
In Vitro Model Systems of Coxsackievirus B3-Induced Myocarditis: Comparison of Commonly Used Cell Lines and Characterization of CVB3-Infected iCell® Cardiomyocytes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus
2.2. Cell Culture
2.3. RNA Isolation and Quantitative RT-PCR (qRT-PCR)
2.4. RNA/RNA In Situ Hybridization (ISH)
2.5. Western Blot
2.6. Immunofluorescence Stainings
2.7. Electron Microscopy
2.8. LIVE/DEAD Staining
2.9. Transfection with pIRES-EGFP-2A and pIRES-EGFP-3C
2.10. Statistical Analyses
3. Results
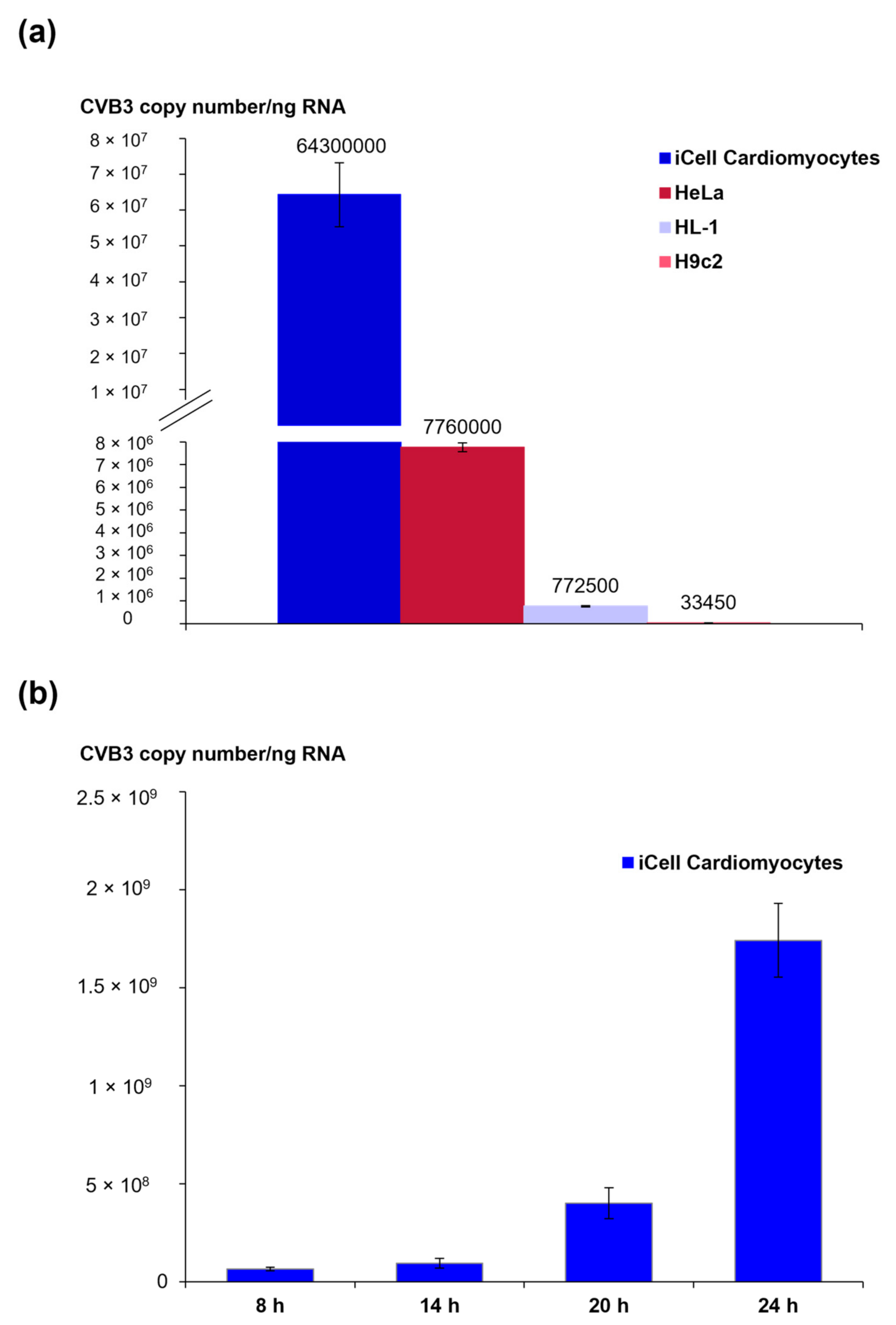
3.1. Viral Load and CVB3 Genome Localization in Different CVB3-Infected Cell Lines
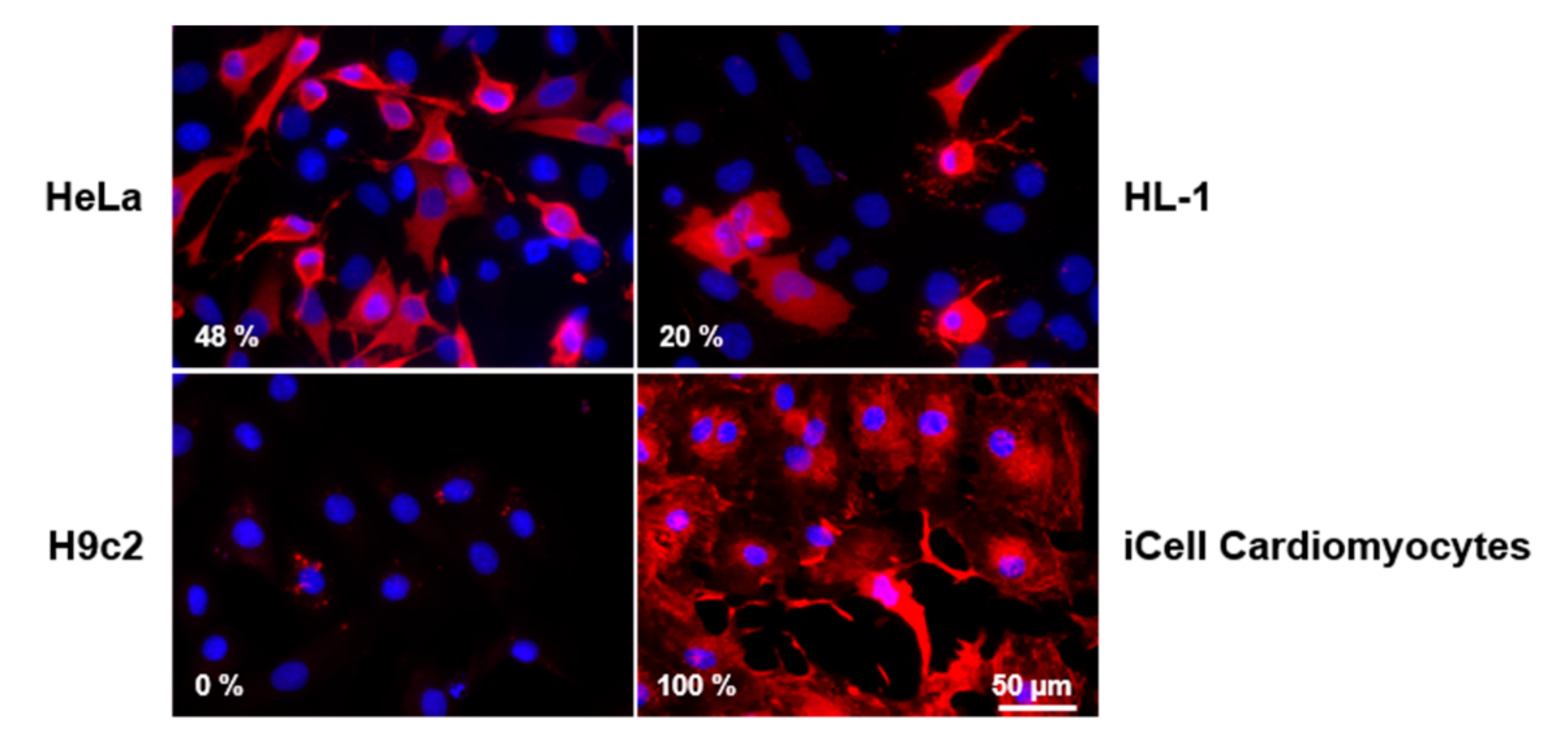
3.2. CVB3 Capsid Protein VP1 Expression and Localization in Different CVB3-Infected Cell Lines
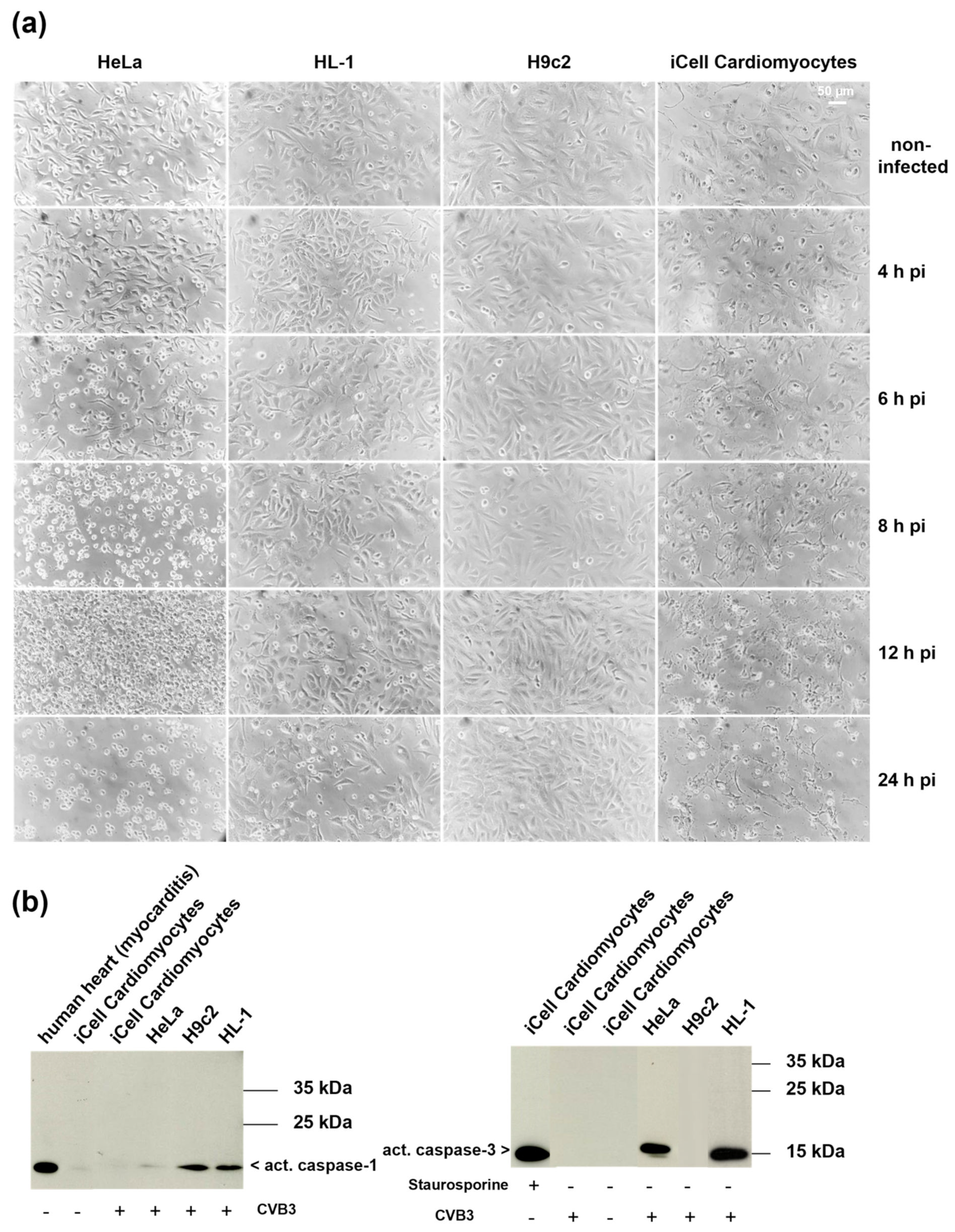
3.3. Morphological Changes in the Course of CVB3 Infection in Different Cell Lines
3.4. Analysis of Cell Death Markers in Different CVB3-Infected Cell Lines
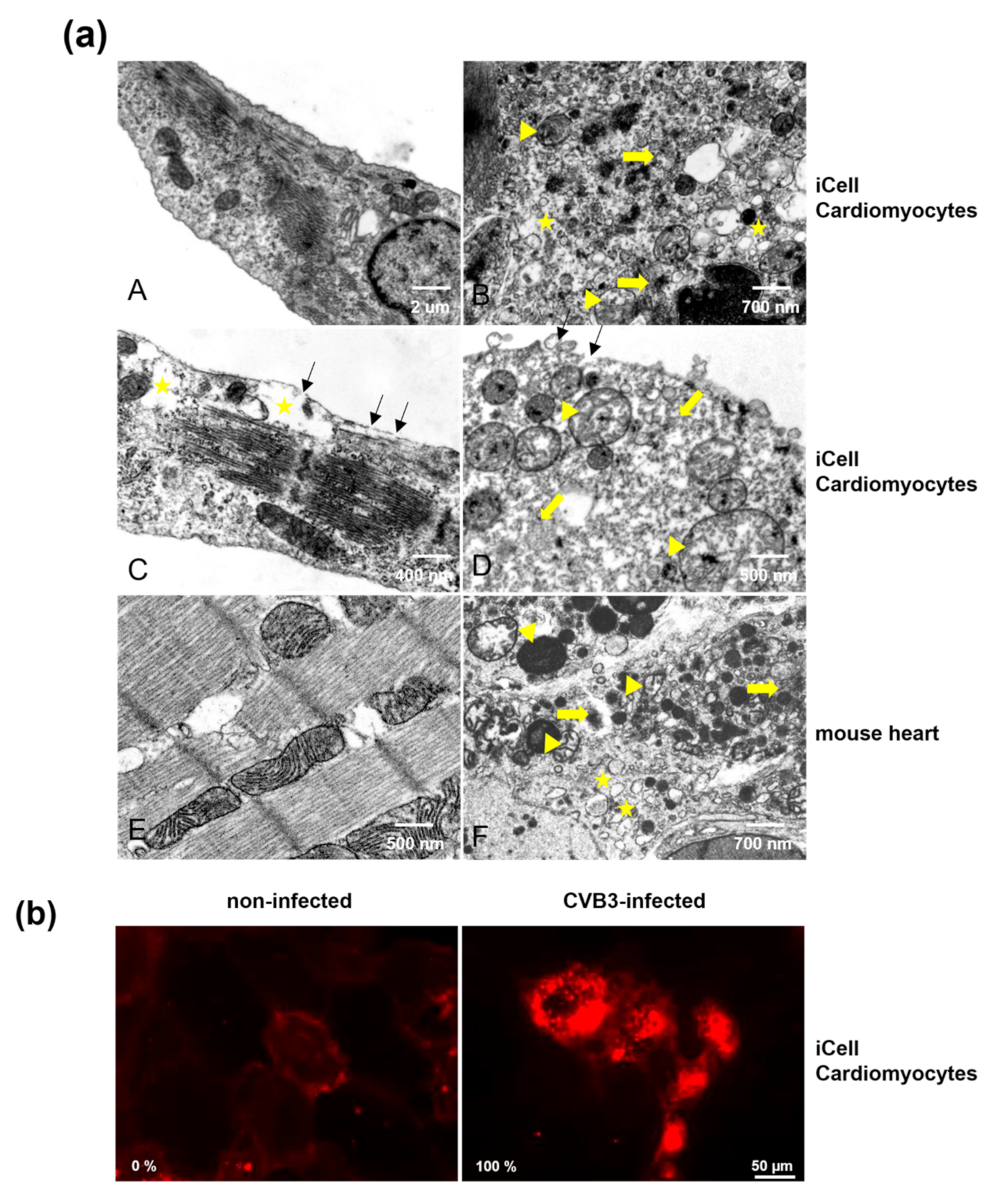
3.5. Comparison of CVB3-Infected iCell® Cardiomyocytes and Murine Heart Tissue of CVB3-Infected Mice by Electron Microscopy
3.6. Analysis of Cell Membrane Integrity of CVB3-Infected iCell® Cardiomyocytes
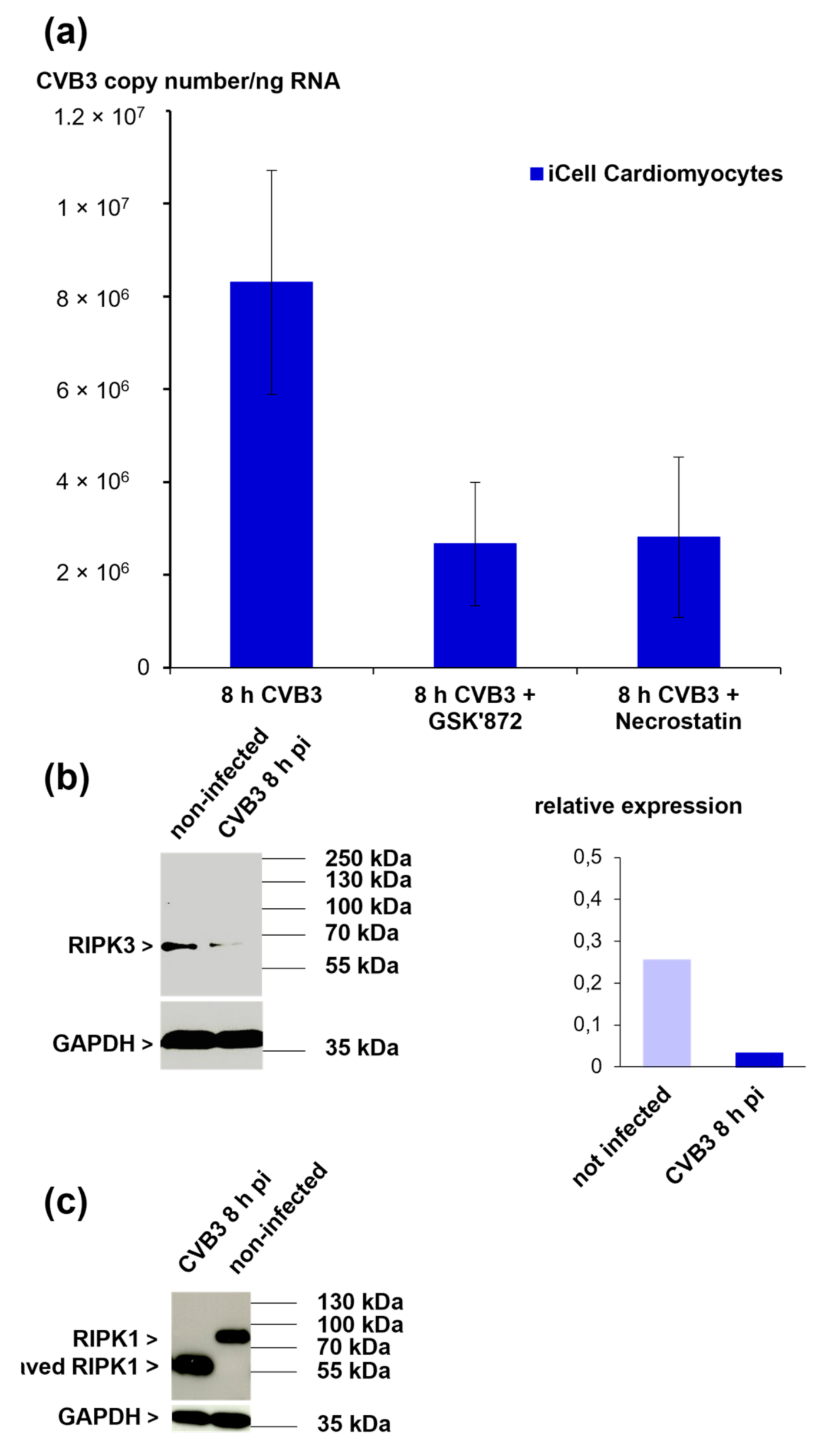
3.7. The Role of RIPK1 and RIPK3 in CVB3-Infected iCell® Cardiomyocytes
3.8. The Viral Proteases 2Apro and 3Cpro Are Not Responsible for the Cleavage of RIPK1 in iCell® Cardiomyocytes
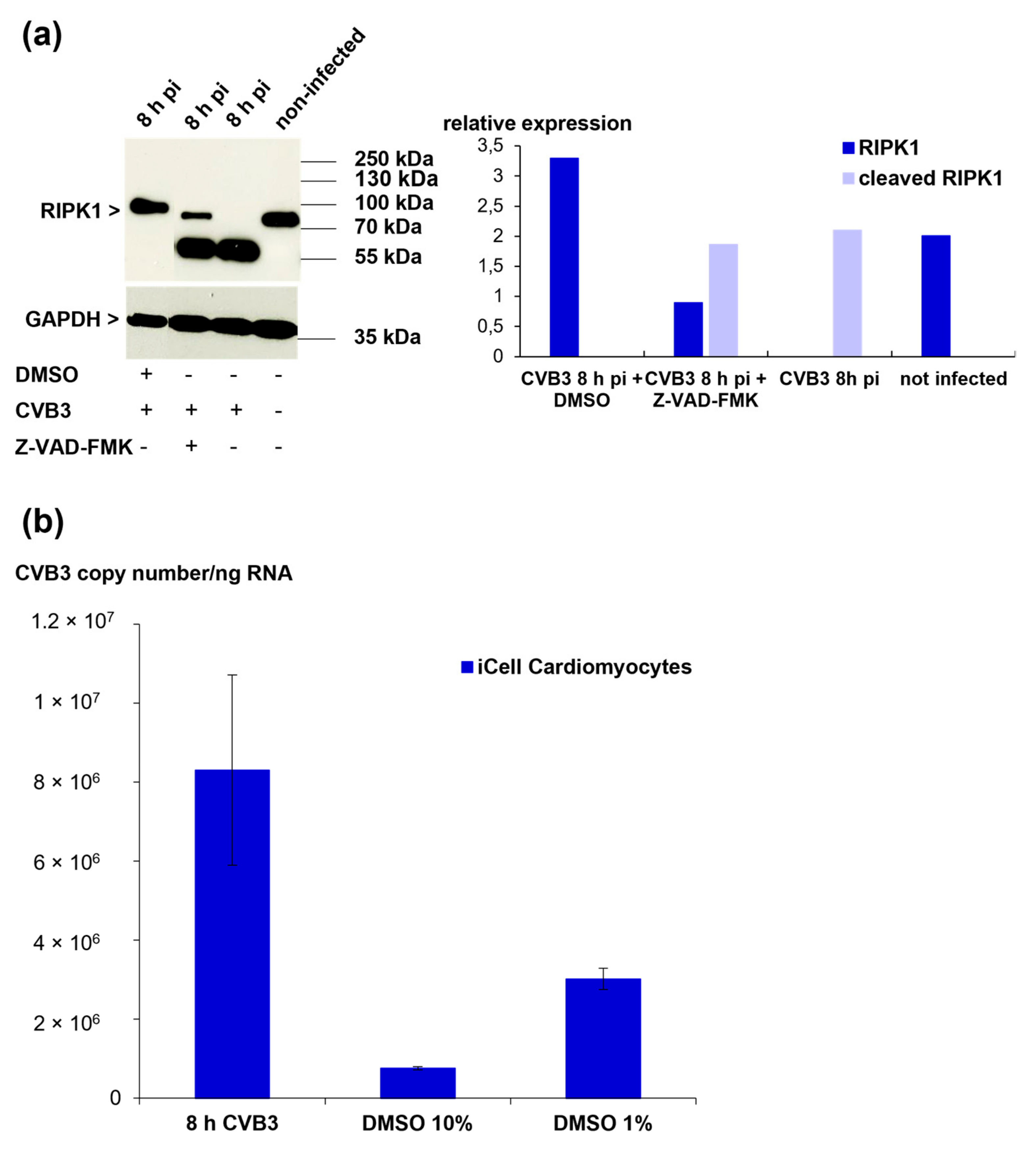
3.9. RIPK1 Cleavage Cannot Be Prevented by Protease Inhibitors but Is Inhibited by Treatment with DMSO
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Caforio, A.L.; Pankuweit, S.; Arbustini, E.; Basso, C.; Gimeno-Blanes, J.; Felix, S.B.; Fu, M.; Helio, T.; Heymans, S.; Jahns, R.; et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2013, 34, 2636–2648. [Google Scholar] [CrossRef]
- Pollack, A.; Kontorovich, A.R.; Fuster, V.; Dec, G.W. Viral myocarditis--diagnosis, treatment options, and current controversies. Nature reviews. Cardiology 2015, 12, 670–680. [Google Scholar]
- Carthy, C.M.; Granville, D.J.; Watson, K.A.; Anderson, D.R.; Wilson, J.E.; Yang, D.; Hunt, D.W.C.; McManus, B.M. Caspase Activation and Specific Cleavage of Substrates after Coxsackievirus B3-Induced Cytopathic Effect in HeLa Cells. J. Virol. 1998, 72, 7669–7675. [Google Scholar] [CrossRef] [Green Version]
- Claycomb, W.C.; Lanson, N.A.; Stallworth, B.S.; Egeland, D.B.; Delcarpio, J.B.; Bahinski, A.; Izzo, N. HL-1 cells: A cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. USA 1998, 95, 2979–2984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Wang, X.; Yu, Y.; Yu, Y.; Xie, Y.; Zou, Y.; Ge, J.; Peng, T.; Chen, R. Coxsackievirus B3-induced calpain activation facilitates the progeny virus replication via a likely mechanism related with both autophagy enhancement and apoptosis inhibition in the early phase of infection: An in vitro study in H9c2 cells. Virus Res. 2014, 179, 177–186. [Google Scholar] [CrossRef]
- Kimes, B.; Brandt, B. Properties of a clonal muscle cell line from rat heart. Exp. Cell Res. 1976, 98, 367–381. [Google Scholar] [CrossRef]
- Bozym, R.A.; Patel, K.; White, C.; Cheung, K.H.; Bergelson, J.M.; Morosky, S.A.; Coyne, C.B. Calcium signals and calpain-dependent necrosis are essential for release of coxsackievirus B from polarized intestinal epithelial cells. Mol. Biol. Cell 2011, 22, 3010–3021. [Google Scholar] [CrossRef] [Green Version]
- Saraste, A.; Arola, A.; Vuorinen, T.; Kyto, V.; Kallajoki, M.; Pulkki, K.; Voipio-Pulkki, L.M.; Hyypia, T. Cardiomyocyte apoptosis in experimental coxsackievirus B3 myocarditis. Cardiovasc. Pathol. 2003, 12, 255–262. [Google Scholar] [CrossRef]
- Nie, J.; Ta, N.; Liu, L.; Shi, G.; Kang, T.; Zheng, Z. Activation of CaMKII via ER-stress mediates coxsackievirus B3-induced cardiomyocyte apoptosis. Cell Biol. Int. 2019, 44, 488–498. [Google Scholar] [CrossRef]
- Wu, R.; Wu, T.; Li, P.; Wang, Q.; Shi, Y.; Zhan, Y.; Zhang, S.; Xia, T.; Wang, Z.; Lv, H. The protection effects of survivin in the cell model of CVB3-induced viral myocarditis. Hear. Vessel. 2020, 35, 1171–1179. [Google Scholar] [CrossRef]
- Zhou, F.; Jiang, X.; Teng, L.; Yang, J.; Ding, J.; He, C. Necroptosis may be a novel mechanism for cardiomyocyte death in acute myocarditis. Mol. Cell. Biochem. 2017, 442, 11–18. [Google Scholar] [CrossRef]
- Vercammen, D.; Beyaert, R.; Denecker, G.; Goossens, V.; Van Loo, G.; Declercq, W.; Grooten, J.; Fiers, W.; Vandenabeele, P. Inhibition of Caspases Increases the Sensitivity of L929 Cells to Necrosis Mediated by Tumor Necrosis Factor. J. Exp. Med. 1998, 187, 1477–1485. [Google Scholar] [CrossRef] [Green Version]
- Belizário, J.; Cordeiro, L.; Enns, S. Necroptotic Cell Death Signaling and Execution Pathway: Lessons from Knockout Mice. Mediat. Inflamm. 2015, 2015, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Vandenabeele, P.; Declercq, W.; Van Herreweghe, F.; Berghe, T.V. The Role of the Kinases RIP1 and RIP3 in TNF-Induced Necrosis. Sci. Signal. 2010, 3, re4. [Google Scholar] [CrossRef]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.; Wang, F.-S.; Wang, X. Mixed Lineage Kinase Domain-like Protein MLKL Causes Necrotic Membrane Disruption upon Phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef] [Green Version]
- Toldo, S.; Kannan, H.; Bussani, R.; Anzini, M.; Sonnino, C.; Sinagra, G.; Merlo, M.; Mezzaroma, E.; De Giorgio, F.; Silvestri, F.; et al. Formation of the inflammasome in acute myocarditis. Int. J. Cardiol. 2014, 171, e119–e121. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, B.; Xiong, S. Involvement of NLRP3 inflammasome in CVB3-induced viral myocarditis. Am. J. Physiol. Circ. Physiol. 2014, 307, H1438–H1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mummery, C.L. Perspectives on the Use of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes in Biomedical Research. Stem Cell Rep. 2018, 11, 1306–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Marceau, C.; Hamaguchi, R.; Burridge, P.W.; Rajarajan, K.; Churko, J.M.; Wu, H.; Sallam, K.I.; Matsa, E.; Sturzu, A.C.; et al. Human Induced Pluripotent Stem Cell–Derived Cardiomyocytes as an In Vitro Model for Coxsackievirus B3–Induced Myocarditis and Antiviral Drug Screening Platform. Circ. Res. 2014, 115, 556–566. [Google Scholar] [CrossRef]
- Klingel, K.; Hohenadl, C.; Canu, A.; Albrecht, M.; Seemann, M.; Mall, G.; Kandolf, R. Ongoing enterovirus-induced myocarditis is associated with persistent heart muscle infection: Quantitative analysis of virus replication, tissue damage, and inflammation. Proc. Natl. Acad. Sci. USA 1992, 89, 314–318. [Google Scholar] [CrossRef] [Green Version]
- Klingel, K.; Rieger, P.; Mall, G.; Selinka, H.C.; Huber, M.; Kandolf, R. Visualization of enteroviral replication in myocardial tissue by ultrastructural in situ hybridization: Identification of target cells and cytopathic effects. Lab. Investig. 1998, 78, 1227–1237. [Google Scholar]
- Harris, K.G.; Morosky, S.A.; Drummond, C.G.; Patel, M.; Kim, C.; Stolz, D.B.; Bergelson, J.M.; Cherry, S.; Coyne, C.B. RIP3 Regulates Autophagy and Promotes Coxsackievirus B3 Infection of Intestinal Epithelial Cells. Cell Host Microbe 2015, 18, 221–232. [Google Scholar] [CrossRef] [Green Version]
- Albrecht, T.; Fons, M.; Boldogh, I.; Rabson, A.S. Effects on Cells. In Medical Microbiology; Baron, S., Ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996. [Google Scholar]
- Wang, Y.; Jia, L.; Shen, J.; Wang, Y.; Fu, Z.; Su, S.-A.; Cai, Z.; Wang, J.-A.; Xiang, M. Cathepsin B aggravates coxsackievirus B3-induced myocarditis through activating the inflammasome and promoting pyroptosis. PLOS Pathog. 2018, 14, e1006872. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.-Y.; Xie, K.-X.; Wang, S.-L.; Yuan, L.-W. Inflammatory caspase-related pyroptosis: Mechanism, regulation and therapeutic potential for inflammatory bowel disease. Gastroenterol. Rep. 2018, 6, 167–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Li, X.; Li, Z.; Zhou, W.; Xing, X.; Huang, L.; Tian, L.; Chen, J.; Chen, C.; Ma, X.; Yang, Z. Overexpression of 4EBP1, p70S6K, Akt1 or Akt2 differentially promotes Coxsackievirus B3-induced apoptosis in HeLa cells. Cell Death Dis. 2013, 4, e803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zhang, J.; Chen, Z.; Yang, L.; Xing, X.; Ma, X.; Yang, Z. Both PI3K- and mTOR-signaling pathways take part in CVB3-induced apoptosis of Hela cells. DNA Cell Biol. 2013, 32, 359–370. [Google Scholar] [CrossRef]
- Błyszczuk, P. Myocarditis in Humans and in Experimental Animal Models. Front. Cardiovasc. Med. 2019, 6, 64. [Google Scholar] [CrossRef] [Green Version]
- Krausslich, H.G.; Wimmer, E. Viral proteinases. Annu. Rev. Biochem. 1988, 57, 701–754. [Google Scholar] [CrossRef]
- Lawson, M.A.; Semler, B.L. Picornavirus protein processing--enzymes, substrates, and genetic regulation. Curr. Top. Microbiol. Immunol. 1990, 161, 49–87. [Google Scholar]
- Blom, N.S.; Hansen, J.; Brunak, S.; Blaas, D. Cleavage site analysis in picornaviral polyproteins: Discovering cellular targets by neural networks. Protein Sci. 1996, 5, 2203–2216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinkert, S.; Klingel, K.; Lindig, V.; Dorner, A.; Zeichhardt, H.; Spiller, O.B.; Fechner, H. Virus-Host Coevolution in a Persistently Coxsackievirus B3-Infected Cardiomyocyte Cell Line. J. Virol. 2011, 85, 13409–13419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feuer, R.; Mena, I.; Pagarigan, R.; Slifka, M.K.; Whitton, J.L. Cell Cycle Status Affects Coxsackievirus Replication, Persistence, and Reactivation In Vitro. J. Virol. 2002, 76, 4430–4440. [Google Scholar] [CrossRef] [Green Version]
- Feuer, R.; Mena, I.; Pagarigan, R.R.; Hassett, D.E.; Whitton, J.L. Coxsackievirus replication and the cell cycle: A potential regulatory mechanism for viral persistence/latency. Med Microbiol. Immunol. 2004, 193, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.P.; Zhao, W.; Wang, H.T.; Wu, K.Y.; Li, T.; Guo, X.K.; Tong, S.Q. Coxsackievirus B3-induced apoptosis and Caspase-3. Cell Res. 2003, 13, 203–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Chen, X.; Gueydan, C.; Han, J. Plasma membrane changes during programmed cell deaths. Cell Res. 2017, 28, 9–21. [Google Scholar] [CrossRef]
- Mohamud, Y.; Shi, J.; Tang, H.; Xiang, P.; Xue, Y.C.; Liu, H.; Ng, C.S.; Luo, H. Coxsackievirus infection induces a non-canonical autophagy independent of the ULK and PI3K complexes. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Dunai, Z.A.; Imre, G.; Barna, G.; Korcsmáros, T.; Petak, I.; Bauer, P.I.; Mihalik, R. Staurosporine Induces Necroptotic Cell Death under Caspase-Compromised Conditions in U937 Cells. PLoS ONE 2012, 7, e41945. [Google Scholar] [CrossRef] [Green Version]
- Van Raam, B.J.; Ehrnhöfer, D.; Hayden, M.; Salvesen, G.S. Intrinsic cleavage of receptor-interacting protein kinase-1 by caspase-6. Cell Death Differ. 2012, 20, 86–96. [Google Scholar] [CrossRef]
- Lin, Y.; Devin, A.; Rodriguez, Y.; Liu, Z.-G. Cleavage of the death domain kinase RIP by Caspase-8 prompts TNF-induced apoptosis. Genes Dev. 1999, 13, 2514–2526. [Google Scholar] [CrossRef]
- Aguilar, J.; Roy, D.; Ghazal, P.; Wagner, E. Dimethyl sulfoxide blocks herpes simplex virus-1 productive infection in vitro acting at different stages with positive cooperativity. Application of micro-array analysis. BMC Infect. Dis. 2002, 2, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viza, D.; Aranda-Anzaldo, A.; Ablashi, D.; Kramarsky, B. HHV-6 inhibition by two polar compounds. Antivir. Res. 1992, 18, 27–38. [Google Scholar] [CrossRef]
- Chan, J.C.; Gadebusch, H.H. Virucidal properties of dimethyl sulfoxide. Appl. Microbiol. 1968, 16, 1625–1626. [Google Scholar] [CrossRef]
- Yun, S.H.; Shin, H.H.; Ju, E.S.; Lee, Y.J.; Lim, B.K.; Jeon, E.S. Inhibition of RNA Helicase Activity Prevents Coxsackievirus B3-Induced Myocarditis in Human iPS Cardiomyocytes. Int. J. Mol. Sci. 2020, 21, 3041. [Google Scholar] [CrossRef] [PubMed]
- Peischard, S.; Ho, H.T.; Theiss, C.; Strutz-Seebohm, N.; Seebohm, G. A Kidnapping Story: How Coxsackievirus B3 and Its Host Cell Interact. Cell. Physiol. Biochem. 2019, 53, 121–140. [Google Scholar]
- Kaiser, W.J.; Sridharan, H.; Huang, C.; Mandal, P.; Upton, J.W.; Gough, P.J.; Sehon, C.A.; Marquis, R.W.; Bertin, J.; Mocarski, E.S. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 2013, 288, 31268–31279. [Google Scholar] [CrossRef] [Green Version]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’en, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kraft, L.; Sauter, M.; Seebohm, G.; Klingel, K. In Vitro Model Systems of Coxsackievirus B3-Induced Myocarditis: Comparison of Commonly Used Cell Lines and Characterization of CVB3-Infected iCell® Cardiomyocytes. Viruses 2021, 13, 1835. https://doi.org/10.3390/v13091835
Kraft L, Sauter M, Seebohm G, Klingel K. In Vitro Model Systems of Coxsackievirus B3-Induced Myocarditis: Comparison of Commonly Used Cell Lines and Characterization of CVB3-Infected iCell® Cardiomyocytes. Viruses. 2021; 13(9):1835. https://doi.org/10.3390/v13091835
Chicago/Turabian StyleKraft, Lisa, Martina Sauter, Guiscard Seebohm, and Karin Klingel. 2021. "In Vitro Model Systems of Coxsackievirus B3-Induced Myocarditis: Comparison of Commonly Used Cell Lines and Characterization of CVB3-Infected iCell® Cardiomyocytes" Viruses 13, no. 9: 1835. https://doi.org/10.3390/v13091835
APA StyleKraft, L., Sauter, M., Seebohm, G., & Klingel, K. (2021). In Vitro Model Systems of Coxsackievirus B3-Induced Myocarditis: Comparison of Commonly Used Cell Lines and Characterization of CVB3-Infected iCell® Cardiomyocytes. Viruses, 13(9), 1835. https://doi.org/10.3390/v13091835