; Werner, T.L.; Becerra, C.; Wang, C.; et al. Phase I dose-escalation trial of the oral AKT inhibitor uprosertib in combination with the oral MEK1/MEK2 inhibitor trametinib in patients with solid tumors. Cancer Chemother. Pharmacol. 2020, 85, 673–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolcher, A.W.; Patnaik, A.; Papadopoulos, K.P.; Rasco, D.W.; Becerra, C.R.; Allred, A.J.; Orford, K.; Aktan, G.; Ferron-Brady, G.; Ibrahim, N.; et al. Phase I study of the MEK inhibitor trametinib in combination with the AKT inhibitor afuresertib in patients with solid tumors and multiple myeloma. Cancer Chemother. Pharmacol. 2015, 75, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Gurney, A.; Axelrod, F.; Bond, C.J.; Cain, J.; Chartier, C.; Donigan, L.; Fischer, M.; Chaudhari, A.; Ji, M.; Kapoun, A.M.; et al. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 11717–11722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J. BMS-345541 Targets Inhibitor of B Kinase and Induces Apoptosis in Melanoma: Involvement of Nuclear Factor B and Mitochondria Pathways. Clin. Cancer Res. 2006, 12, 950–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, N.; Buchbinder, E.I.; Granter, S.R.; Rodig, S.J.; Giobbie-Hurder, A.; Becerra, C.; Tsiaras, A.; Gjini, E.; Fisher, D.E.; Hodi, F.S. A phase I trial of panobinostat (LBH 589) in patients with metastatic melanoma. Cancer Med. 2016, 5, 3041–3050. [Google Scholar] [CrossRef] [PubMed]
- Louveau, B.; Resche-Rigon, M.; Lesimple, T.; Da Meda, L.; Pracht, M.; Baroudjian, B.; Delyon, J.; Amini-Adle, M.; Dutriaux, C.; Reger de Moura, C.; et al. Phase I-II Open-Label Multicenter Study of Palbociclib + Vemurafenib in. Clin. Cancer Res. 2021, 27, 3876–3883. [Google Scholar] [CrossRef]
- Yadav, V.; Burke, T.F.; Huber, L.; Van Horn, R.D.; Zhang, Y.; Buchanan, S.G.; Chan, E.M.; Starling, J.J.; Beckmann, R.P.; Peng, S.B. The CDK4/6 Inhibitor LY2835219 Overcomes Vemurafenib Resistance Resulting from MAPK Reactivation and Cyclin D1 Upregulation. Mol. Cancer Ther. 2014, 13, 2253–2263. [Google Scholar] [CrossRef] [Green Version]
- Drilon, A.; Ou, S.-H.I.; Cho, B.C.; Kim, D.-W.; Lee, J.; Lin, J.J.; Zhu, V.W.; Ahn, M.-J.; Camidge, D.R.; Nguyen, J.; et al. Repotrectinib (TPX-0005) Is a Next-Generation ROS1/TRK/ALK Inhibitor That Potently Inhibits ROS1/TRK/ALK Solvent- Front Mutations. Cancer Discovery 2018, 8, 1227–1236. [Google Scholar] [CrossRef] [Green Version]
- Drilon, A. TRK inhibitors in TRK fusion-positive cancers. Ann. Oncol. 2019, 30, viii23–viii30. [Google Scholar] [CrossRef] [Green Version]
- Couts, K.L.; Bemis, J.; Turner, J.A.; Bagby, S.M.; Murphy, D.; Christiansen, J.; Hintzsche, J.D.; Le, A.; Pitts, T.M.; Wells, K.; et al. ALK Inhibitor Response in Melanomas Expressing EML4-ALK Fusions and Alternate ALK Isoforms. Mol. Cancer Ther. 2018, 17, 222–231. [Google Scholar] [CrossRef] [Green Version]
- Janku, F.; Sakamuri, D.; Kato, S.; Huang, H.J.; Call, S.G.; Naing, A.; Holley, V.R.; Patel, S.P.; Amaria, R.N.; Falchook, G.S.; et al. Dose-escalation study of vemurafenib with sorafenib or crizotinib in patients with BRAF-mutated advanced cancers. Cancer 2021, 127, 391–402. [Google Scholar] [CrossRef]
- Hong, D.S.; Kurzrock, R.; Naing, A.; Wheler, J.J.; Falchook, G.S.; Schiffman, J.S.; Faulkner, N.; Pilat, M.J.; O’Brien, J.; LoRusso, P. A phase I, open-label, single-arm, dose-escalation study of E7107, a precursor messenger ribonucleic acid (pre-mRNA) splicesome inhibitor administered intravenously on days 1 and 8 every 21 days to patients with solid tumors. Invest. New Drugs 2014, 32, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Eskens, F.A.; Ramos, F.J.; Burger, H.; O’Brien, J.P.; Piera, A.; de Jonge, M.J.; Mizui, Y.; Wiemer, E.A.; Carreras, M.J.; Baselga, J.; et al. Phase I pharmacokinetic and pharmacodynamic study of the first-in-class spliceosome inhibitor E7107 in patients with advanced solid tumors. Clin. Cancer Res. 2013, 19, 6296–6304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef] [Green Version]
- Su, F.; Viros, A.; Milagre, C.; Trunzer, K.; Bollag, G.; Spleiss, O.; Reis-Filho, J.S.; Kong, X.; Koya, R.C.; Flaherty, K.T.; et al. RAS Mutations in Cutaneous Squamous-Cell Carcinomas in Patients Treated with BRAF Inhibitors. N. Engl. J. Med. 2012, 366, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.K.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef] [Green Version]
- Amann, V.C.; Ramelyte, E.; Thurneysen, S.; Pitocco, R.; Bentele-Jaberg, N.; Goldinger, S.M.; Dummer, R.; Mangana, J. Developments in targeted therapy in melanoma. Eur. J. Surgical Oncol. J. Eur. Soc. Surgical Oncol. Br. Asso. Surgical Oncol. 2017, 43, 581–593. [Google Scholar] [CrossRef]
- Solit, D.B.; Garraway, L.A.; Pratilas, C.A.; Sawai, A.; Getz, G.; Basso, A.; Ye, Q.; Lobo, J.M.; She, Y.; Osman, I.; et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature 2006, 439, 358–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroyakovskiy, D.; Dummer, R.; Grange, F.; Mortier, L.; Chiarion-Sileni, V.; et al. Three-year estimate of overall survival in COMBI-v, a randomized phase 3 study evaluating first-line dabrafenib (D) + trametinib (T) in patients (pts) with unresectable or metastatic BRAF V600E/K–mutant cutaneous melanoma. Ann. Oncol. 2016, 27, vi575. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: A multicentre, double-blind, phase 3 randomised controlled trial. Lancet 2015, 386, 444–451. [Google Scholar] [CrossRef]
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion Sileni, V.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef]
- Ascierto, P.A.; McArthur, G.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Di Giacomo, A.M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Cobimetinib combined with vemurafenib in advanced BRAF(V600)-mutant melanoma (coBRIM): Updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016, 17, 1248–1260. [Google Scholar] [CrossRef]
- Dreno, B.; Ascierto, P.A.; McArthur, G.A.; Atkinson, V.; Liszkay, G.; Di Giacomo, A.M.; Mandalà, M.; Demidov, L.V.; Stroyakovskiy, D.; Thomas, L.; et al. Efficacy and safety of cobimetinib (C) combined with vemurafenib (V) in patients (pts) with BRAFV600 mutation–positive metastatic melanoma: Analysis from the 4-year extended follow-up of the phase 3 coBRIM study. J. Clin. Oncol. 2018, 36, 9522. [Google Scholar] [CrossRef]
- Schadendorf, D.; Long, G.V.; Stroiakovski, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion-Sileni, V.; Schachter, J.; Garbe, C.; Dutriaux, C.; et al. Three-year pooled analysis of factors associated with clinical outcomes across dabrafenib and trametinib combination therapy phase 3 randomised trials. Eur. J. Cancer 2017, 82, 45–55. [Google Scholar] [CrossRef]
- Long, G.V.; Grob, J.J.; Nathan, P.; Ribas, A.; Robert, C.; Schadendorf, D.; Lane, S.R.; Mak, C.; Legenne, P.; Flaherty, K.T.; et al. Factors predictive of response, disease progression, and overall survival after dabrafenib and trametinib combination treatment: A pooled analysis of individual patient data from randomised trials. Lancet Oncol. 2016, 17, 1743–1754. [Google Scholar] [CrossRef]
- Nathanson, K.L.; Martin, A.M.; Wubbenhorst, B.; Greshock, J.; Letrero, R.; D’Andrea, K.; O’Day, S.; Infante, J.R.; Falchook, G.S.; Arkenau, H.T.; et al. Tumor genetic analyses of patients with metastatic melanoma treated with the BRAF inhibitor dabrafenib (GSK2118436). Clin. Cancer Res. 2013, 19, 4868–4878. [Google Scholar] [CrossRef] [Green Version]
- Atefi, M.; von Euw, E.; Attar, N.; Ng, C.; Chu, C.; Guo, D.; Nazarian, R.; Chmielowski, B.; Glaspy, J.A.; Comin-Anduix, B.; et al. Reversing melanoma cross-resistance to BRAF and MEK inhibitors by co-targeting the AKT/mTOR pathway. PLoS ONE 2011, 6, e28973. [Google Scholar] [CrossRef]
- Maertens, O.; Johnson, B.; Hollstein, P.; Frederick, D.T.; Cooper, Z.A.; Messiaen, L.; Bronson, R.T.; McMahon, M.; Granter, S.; Flaherty, K.; et al. Elucidating distinct roles for NF1 in melanomagenesis. Cancer Discov. 2013, 3, 338–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulikakos, P.I.; Persaud, Y.; Janakiraman, M.; Kong, X.; Ng, C.; Moriceau, G.; Shi, H.; Atefi, M.; Titz, B.; Gabay, M.T.; et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 2011, 480, 387–390. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, C.A.; Cullinane, C.; Kirby, L.; Abuhammad, S.; Lelliott, E.J.; Waldeck, K.; Young, R.J.; Brajanovski, N.; Cameron, D.P.; Walker, R.; et al. Palbociclib synergizes with BRAF and MEK inhibitors in treatment naïve melanoma but not after the development of BRAF inhibitor resistance: Palbociclib and MAPK inhibitor efficacy in drug-resistant melanoma. Int. J. Cancer 2018, 142, 2139–2152. [Google Scholar] [CrossRef] [Green Version]
- Misek, S.A.; Appleton, K.M.; Dexheimer, T.S.; Lisabeth, E.M.; Lo, R.S.; Larsen, S.D.; Gallo, K.A.; Neubig, R.R. Rho-mediated signaling promotes BRAF inhibitor resistance in de-differentiated melanoma cells. Oncogene 2020, 39, 1466–1483. [Google Scholar] [CrossRef]
- Teixidó, C.; González-Cao, M.; Karachaliou, N.; Rosell, R. Predictive factors for immunotherapy in melanoma. Ann. Transl. Med. 2015, 3, 208. [Google Scholar] [CrossRef]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015. [Google Scholar] [CrossRef] [PubMed]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.; Weber, J.S.; et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann. Oncol. 2019, 30, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Moser, J.C.; Wei, G.; Colonna, S.V.; Grossmann, K.F.; Patel, S.; Hyngstrom, J.R. Comparative-effectiveness of pembrolizumab vs. nivolumab for patients with metastatic melanoma. Acta Oncol. 2020, 59, 434–437. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- Gogas, H.; Dréno, B.; Larkin, J.; Demidov, L.; Stroyakovskiy, D.; Eroglu, Z.; Francesco Ferrucci, P.; Pigozzo, J.; Rutkowski, P.; Mackiewicz, J.; et al. Cobimetinib plus atezolizumab in BRAF. Ann. Oncol 2021, 32, 384–394. [Google Scholar] [CrossRef]
- Zhang, T.; Dutton-Regester, K.; Brown, K.M.; Hayward, N.K. The genomic landscape of cutaneous melanoma. Pigment. Cell Melanoma Res. 2016, 29, 266–283. [Google Scholar] [CrossRef]
- Reuben, A.; Spencer, C.N.; Prieto, P.A.; Gopalakrishnan, V.; Reddy, S.M.; Miller, J.P.; Mao, X.; De Macedo, M.P.; Chen, J.; Song, X.; et al. Genomic and immune heterogeneity are associated with differential responses to therapy in melanoma. NPJ Genom. Med. 2017, 2. [Google Scholar] [CrossRef]
- Teixido, C.; Reguart, N. Using biomarkers to determine optimal combinations with immunotherapy (biomarker discovery perspective). Future Oncol. 2020, 16, 1677–1681. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Karachaliou, N.; Gonzalez-Cao, M.; Crespo, G.; Drozdowskyj, A.; Aldeguer, E.; Gimenez-Capitan, A.; Teixido, C.; Molina-Vila, M.A.; Viteri, S.; De Los Llanos Gil, M.; et al. Interferon gamma, an important marker of response to immune checkpoint blockade in non-small cell lung cancer and melanoma patients. Ther. Adv. Med. Oncol. 2018, 10, 1758834017749748. [Google Scholar] [CrossRef] [PubMed]
- Karachaliou, N.; Pilotto, S.; Teixidó, C.; Viteri, S.; González-Cao, M.; Riso, A.; Morales-Espinosa, D.; Molina, M.A.; Chaib, I.; Santarpia, M.; et al. Melanoma: Oncogenic drivers and the immune system. Ann. Transl. Med. 2015, 3, 265. [Google Scholar] [CrossRef]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Invest. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Puzanov, I.; Dummer, R.; Schadendorf, D.; Hamid, O.; Robert, C.; Hodi, F.S.; Schachter, J.; Pavlick, A.C.; Lewis, K.D.; et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): A randomised, controlled, phase 2 trial. Lancet Oncol. 2015, 16, 908–918. [Google Scholar] [CrossRef] [Green Version]
- Weichenthal, M.; Ugurel, S.; Leiter, U.M.; Satzger, I.; Kähler, K.C.; Welzel, J.; Pföhler, C.; Feldmann-Böddeker, I.; Meier, F.E.; Terheyden, P.; et al. Salvage therapy after failure from anti-PD-1 single agent treatment: A Study by the German ADOReg melanoma registry. J. Clin. Oncol. 2019, 37, 9505. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Dafni, U.; Michielin, O.; Lluesma, S.M.; Tsourti, Z.; Polydoropoulou, V.; Karlis, D.; Besser, M.J.; Haanen, J.; Svane, I.M.; Ohashi, P.S.; et al. Efficacy of adoptive therapy with tumor-infiltrating lymphocytes and recombinant interleukin-2 in advanced cutaneous melanoma: A systematic review and meta-analysis. Ann. Oncol. 2019, 30, 1902–1913. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef]
- Sarnaik, A.A.; Hamid, O.; Khushalani, N.I.; Lewis, K.D.; Medina, T.; Kluger, H.M.; Thomas, S.S.; Domingo-Musibay, E.; Pavlick, A.C.; Whitman, E.D.; et al. Lifileucel, a Tumor-Infiltrating Lymphocyte Therapy, in Metastatic Melanoma. J. Clin. Oncol. 2021, JCO2100612. [Google Scholar] [CrossRef]
- Johnson, L.A.; Morgan, R.A.; Dudley, M.E.; Cassard, L.; Yang, J.C.; Hughes, M.S.; Kammula, U.S.; Royal, R.E.; Sherry, R.M.; Wunderlich, J.R.; et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 2009, 114, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Linette, G.P.; Stadtmauer, E.A.; Maus, M.V.; Rapoport, A.P.; Levine, B.L.; Emery, L.; Litzky, L.; Bagg, A.; Carreno, B.M.; Cimino, P.J.; et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 2013, 122, 863–871. [Google Scholar] [CrossRef]
- Morgan, R.A.; Chinnasamy, N.; Abate-Daga, D.; Gros, A.; Robbins, P.F.; Zheng, Z.; Dudley, M.E.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. 2013, 36, 133–151. [Google Scholar] [CrossRef] [Green Version]
- Robbins, P.F.; Kassim, S.H.; Tran, T.L.; Crystal, J.S.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Dudley, M.E.; Wunderlich, J.R.; Sherry, R.M.; et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: Long-term follow-up and correlates with response. Clin. Cancer Res. 2015, 21, 1019–1027. [Google Scholar] [CrossRef] [Green Version]
- Poirot, L.; Philip, B.; Schiffer-Mannioui, C.; Le Clerre, D.; Chion-Sotinel, I.; Derniame, S.; Potrel, P.; Bas, C.; Lemaire, L.; Galetto, R.; et al. Multiplex Genome-Edited T-cell Manufacturing Platform for "Off-the-Shelf" Adoptive T-cell Immunotherapies. Cancer Res. 2015, 75, 3853–3864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, B.M.; McNeel, D.G. Antigen loss and tumor-mediated immunosuppression facilitate tumor recurrence. Expert Rev. Vaccines 2012, 11, 1315–1317. [Google Scholar] [CrossRef]
- Chiappetta, C.; Proietti, I.; Soccodato, V.; Puggioni, C.; Zaralli, R.; Pacini, L.; Porta, N.; Skroza, N.; Petrozza, V.; Potenza, C.; et al. BRAF and NRAS Mutations are Heterogeneous and Not Mutually Exclusive in Nodular Melanoma. Applied Immunohistochem. Mol. Morphol. 2015, 23, 172–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, J.; Curtin, J.A.; Pinkel, D.; Bastian, B.C. Congenital melanocytic nevi frequently harbor NRAS mutations but no BRAF mutations. J.Invest. Dermatol. 2007, 127, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Network, C.G.A. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [Green Version]
- Singh, H.; Longo, D.L.; Chabner, B.A. Improving Prospects for Targeting RAS. J. Clin. Oncol. 2015, 33, 3650–3659. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Schadendorf, D.; Ascierto, P.A.; Arance, A.; Dutriaux, C.; Di Giacomo, A.M.; Rutkowski, P.; Del Vecchio, M.; Gutzmer, R.; Mandala, M.; et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 435–445. [Google Scholar] [CrossRef]
- Falchook, G.S.; Lewis, K.D.; Infante, J.R.; Gordon, M.S.; Vogelzang, N.J.; DeMarini, D.J.; Sun, P.; Moy, C.; Szabo, S.A.; Roadcap, L.T.; et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: A phase 1 dose-escalation trial. Lancet Oncol. 2012, 13, 782–789. [Google Scholar] [CrossRef] [Green Version]
- Kirkwood, J.M.; Bastholt, L.; Robert, C.; Sosman, J.; Larkin, J.; Hersey, P.; Middleton, M.; Cantarini, M.; Zazulina, V.; Kemsley, K.; et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin. Cancer Res. 2012, 18, 555–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Carvajal, R.D.; Dummer, R.; Hauschild, A.; Daud, A.; Bastian, B.C.; Markovic, S.N.; Queirolo, P.; Arance, A.; Berking, C.; et al. Efficacy and safety of nilotinib in patients with KIT-mutated metastatic or inoperable melanoma: Final results from the global, single-arm, phase II TEAM trial. Ann. Oncol. 2017, 28, 1380–1387. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, C.D.; Griewank, K.G.; Crosby, M.B.; Garrido, M.C.; Vemula, S.; Wiesner, T.; Obenauf, A.C.; Wackernagel, W.; Green, G.; Bouvier, N.; et al. Mutations in GNA11 in Uveal Melanoma. N. Engl. J. Med. 2010, 363, 2191–2199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalili, J.S.; Yu, X.; Wang, J.; Hayes, B.C.; Davies, M.A.; Lizee, G.; Esmaeli, B.; Woodman, S.E. Combination Small Molecule MEK and PI3K Inhibition Enhances Uveal Melanoma Cell Death in a Mutant GNAQ- and GNA11-Dependent Manner. Clin. Cancer Res. 2012, 18, 4345–4355. [Google Scholar] [CrossRef] [Green Version]
- Newell, F.; Kong, Y.; Wilmott, J.S.; Johansson, P.A.; Ferguson, P.M.; Cui, C.; Li, Z.; Kazakoff, S.H.; Burke, H.; Dodds, T.J.; et al. Whole-genome landscape of mucosal melanoma reveals diverse drivers and therapeutic targets. Nat. Commun. 2019, 10, 3163. [Google Scholar] [CrossRef]
- Darman, R.B.; Seiler, M.; Agrawal, A.A.; Lim, K.H.; Peng, S.; Aird, D.; Bailey, S.L.; Bhavsar, E.B.; Chan, B.; Colla, S.; et al. Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3′ Splice Site Selection through Use of a Different Branch Point. Cell Rep. 2015, 13, 1033–1045. [Google Scholar] [CrossRef] [Green Version]
- Maguire, S.L.; Leonidou, A.; Wai, P.; Marchiò, C.; Ng, C.K.; Sapino, A.; Salomon, A.V.; Reis-Filho, J.S.; Weigelt, B.; Natrajan, R.C. SF3B1 mutations constitute a novel therapeutic target in breast cancer. J. Pathol. 2015, 235, 571–580. [Google Scholar] [CrossRef] [Green Version]
- Nakagawara, A. Trk receptor tyrosine kinases: A bridge between cancer and neural development. Cancer Letters 2001, 169, 107–114. [Google Scholar] [CrossRef]
- Wiesner, T.; He, J.; Yelensky, R.; Esteve-Puig, R.; Botton, T.; Yeh, I.; Lipson, D.; Otto, G.; Brennan, K.; Murali, R.; et al. Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nature Commun. 2014, 5, 3116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, I.; Jorgenson, E.; Shen, L.; Xu, M.; North, J.P.; Shain, A.H.; Reuss, D.; Wu, H.; Robinson, W.A.; Olshen, A.; et al. Targeted Genomic Profiling of Acral Melanoma. J. National Cancer Inst. 2019, 111, 1068–1077. [Google Scholar] [CrossRef]
- Lezcano, C.; Shoushtari, A.N.; Ariyan, C.; Hollmann, T.J.; Busam, K.J. Primary and Metastatic Melanoma With NTRK Fusions. Am. J. Surgical Pathol. 2018, 42, 1052–1058. [Google Scholar] [CrossRef] [PubMed]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Hong, D.S.; DuBois, S.G.; Kummar, S.; Farago, A.F.; Albert, C.M.; Rohrberg, K.S.; van Tilburg, C.M.; Nagasubramanian, R.; Berlin, J.D.; Federman, N.; et al. Larotrectinib in patients with TRK fusion-positive solid tumours: A pooled analysis of three phase 1/2 clinical trials. Lancet Oncol. 2020, 21, 531–540. [Google Scholar] [CrossRef]
- Dey, A.; Wong, E.; Kua, N.; Ling Teo, H.; Tergaonkar, V.; Lane, D. Hexamethylene Bisacetamide (HMBA) simultaneously targets AKT and MAPK pathway and represses NF-κB activity: Implications for cancer therapy. Cell Cycle 2008, 7, 3759–3767. [Google Scholar] [CrossRef]
- Amiri, K.I.; Horton, L.W.; LaFleur, B.J.; Sosman, J.A.; Richmond, A. Augmenting Chemosensitivity of Malignant Melanoma Tumors via Proteasome Inhibition: Implication for Bortezomib (VELCADE, PS-341) as a Therapeutic Agent for Malignant Melanoma. Cancer Res. 2004, 64, 4912–4918. [Google Scholar] [CrossRef] [Green Version]
- Takács, A.; Lajkó, E.; Láng, O.; Istenes, I.; Kőhidai, L. Alpha-lipoic acid alters the antitumor effect of bortezomib in melanoma cells in vitro. Sci Rep. 2020, 10, 14287. [Google Scholar] [CrossRef]
- Croghan, G.A.; Suman, V.J.; Maples, W.J.; Albertini, M.; Linette, G.; Flaherty, L.; Eckardt, J.; Ma, C.; Markovic, S.N.; Erlichman, C. A study of paclitaxel, carboplatin, and bortezomib in the treatment of metastatic malignant melanoma: A phase 2 Consortium study. Cancer 2010, 116, 3463–3468. [Google Scholar] [CrossRef] [Green Version]
- Ianaro, A.; Tersigni, M.; Belardo, G.; Martino, S.D.; Napolitano, M.; Palmieri, G.; Sini, M.; Maio, A.D.; Ombra, M.; Gentilcore, G.; et al. NEMO-binding domain peptide inhibits proliferation of human melanoma cells. Cancer Letters 2009, 274, 331–336. [Google Scholar] [CrossRef]
- Liu, J.; Pan, S.; Hsieh, M.H.; Ng, N.; Sun, F.; Wang, T.; Kasibhatla, S.; Schuller, A.G.; Li, A.G.; Cheng, D.; et al. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc. Natl. Acad. Sci. USA 2013, 110, 20224–20229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laeremans, H.; Hackeng, T.M.; van Zandvoort, M.A.M.J.; Thijssen, V.L.J.L.; Janssen, B.J.A.; Ottenheijm, H.C.J.; Smits, J.F.M.; Blankesteijn, W.M. Blocking of Frizzled Signaling With a Homologous Peptide Fragment of Wnt3a/Wnt5a Reduces Infarct Expansion and Prevents the Development of Heart Failure After Myocardial Infarction. Circulation 2011, 124, 1626–1635. [Google Scholar] [CrossRef] [Green Version]
- Chartier, C.; Raval, J.; Axelrod, F.; Bond, C.; Cain, J.; Dee-Hoskins, C.; Ma, S.; Fischer, M.M.; Shah, J.; Wei, J.; et al. Therapeutic Targeting of Tumor-Derived R-Spondin Attenuates -Catenin Signaling and Tumorigenesis in Multiple Cancer Types. Cancer Res. 2016, 76, 713–723. [Google Scholar] [CrossRef] [Green Version]
- Widlund, H.R.; Horstmann, M.A.; Price, E.R.; Cui, J.; Lessnick, S.L.; Wu, M.; He, X.; Fisher, D.E. β-Catenin–induced melanoma growth requires the downstream target Microphthalmia-associated transcription factor. J. Cell Biol. 2002, 158, 1079–1087. [Google Scholar] [CrossRef] [Green Version]
- Christodoulou, E.; Rashid, M.; Pacini, C.; Droop, A.; Robertson, H.; Groningen, T.V.; Teunisse, A.F.A.S.; Iorio, F.; Jochemsen, A.G.; Adams, D.J.; et al. Analysis of CRISPR-Cas9 screens identifies genetic dependencies in melanoma. Pigment. Cell Melanoma Res. 2021, 34, 122–131. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UV Exposure | Categories | Melanoma Subtype | Key Molecular Genes | |
|---|---|---|---|---|
| Low UV/CSD | I | Superficial spreading melanoma | BRAFV600 mut CDKN2A mut NRAS mut | TERT mut PTEN mut TP53 mut |
| High UV/CSD | II | Lentigo maligna melanoma | NRAS mut BRAFnon-V600E mut KIT mut TERT mut | CDKN2A mut PTEN mut TP53 mut |
| III | Desmoplastic melanoma | NF1 mut NFKBIE mut | NRAS mut PIK3CA mut | |
| Low or no UV/CSD | IV | Spitz melanoma | ALK rearr NTRK1 rearr NRTK3 rearr | CDKN2A mut HRAS mut |
| V | Acral melanoma | KIT mut NRAS or BRAF mut ALK rearr NRTK3 rearr | CDKN2A mut CCND1 amp TERT mut | |
| VI | Mucosal melanoma | KIT mut NRAS or BRAF mut CDKN2A mut SF3B1 mut | CCND1 amp CDK4 mut MDM2 amp | |
| VII | Melanoma in congenital nevus | NRAS mut | BRAFV600E mut | |
| VIII | Melanoma in blue nevus | GNA11 mut GNAQ mut CYSLTR2 mut | BAP1 mut EIFAX mut SF3B1 mut | |
| IX | Uveal melanoma | GNA11 mut GNAQ mut CYSLTR2 mut PLCB4 mut | BAP1 mut EIFAX mut SF3B1 mut | |
| Trial Name | Phase | Patients | Treatment Groups | Primary Endpoint | n | ORR | PFS | OS | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Anti-CTLA-4 | |||||||||
| MDX010-020 (NCT00094653) | III | Untreaded MM | Ipi 3 + gp100 vs. Ipi 3 vs. gp100 (3:1:1) | OS | 676 | 6 vs. 11 vs. 2 | 2.8 vs. 2.9 vs. 2.8 | 10.0 vs. 10.1 vs. 6.4 | [48] |
| Anti-PD1 +/− anti-CTLA-4 | |||||||||
| CM-066 (NCT01721772) | III | Untreated BRAF- wild type MM | Niv 3 q2w + Placebo vs. Placebo + Dacarbazine 1000 q3w (1:1) | OS | 418 | 40 vs. 14 | 5.1 vs. 2.2 | 37.5 vs. 11.2 | [49] |
| CM-067 (NCT01844505) | III | Untreated MM | Niv 1 + Ipi 3 (q3w) x4 − Niv 3; Niv 3 alone q2w, vs. Ipi 3 q3w x4 (1:1:1) | PFS and OS co-primary | 945 | 58 vs. 44 vs. 19 | 11.5 vs. 6.9 vs. 2.9 | [50] | |
| CM-511 (NCT02714218) | III | Untreated MM | Niv 1 + Ipi 3 (q3w) x4 − Niv 3 vs. Niv 3 +Ipi 1 (q3w) x4 − Niv 3 (1:1) | TRAE rate (grade 3–5) | 360 | 48 vs. 34 | 8.9 vs. 9.9 | NR vs. NR | [51] |
| KN-006 (NCT1866319) | III | MM ≤ 1 line (anti-PD1/PD-L1+/− anti-CTLA-4 included) | Pem 10 q2w vs. Pem 10 q3w vs. Ipi 3 q3w (1:1:1) | PFS and OS (co-primary) | 834 | 34 vs. 33 vs. 12 | 8.4 vs. 3.4 | 32.7 vs. 15.9 | [52] |
| BRAFi monotherapy | |||||||||
| BRIM-3 (NCT01006980) | III | Untreated MM | Vem 960 mg bd vs. DTIC (1:1) | PFS and OS (co-primary) | 675 | 48 vs. 5 | 5.3 vs. 1.6 | 13.6 vs. 9.7 | [9] |
| BREAK3 (NCT01227889) | III | Untreated BRAFV600E MM | Dab 150bd vs. DTIC (3:1) | ORR | 250 | 50 vs. 6 | 6.9 vs. 2.7 | 20 vs. 15.6 | [53] |
| Combined BRAFi + MEKi | |||||||||
| COMBI-v (NCT01597908) | III | Untreated BRAF V600E/K MM | Dab 150 bd + Tra 2 od vs. Vem 960 bd (1:1) | OS | 704 | 64 vs. 51 | 11.4 vs. 7.3 | NR vs. 17.2 | [54] |
| COMBI-d (NCT01584648) | III | Untreated BRAF V600E/K MM | Dab 150 bd + Tra 2 od vs. Dab 150 bd (1:1) | PFS | 423 | 69 vs. 53 | 11.0 vs. 8.8 | 25.1 vs. 18.7 | [55] |
| CoBRIM (NCT01689519) | III | Untreated BRAFV600 MM | Cob 60 od d1-21 + Vem 960 bd vs. Vem 960 bd + Placebo (1:1) | PFS | 495 | 68 vs. 45 | 12.3 vs.7.2 | 22.3 vs. 17.4 | [56] |
| COLUMBUS (NCT01909453) | III | Untreated BRAF V600E/K MM | Enc 450 od + Bin 45 mg bd vs. Enc 300 mg od vs. Vem 960 mg bd (1:1:1) | PFS | 577 | 64 vs.52 vs. 41 | 14.9 vs. 9.6 vs. 6.3 | 33.6 vs. 23.5 vs. 16.9 | [57] |
| Triplet therapy (ICI + BRAFi + MEKi) | |||||||||
| IMSpire150 (NCT02908672) | III | Untreated BRAFV600 MM | Ate 840 d1,15 + Vem 720 bd + Cob 60 od d1-21 vs. Placebo + Vem 960 bd + Cob 60 od d1-21 (all: q4w) | PFS | 514 | 66 vs. 65 | 15.1 vs. 10 | Not yet reported | [58] |
| COMBI-I (NCT02967692) | III | Untreated BRAFV600 MM | Spa 400 mg + Dab 150 bd + Tra 2 od vs. Placebo + Dab 150 + Tra 2 (q4w) | PFS | 532 | 69 vs. 64 | 16.2 vs. 12.0 | NR vs. NR | [59] |
| The Key Type of Target Mechanism | Potential Agen/Drugs | Phase | Potential Clinical Indication | ORR (%) | References |
|---|---|---|---|---|---|
| VEGFR1–VEGFR3, C-KIT, PDGFR | Axitinib | II Ib | Monotherapy in advanced melanoma In mucosal melanoma in combination with toripalimab (anti-PD1) | 18.8 48.3 | [60] [61] |
| VEGFR1–VEGFR3; FGFR1-FGFR3; PDGFR; C-KIT; and RET | Lenvatinib | I Ib/II | Monotherapy in advanced melanoma In advance melanoma in combination with pembrolizumab | 17.2 48 | [62] [63] |
| C-KIT inhibitor | Imatinib, Nilotinib, Dasatinib | II | Studied in mucosal, acral, and chronically sun-damaged melanomas | 23.3–26.2 | [64,65,66] |
| IGF-1 inhibitor | Linsitinib | I | In combination with erlotinib in solid tumors | 1/1 | [67] |
| EGF inhibitor | Gefitinib, Erlotinib | II, I | Minimal clinical efficacy as a single-agent therapy for unselected patients with metastatic melanoma. In combination with pictilisib (PI3K inhibitor) in solid tumors | 3.5–4 | [68,69] |
| VEGF inhibitor | Bevacizumab | II | In combination with dacarbazine for the treatment of unresectable/metastatic melanoma In combination with temozolomide as the first line of treatment metastatic uveal melanoma | 18.9 0 | [70] [71] |
| MEK inhibitor | Pimasertib Selumetinib | II I | Monotherapy in NRAS-mutated melanoma Monotherapy in comparison to temozolamide in chemo-naive stage unresectable III/Vmelanoma | 23 5.8 | [72,73,74] |
| PI3K/mTOR dual inhibitor | Voxtalisib | Ib | Tested in combination with pimasertib in melanoma patients with genetic alteration in PTEN, BRAF, NRAS, KRAS, PI3KCA, ERBB1/2, RET, MET, KIT, GNAQ, GNA11, but with limited antitumor activity and tolerance | 6 | [75] |
| PI3K inhibitor | Pictilisib | I | In combination with erlotinib in solid tumors or alone | 3.5–22 | [68,76] |
| mTOR inhibitor | Everolimus | I | In combination with VEGFR kinase inhibitor (vatalanib) for patients with advanced solid tumors | 12.9 | [77] |
| Temsirolimus | II | Tested in combination with sorafenib Clinical activity of combination therapy with temsirolimus plus bevacizumab, which may be greater in patients with BRAF wild-type melanoma | 5 17.7 | [78,79] | |
| AKT inhibitor | Uprosertib (GSK2141795) | I | In combination with trametinib in patients with advanced BRAF wild-type melanoma and triple-negative breast cancer | <5 | [80] |
| AKT inhibitor | Afuresertib | I | In combination with the MEK inhibitor trametinib in patients with solid tumors and multiple myeloma | 5 | [81] |
| Wnt inhibitor | Vantictumab (OMP-18R5) | NA | Have shown antitumor growth in xenograft models, particularly in combination with standard chemotherapeutic agents, have not reached clinical trial | NA | [82] |
| LGK974 | I | Monotherapy or in combination with PDR001 in patients with solid tumors (recruiting) | NA | NA | |
| IKK inhibitor | BMS-345541 | NA | A proposed target drug but have not reached clinical trial | NA | [83] |
| MITF promoter: HDAC inhibitors | Panobinostat | I | Tested in patients with metastatic melanoma | 0 | [84] |
| CDK4/6 inhibitor | Palbociclib | II I/II | Monotherapy in patients with advanced acral lentiginous melanoma with CDK pathway gene aberrations (CDK4 or/and CCND1 amplification or/and CDKN2A loss) In combination with vemurafenib in BRAFV600-mutated advanced melanoma patients harboring CDKN2A loss and RB1 expression | 20 27.8 | [85] |
| CDK4/6 inhibitor | Abemaciclib | NA | Effective in BRAF-resistant melanoma cells, preclinical data | NA | [86] |
| NTRK inhibitors | Selitrectinib (BAY 2731954, LOXO-195); Repotrectinib | NA | The second generation of NTRK designed to address on-target resistance, preclinical data on ROS1-, NTRK1-3-, or ALK-rearranged malignancies | NA | [87,88] |
| ALK inhibitors | Ceritinib Crizotinib | NA I | In vivo and in vitro studies showed that mucosal melanomas expressing EML4-ALK fusions are sensitive to ALK inhibitors Crizotinib in combination with vemurafenib in advanced BRAF-mutated tumors, mostly melanoma | 11 29 | [89] [90] |
| SF3B1 inhibitors | E7107 | I | Monotherapy in solid tumors | 0 | [91,92] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teixido, C.; Castillo, P.; Martinez-Vila, C.; Arance, A.; Alos, L. Molecular Markers and Targets in Melanoma. Cells 2021, 10, 2320. https://doi.org/10.3390/cells10092320
Teixido C, Castillo P, Martinez-Vila C, Arance A, Alos L. Molecular Markers and Targets in Melanoma. Cells. 2021; 10(9):2320. https://doi.org/10.3390/cells10092320
Chicago/Turabian StyleTeixido, Cristina, Paola Castillo, Clara Martinez-Vila, Ana Arance, and Llucia Alos. 2021. "Molecular Markers and Targets in Melanoma" Cells 10, no. 9: 2320. https://doi.org/10.3390/cells10092320
APA StyleTeixido, C., Castillo, P., Martinez-Vila, C., Arance, A., & Alos, L. (2021). Molecular Markers and Targets in Melanoma. Cells, 10(9), 2320. https://doi.org/10.3390/cells10092320